
Abstract
Background
Triple-negative breast cancer (TNBC) remains a difficult breast cancer subtype to treat as it exhibits a particularly aggressive behavior. The dysregulation of distinct signaling pathways underlies this aggressive behavior, with an overactivation of MAP kinase interacting kinases (MNKs) promoting tumor cell behavior, and driving proliferation and migration. Therefore, MNK1 is an excellent target to impair the progression of TNBC and indeed, an MNK1-specific aptamer has proved to be efficient in inhibiting TBNC cell proliferation in vitro. Although polyethyleneimine-coated iron oxide nanoparticles (PEI–IONPs) have been used as transfection and immunomodulating agents, no study has yet addressed the benefits of using these nanoparticles as a magnetic carrier for the delivery of a functional aptamer.
Results
Here, we tested the antitumor effect of a PEI–IONP complexed to the functional MNK1b-specific aptamer in vitro and in vivo. We demonstrated that these apMNKQ2@PEI–IONP nanoconjugates delivered three times more apMNKQ2 to MDA-MB-231 cells than the aptamer alone, and that this enhanced intracellular delivery of the aptamer had consequences for MNK1 signaling, reducing the amount of MNK1 and its target the phospho(Ser209)-eukaryotic initiation factor 4E (eIF4E). As a result, a synergistic effect of the apMNKQ2 and PEI–IONPs was observed that inhibited MDA-MB-231 cell migration, probably in association with an increase in the serum and glucocorticoid-regulated kinase-1 (SGK1) and the phospho(Thr346)-N-myc down-regulated gene 1 (NDRG1). However, intravenous administration of the apMNKQ2 alone did not significantly impair tumor growth in vivo, whereas the PEI–IONP alone did significantly inhibit tumor growth. Significantly, tumor growth was not inhibited when the apMNKQ2@PEI–IONP nanocomplex was administered, possibly due to fewer IONPs accumulating in the tumor. This apMNKQ2-induced reversion of the intrinsic antitumor effect of the PEI–IONPs was abolished when an external magnetic field was applied at the tumor site, promoting IONP accumulation.
Conclusions
Electrostatic conjugation of the apMNKQ2 aptamer with PEI–IONPs impedes the accumulation of the latter in tumors, which appears to be necessary for PEI–IONPs to exert their antitumor activity.
Graphical Abstract

Similar content being viewed by others
Background
Breast cancer (BC) is widespread among women, accounting for almost one-third of diagnosed cancer cases in females worldwide (Łukasiewicz et al. 2021). As for most cancers, 70–90% of all BC-related deaths are related to the appearance of metastasis(Frisk et al. 2012; Pelletier et al. 2008; Dillekås et al. 2019), which complicates BC management. Another difficulty when managing BC is the complex genetic and epigenetic background underlying the development of these tumors and the molecular pathways involved in BC progression (Perou et al. 2000). The expression of hormone receptors (the estrogen and progesterone receptor) and epidermal growth factor receptor (HER2) expression define different BC subtypes, with the triple-negative being the most difficult to treat (Gou et al. 2022). Triple-negative breast cancer (TNBC) is the BC subtype most resistant to current treatments due to its propensity to metastasize and its molecular heterogeneity (Schmid et al. 2020), accounting for 15–20% of all BC cases (Perou et al. 2000).
Dysregulation of a variety of molecular signaling pathways has been documented in BC, contributing to its development and resistance to therapy. For instance, the phosphatidylinositol-3-kinase (PI3K), protein kinase B (PKB or AKT) and the mammalian target of the rapamycin (mTOR) pathways have been linked to cancer cell proliferation, drug resistance, angiogenesis and metabolism(Pugazhenthi et al. 2000; Liu et al. 2020; Ortega et al. 2020). The Janus kinase (JAK), and the signal transducer and activator of the transcription 3 (STAT3) pathways also modulate cancer cell growth and death (Manore et al. 2022; Banerjee and Resat 2016). Moreover, the extracellular signal-regulated kinase (ERK) and the mitogen-activated protein kinase (MAPK) pathways transduce an external signal from mitogens into several signaling events, ultimately driving cell division, proliferation, survival and differentiation(Wee and Wang 2017; Brock et al. 2021).
Recently, the MAPK-interacting serine/threonine-protein kinases 1 and 2 (MNK1/2) were shown to be essential kinases in several critical signaling pathways. MNK1/2 overexpression or phosphorylation positively correlates with the progression and prognosis of several cancers, including TNBC (Pinto-Díez et al. 2018; Hou et al. 2017; Fan et al. 2017; Zheng et al. 2014). Indeed, MNK1/2 trigger oncogenesis by promoting myeloid cell leukemia 1(MCL-1) (Wendel et al. 2007), vascular endothelial growth factor (VEGF)(Korneeva et al. 2010), metalloproteinase-3 (MMP3) (Furic et al. 2010) or cyclin D1 expression(Wheater et al. 2010), regulating the production of pro-inflammatory cytokines [e.g., tumor necrosis factor-α (TNFα), Chemokine (C–C motif) ligand 5 (CCL5, also known as RANTES), and inteleukin-17 (IL-17)] (Joshi and Platanias 2012), or they can even mediate resistance to drugs that combat cancers(Adesso et al. 2013; Geter et al. 2017). More recently, MNK1/2 proved to be involved in the migration, invasion and metastasis of distinct cancer cells (Wang et al. 2018; Zhan et al. 2017; Beggs et al. 2015). MNK1/2 are derived from two genes in mammals, and each gene transcript is alternatively spliced in humans to produce two proteins. While the shorter variant (MNK1b/2b) lacks an MAPK-binding domain and has stronger basal kinase activity, the longer isoforms (MNK1a/MNK2a) do contain an MAPK-binding domain, and therefore, their activity is tightly regulated by upstream kinases (O’Loghlen et al. 2004; Scheper et al. 2003, 2001). Indeed, the longer MNK1a/2a are key intermediates in several cell signaling pathways, and they are downstream targets of the ERK and p38 MAP kinases.
The influence of MNK1/2 on cell signaling seems to depend on how these kinases regulate mRNA translation through the phosphorylation of the cap-binding protein eIF4E (eukaryotic translation initiation factor 4E) (Waskiewicz et al. 1997; Scheper and Proud 2002). However, Ser209 eIF4E phosphorylation by MNK1/2 appears irrelevant for cap-dependent translation or general protein synthesis, as evident in MNK1/2-deficient mice, suggesting other signaling outputs might be involved in the effect of MNK on tumor cell progression (Ueda et al. 2004). MNK1/2-mediated phosphorylation of eIF4E appears to be essential for the translation of mRNAs containing 5’ untranslated terminal regions (UTRs), which adopt extensive secondary structures (Koromilas et al. 1992). Nonetheless, eIF4E Ser209 phosphorylation by MNK1/2 can regulate mRNA translation and the nuclear export of mRNAs through binding to an eIF4E-sensitive element (4E-SE) in the 3’UTRs (Culjkovic-Kraljacic et al. 2012; Culjkovic et al. 2005). Thus, MNK1/2, and their longer variants in particular, are attractive targets for cancer therapies, which has promoted recent efforts into developing MNK1/2 inhibitors (Xu et al. 2022).
Since numerous experiments have indicated that phosphorylation of eIF4E, the main MNK1/2 target, is insufficient to explain the full effects of MNK1/2 on tumor progression, it is only logical to think that other downstream signaling pathways influence these events. For example, MNK1 can repress the expression of N-myc down-regulated gene 1 (NDRG1) transcripts, such that its expression is enhanced in MNK1 inhibitor-treated MDA-MB-231 cells (Tian et al. 2017). Because NDRG1 is a potent tumor suppressor and a negative regulator of metastasis (Lian et al. 2022; Wei et al. 2021; Chekmarev et al. 2382), the pro-metastatic influence of MNK1 has been in part associated with the down-regulation of NDRG1. The serum and glucocorticoid-regulated kinase-1 (SGK1) is another factor that appears to act upstream of MNK1 and NDRG1 to negatively regulate the migration/invasion of human BC cells through two independent pathways: either by repressing MNK1 through its phosphorylation, or by phosphorylating and activating NDRG1 (Tian et al. 2017). Subsequently, SGK1 was seen to also induce NDRG1 expression, mediated by the activating protein-1 (AP1) transcription factor (Godbole et al. 2018). Therefore, MNK1, SGK1 and NDRG1 are interconnected in regulating BC cell motility and thus, their activation/expression can be affected individually if one is targeted therapeutically. Then, we assessed their activation status as a readout for MNK1 inhibition.
Aptamers are short, single-stranded RNA or DNA (ssRNA or ssDNA) oligonucleotides that recognize specific molecules such as proteins, peptides and carbohydrates (Gao et al. 2022; Jin et al. 2017; Cruz-Hernández et al. 1056). The great versatility of aptamers is derived from their conformational diversity, folding into helices and single-stranded loops (Xiao et al. 2021). Selected from an extensive oligonucleotide library through sequential evolution of ligands by exponential enrichment (SELEX), aptamers may not only contain a moiety targeting cell membrane-anchored targets but also, aptamers may be intracellular species that also achieve functional targeting (Gao et al. 2022; Xiao et al. 2021). However, these aptamers may require a carrier to protect them from harsh environments (e.g., the bloodstream or tumor microenvironment), where single-stranded oligonucleotides may suffer enzymatic and chemical degradation, although this may also facilitate the aptamer’s entry into intracellular compartments.
Iron oxide nanoparticles (IONPs) are proving to be reliable platforms as non-viral substitutes for the delivery of genes, or any DNA/RNA molecule, to cells. Compared to other inorganic NPs, IONPs exhibit various advantages that include their magnetic responsiveness, ease of production, variety of simple surface modifications and good biocompatibility (Luther et al. 2020; Hola et al. 2015). The magnetic responsiveness of IONPs allows them to be physically targeted to sites of interest, facilitating correct cargo delivery. Indeed, in vitro (Liu et al. 2022; Cao et al. 1692) and in vivo (Gong et al. 2020; Sizikov et al. 2021) transfection has been accomplished by applying a magnetic field to the cells or tissue of interest. Various polymers can be used to coat IONPs, providing both stability and the capacity to bind RNA/DNA. Polyethyleneimine (PEI) is a well-known polycation with good transfection efficiency (He et al. 2012) and it has been exploited as a promising platform for gene delivery in recent years (Gong et al. 2020; Hoang et al. 2015; Chen et al. 2012). However, PEI-coated IONPs (PEI–IONPS) have intrinsic biological properties and they exert a variety of effects on tumor cells, e.g., inhibiting cell migration (Mulens-Arias et al. 2015a), impairing tumor angiogenesis (Mulens-Arias et al. 2019) and activating immune responses (Mulens-Arias et al. 2015b). Indeed, we have previously described the underlying molecular pathways involved in PEI–IONP-induced anti-tumor effects on pancreatic tumor cells, in which we observed a profound decrease of pro-migration factors such as metalloproteinases and the increase of anti-migration/proliferation pathway microRNA-21–PTEN–PDCD4–Sprouty-1 (Mulens-Arias et al. 2015a). Therefore, PEI–IONPs must be considered an intrinsically active nanocarrier independent of any cargo they may carry.
Although the utility of IONP and aptamer conjugates as nanosensors (Wang et al. 2021; Ma et al. 2018) or photodynamic therapy-driven nanoformulations (Zhao et al. 2020) has been considered, to our knowledge IONPs have yet to be used to deliver functional aptamers to specific cells. Thus, we studied whether PEI–IONPs might be suitable carriers for the intracellular delivery of a functional DNA aptamer targeting MNK1 in BC cells. We found that these PEI–IONPs delivered the MNK1 specific aptamer to the TNBC MDA-MB-231 cell line, modulating the MNK1 signaling pathway in vitro and synergizing with the PEI–IONPs in inhibiting cell migration. However, the MNK1-specific aptamer complexed with the PEI–IONPs failed to dampen the growth of MDA-MB-231-induced tumors when administered in vivo. Surprisingly, not only was the MNK1 specific aptamer ineffective but also, it abolished the intrinsic antitumor effect of the PEI–IONPs. This study highlights the need to better understand the suitability of IONPs to deliver cargo, as it can be counterproductive and lead to undesired behavior in vivo.
Materials and methods
Reagents
PEI (25 kDa, 408727), Zonyl FSA (65530-69-0), FeCl2.4H2O (44939), and FeCl3.6H2O (236489) were purchased from Sigma-Aldrich. Antibodies against the following proteins were used to probe the western blots: phospho-MNK1 (Thr197/202: Cell Signaling #2111S); MNK1 (Cell Signaling #2195S); phospho-eIF4E (ser209: Cell Signaling #9741S); eIF4E (Cell Signaling #2067S); phospho-NDRG1 (Thr346, D98G11 XP® Rabbit mAb: Cell Signaling #5482S); NDRG1 (D8G9, XP® Rabbit mAb: Cell Signaling #9485S); SGK1 (D27C11 Rabbit mAb: Cell Signaling #12103S); phospho-SGK1 (Ser78, D36D11 Rabbit mAb: Cell Signaling #5599S). Horseradish peroxidase (HRP)-conjugated goat anti-rabbit, goat anti-mouse and rabbit anti-goat (all from DAKO) were used as secondary antibodies. The apMNKQ2 aptamer specific to MNK1b was obtained from IBA Life Sciences (Goettingen, Germany).
PEI–IONP synthesis
PEI–IONPs were synthesized as reported previously (Mulens-Arias et al. 2015a, b), with some minor modifications. Briefly, a mixture of Fe2+ and Fe3+ chlorides was mixed in an oxygen-free environment with a PEI (25 kDa) and Zonyl FSA solution in NH4OH at 4 °C, stirring and heating (90 °C, 2 h) the mixture. The product was washed three times with distilled water, dialyzed for at least 72 h against distilled ultra-pure water and filtered through a 0.4 μm pore-size filter. For chemical and physical characterization, images were obtained on a 200-keV JEOL-2000 FXII transmission electron microscope. The hydrodynamic diameter and zeta potential were measured by dynamic light scanning (DLS: Malvern NanoZS), on PEI–IONPs were diluted to ∼50 μg/mL in distilled water. Inductively coupled plasma–optical emission spectrometry (ICP–OES) was used to determine the Fe concentration in the PEI–IONP solutions. The colloidal properties of the samples (NPs in different media) were studied in a Zetasizer Nano S apparatus (Malvern Instruments) and their electrophoretic mobility was measured as a function of pH at 25 ºC, using 10−2 M KNO3 as an electrolyte, and HNO3 and KOH to change the pH of the suspensions.
Cell internalization
MDA-MB-231 cells (ATCC, HTB-26) were seeded (5 × 105) and cultured for 24 h in DMEM supplemented with 10% fetal bovine serum (FBS), l-glutamine and penicillin/streptomycin, before adding AlexaFluor488-apMNKQ2 (1 μM) or the AlexaFluor488-apMNKQ2@PEI–IONP conjugate [(apMNKQ2) 0.5 μM or 1 μM]. The cells were then cultured for 24 h, washed, detached and analyzed by flow cytometry (FC500: Beckman-Coulter).
Iron content
MDA-MB-231, RAW264.7 and SVEC4-10 cells were seeded in a 6-well plate at a density of 1 × 105 cells per well, and cultured for 24 h at 37 °C in DMEM supplemented with 10% FBS, l-glutamine and penicillin/streptomycin. The cells were then incubated with the IONPs (64 μM) or the apMNKQ2@PEI–IONP nanoconjugate (1 μM apMNKQ2) in the same culture conditions for 24 h. Subsequently, the cells were washed three times with phosphate buffer saline (PBS) to remove the non-internalized IONPs, harvested and counted in a Neubauer chamber. The samples were digested in HNO3 (1 mL) for 1 h at 90 °C and the amount of iron per cell was measured by ICP–OES (Perkin Elmer-2400).
Cytotoxicity
MDA-MB-231 cells (2 × 104) were cultured for up to 24 h in 96-well plates with the aptamer (apMNKQ2), PEI–IONPs or apMNKQ2@PEI–IONP complex in DMEM supplemented with 10% FBS, l-glutamine and penicillin/streptomycin. For the last 4 h, the MTT reagent was added (1/10 final dilution: MTT Cell Viability Reagent, Roche 11465007001), after which an equal volume of lysis buffer (10% sodium dodecyl sulfate, 0.01 M HCl) was added and the plates were incubated overnight. Absorbance was measured at 596 nm with a 655 nm wavelength as reference. The absorbance of complete DMEM incubated with MTT reagent was used as a background control and cell viability was calculated according to the formula:
Viability (%) = [A535 nm IONP-treated−A655 nm IONP-treated]/[A535 nm Untreated−A655 nm Untreated]*100%
ELISA
Secreted human TIMP-1 (tissue inhibitor of metalloproteinase 1) was quantified by ELISA (ELISA DuoSet, DY970-05, R&D Systems) in triplicate samples, according to manufacturer’s instructions.
Western blotting
After treatment, the cells were washed with PBS, scraped and lysed in a buffer containing 1% Triton X-100, and the protease and phosphatase inhibitors leupeptin (1 mg/mL), NaF (5 nm), sodium orthovanadate (1 mm), PMSF (1 mm), EDTA (1 mm), aprotinin (1 μg/mL) and okadaic acid (2 nm). Equal amounts of protein lysates (20 μg/well) were loaded onto gels, resolved by SDS–PAGE and then transferred to 0.2 μm PVDF membranes that were blocked for 30 min at room temperature with Tris-buffered saline (TBS)/0.05% Tween 20/5% bovine serum albumin (BSA). The membranes were then probed overnight with primary antibodies diluted in TBS/0.05% Tween 20/1% BSA, washed and incubated for 1 h at room temperature with HRP-conjugated secondary antibodies diluted 1/1000 in TBS/0.05% Tween 20/1% BSA. After extensive washing with TBS/0.05% Tween 20, the protein bands were visualized by ECL in Western Blotting Detection Reagent (RPN2106, Amersham) for up to 30 min. We used ImageJ software (US National Institutes of Health) to quantify the band intensity, in all cases using β-actin as a loading control.
MDA-MB-231 cell migration
Cells (1 × 104) were incubated on the 8 μm-pore inserts in cell culture medium with 2% FBS, alone or containing the apMNKQ2, the PEI–IONP or the nanoconjugates at an [apMNKQ2] of 2 or 4 μM (or equivalent PEI–IONP concentrations). The inserts were immersed in medium supplemented with 10% FBS for 24 h to allow the cells to migrate and they were then transferred to 4% paraformaldehyde (PFA) in PBS for 20 min after carefully removing the cells attached to the inner part of the bottom chamber with a cotton swap. The cells on the outer part of the bottom chamber were stained with crystal blue dye, and the number of cells per 20 × micrograph (ten images) was quantified. The proportion of migrating cells was calculated by normalization to the untreated cells.
qPCR-RT
RNA was extracted from the cells using the PureLink RNA Mini Kit (12183018A, Applied Biosystems) and cDNA was produced from the RNA (30 ng/sample) with the MultiScribe reverse transcription-based kit (4374966, High Capacity cDNA Reverse Transcription kit, Applied Biosystems). A two-step RT-PCR program (95 °C for 15 s, 60 °C for 60 s, 40 cycles) was used to amplify the relevant genes using specific primers (listed in Additional file 1: Table S1: all from Sigma–Aldrich), and gene expression was quantified according to the 2ΔΔCt method, using β-actin mRNA expression as a control for normalization.
Animal experimentation
Six-week-old female nude mice (Hsd, Athymic Nude-Foxn1nu: Envigo) were maintained at the CNB animal facility. All animal studies were approved by the Ethics in Animal Experimentation Committee at the National Center for Biotechnology (CEEA–CNB), the Spanish Scientific Research Council (CSIC) Ethics Committee, and by the Division of Animal Protection of the Comunidad Autómoma de Madrid (CAM, PROEX 185/19), and they were carried out in compliance with national and European Union legislation (Directive 2010/63/EU of the European Parliament and of the Council of 22 September 2010 on the protection of animals used for scientific purposes). MDA-MB-231 cells (1 × 106) were injected subcutaneously into the right flank of the mice and once the tumors formed had reached ~ 200 mm3, the mice were randomized into six groups and administered the corresponding treatment (twice weekly, up to five doses, 100 μL, by retro-orbital injection): Saline, n = 5; PEI–IONP (~ 0.3 mg/kg), n = 4; PEI–IONP (~ 0.3 mg/kg) + magnet, n = 6; apMNKQ2 (1 mg/kg), n = 7; apMNKQ2–IONP (aptamer dose, 1 mg/kg), n = 7; apMNKQ2–IONP (aptamer dose, 1 mg/kg) + magnet, n = 6. The tumor volume was monitored with a caliper and calculated as follows: V = (xy2)/2, where x is the longest and y is the shortest of the perpendicular diameters. The mice were sacrificed 18 days after the first administration, and the tumor was preserved to determine its iron content and for histological assessment. Blood was withdrawn before sacrifice from 1 to 3 mice per group for systemic toxicity analysis.
Histological studies
Tumors were snap-frozen in tissue-freezing medium (O.C.T.) and frozen Sects. (6 µm) were acetone-fixed and stained with anti-CD31 (vascular density) or anti-F4/80 (macrophage infiltration) antibody. Immunohistochemistry images were acquired on an Olympus-IX70-Inverted microscope (magnification × 10). 10 tumor sections were imaged per tumor and all tumor sections of from the same treatment group were pooled and compared. CD31-positive and F4/80-positive region in each image was measured using ImageJ software.
Statistical analysis
The data were analyzed and displayed with GraphPad Prism and FlowJo software. The data are presented as the mean ± SEM, and the number of samples and independent experiments are indicated in each figure caption. The data were analyzed with a one-tailed Mann–Whitney U test under the assumption of a non-Gaussian distribution (non-parametric test), with a 95% confidence and *p < 0.05. A two-way ANOVA was performed to compare tumor growth.
Results and discussion
PEI–IONP synthesis, apMNKQ2 conjugation and chemical–physical characterization
Due to the strong negative charge of the oligonucleotides, a positively charged carrier will likely deliver the oligonucleotide to the cells correctly. PEI has been proposed as a suitable coating for IONPs and its ability to conjugate nucleic acids has been tested, from plasmids (Thomas and Klibanov 2003; Huang et al. 2010) to siRNAs (Mohammad Gholinia Sarpoli et al. 2022), protecting the nucleic acid from a harsh environment. We previously synthesized and characterized PEI–IONPs that combine the transfection efficiency of the polycation and the possibility of magnetically targeting the nanocarriers to the desired site of action (Hoang et al. 2015; Mulens-Arias et al. 2015a, b). Since PEI–IONPs can easily complex aptamers, we synthesized PEI–IONPs in an oxygen-free environment according to our existing protocol, and based on reducing Fe2+ and Fe3+ in an alkaline milieu (Fig. 1a) (Mulens-Arias et al. 2015b). We introduced 25 kDa PEI into the reaction and provided the IONPs synthesized with a stabilizer and with Zonyl FSA as a surfactant. As expected, the PEI–IONPs had a mean iron oxide core diameter of 11.05 ± 1.97 nm according to TEM imaging (Fig. 1a). The z-potential ~ + 23 mV in the aptamer selection buffer (20 mM Tris–HCl [pH 7.4], 150 mM NaCl, 1 mM KCl) indicated the presence of the polycation at the IONP surface (Fig. 1c, left) and further characterization revealed the maghemite nature of the PEI–IONP, as indicated elsewhere (Mulens-Arias et al. 2015b).

Synthesis of the PEI–IONPs and the apMNKQ2@PEI–IONP complexes. a TEM images of the IONPs synthesized (upper) and a histogram depicting the PEI–IONP diameter distribution calculated from the TEM images (bottom). Scale bar: 40 nm. b apMNKQ2 concentration remaining in solution after centrifugation (blue dots) and the calculated aptamer loading (brown dots). The Brown dashed line depicts the best-fit curve with a KD ~ 22.01 μM (results of an experiment performed twice in triplicate). c Z-potential (left) and hydrodynamic diameter (right) of the PEI–IONPs and apMNKQ2@PEI–IONP complexes in standard reaction buffer or in 10% mouse serum in standard reaction buffer. d Cumulative aptamer release in standard buffer over 24 h from an initial concentration of aptamer in the complex of 250 ng/μl (results of an experiment performed in triplicate)
Given the strong positive surface charge of the PEI–IONPs, we proceeded to complex the apMNKQ2 DNA aptamer to these NPs previously demonstrated to exert an anti-proliferative effect on the human MDA-MB-231 BC cell line (García-Recio et al. 2016), producing a nanocomplex for TNBC treatment in vitro and in vivo. The apMNKQ2 recognizes both splice variants of MNK1, MNK1a and MNK1b, and it may, therefore, affect both kinases irrespective of the upstream factors (García-Recio et al. 2016). We then incubated AlexaFluor488-conjugated apMNKQ2 (2 μM) with an increasing concentration of PEI–IONPs to track the aptamer in vitro and to determine the optimal [Fe]:[apMNKQ2] (μM:μM) ratio to ensure that all the aptamer is complexed (Fig. 1b). After a 1 h incubation, the reaction tubes were centrifuged and the apMNKQ2 concentration in the supernatant was determined in a nanodrop. The apMNKQ2 remaining decreased as the PEI–IONP concentration increased (indicated as the equivalent [Fe], Fig. 1b) and at a [Fe]:[apMNKQ2] ratio of 64 virtually no aptamer remained unbound, suggesting that all the aptamer had complexed with the NPs.
We detected a slight decrease in the PEI–IONP z-potential from + 23 to + 21.8 mV when complexed with the aptamer in buffer (20 mmol/L Tris–HCl [pH 7.4], 1 mmol/L MgCl2, 150 mmol/L NaCl and 5 mmol/L KCl: Fig. 1c, left). However, a more pronounced increase in the hydrodynamic diameter was observed, from 191 nm (PEI–IONP) to 2567.77 nm (apMNKQ2@PEI–IONP), indicating complex formation in the same buffer (Fig. 1c, right). To further examine the stability of the complex we performed an in vitro release assay over 24 h in the reaction buffer. The apMNKQ2@PEI–IONP nanoconjugates barely released a cumulative ~ 1.15% of the complexed aptamer, highlighting the stability of the complex in solution (Fig. 1d). Thus, the PEI–IONPs can complex all the aptamer present in the reaction, at a [Fe]:[apMNKQ2] of 64 and with a calculated dissociation constant ~ 22.01 μM. This [Fe]:[apMNKQ2] ratio was applied to produce the apMNKQ2@PEI–IONP complexes used in all the ensuing in vitro and in vivo experiments.
The relevance of PEI-coated magnetic IONPs in complexing DNA/RNA has been studied extensively over the past two decades. For instance, in an attempt to find the best magnetic NPs to isolate the DNA content from a sample, it was found that a PEI coating yielded the most efficient DNA extraction of calf thymus DNA (Szymczyk et al. 2022). This ability of PEI to complex DNA/RNA has become equally valuable for non-viral gene transfection in vitro or in vivo. That was the case when complexing a neuregulin-1 (NRG1) encoding DNA plasmid with PEI–IONPs to magnetically transfect adipose-derived stem cells, for which only a 15 min incubation and a nitrogen/phosphorous ratio of 8 was needed to obtain the nanocomplex (Cheng et al. 2022). Furthermore, PEI–IONPs have been used as a vaccine core to deliver malaria DNA in vitro and in vivo (Al-Deen et al. 2017). These are only a few examples, yet to our knowledge, this is the first time a PEI–IONP has been complexed with an aptamer endowed with functional activity.
PEI–IONPs deliver the apMNKQ2 intracellularly
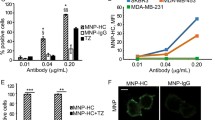
Having demonstrated that PEI–IONPs can complex the apMNKQ2, we determined whether these nanoconjugates adequately delivered the aptamer intracellularly. As apMNKQ2 exhibited antitumoral potential against human TNBC MDA-MB-231 cells in vitro (García-Recio et al. 2016), we tested the internalization and functional activity of our nanocomplex in these cells. Based on flow cytometry, when MDA-MB-231 cells (5 × 105) were incubated for 24 h with AlexaFluor488–apMNKQ2@PEI–IONPs [(apMNKQ2) = 1 μM], > threefold more of the nanocomplex was internalized by the cells than the equivalent fluorescent aptamer alone (Fig. 2a). Even when cells were treated with an equivalent of 0.5 μM apMNKQ2 complexed to PEI–IONPs, > twofold more internalization was still detected than with the aptamer alone, indicating that PEI–IONPs facilitate entry of the aptamer into the cells.

ApMNKQ2@PEI–IONP internalization and functional activity in MDA-MB-231 cells. a Mean fluorescent intensity of AlexaFluor488–apMNKQ2-labelled MDA-MB-231 cells incubated with different formulations for 24 h. b Cell viability was measured after 24 h-treatment with apMNKQ2@PEI–IONP (green) or apMNKQ2 (red) with different apMNKQ2 concentrations using the MTT assay. The viability of cells treated with the PEI–IONPs alone (at an equivalent concentration) is shown for comparison: Mann–Whitney test, one-tailed, p < 0.05, n = 3. c MNK1 expression in MDA-MB-231 cell lysates treated for 24 h with apMNKQ2 alone or complexed with PEI–IONPs at different concentrations. The graph reflects the results of three independent experiments and above a representative western blot is shown. d Phospho(Ser209)-eIF4E expression in MDA-MB-231 cell lysates treated for 24 h with apMNKQ2 alone or complexed with PEI–IONP at different concentrations. The graph reflects the results of three independent experiments and a representative western blot is shown above
Furthermore, MDA-MB-231 cells treated with 1 μM apMNKQ2@PEI–IONP accumulated ~ 10.94-fold more iron per cell than untreated cells (2.08 pg/cell vs. 0.19 pg/mL), which was even more than that in the cells exposed to PEI–IONPs alone (1.17 pg/cell: Additional file 1: Fig. S1a). Hence, the apMNKQ2@PEI–IONP nanoconjugate facilitates the internalization of more aptamer and PEI–IONP into the MDA-MB-231 cells. Similar findings were also observed in the macrophage-like murine RAW264.7 cell line (Additional file 1: Fig. S1b) and in murine SVEC4-10 endothelial cells (Additional file 1: Fig. S1c).
As expected, apMNKQ2 administered alone is toxic to MDA-MB-231 cells in vitro in a concentration dependent manner: 97.26% cell viability at 1 μM; 59.32% viability at 4 μM (Fig. 2b). A similar effect on viability was observed with the apMNK@PEI–IONP nanoformulation, indicating that such toxicity was mainly due to the effect of apMNKQ2 (Fig. 2b). Indeed, at a concentration of 4 μM apMNKQ2 produced 33.80% cytotoxicity in the MDA-MB-231 cells after a 48 h exposure, which did not differ significantly to the cell toxicity produced by the equivalent apMNKQ2@PEI–IONP (28.9% cell death; Additional file 1: Fig. S2). Significantly, MDA-MB-231 cell viability was considerably higher when exposed to the PEI–IONPs alone at a concentration equivalent to that of 4 μM apMNKQ2 (80.65%), as opposed to the viability in the presence of apMNKQ2 either alone (59.32%) or when complexed with the IONPs (61.24%: Fig. 2b).
We then determined whether apMNKQ2 exerts a biological effect on MNK1-associated signaling pathways. The apMNKQ2 ssDNA folds into a G-quadruplex conformation that has been previously tested for its biological activity in MDA-MB-231 cells (García-Recio et al. 2016). Treatment with the parental aptamer, apMNK2F, affected mRNA translation, proliferation and migration of these cells, although independently of the phosphorylation status of the main MNK1 target, eIF4E. Surprisingly, we detected a slight decrease in MNK1 expression in cells treated with apMNKQ2, both alone or as part of the nanocomplex, with a maximum decrease at a [apMNKQ2] = 3 μM: 0.55 in cells treated with apMNKQ2 and 0.58 in apMNKQ2@PEI–IONP-treated cells (Fig. 2c). The decrease in MNK1 was coupled with a tendency towards less phospho(Ser209)-eIF4E in the cells relative to the untreated cells, with the lowest levels reached in cells exposed to 4 μM apMNKQ2@PEI–IONP (Fig. 2d).
In characterizing the parenteral MNK1-specific aptamers, no evidence of a decrease in MNK1 was reported upon aptamer treatment (García-Recio et al. 2016). Although MNK1 aptamers reduced the activity of the MNK1 kinase and its protein translation rate in MDA-MB-231 cells, the phosphorylation level of eIF4E was not affected. This was justified by considering that the phosphorylation of this initiation factor mainly depended on MNK2 in this cell line. However, we did detect an effect of MNK1 on phospho(Ser209)-eIF4E with in the presence of apMNKQ2, which might be related to the concentration of the MNK1 aptamer used in the in vitro experiments. While the effect of up to 20 nM apMNKQ2 was addressed previously, here the biological activity of apMNKQ2 was assessed from 1 to 4 μM (50–200-fold higher). In addition, we also recently showed a similar effect after transfection with 400 nM of apMNKQ2 (Pinto-Díez et al. 2022), indicating that apMNKQ2 behaves distinctly to the parental apMNK2F.
Synergism of the PEI–IONPs and apMNKQ2 in repressing MDA-MB-231 cell migration in vitro
We previously demonstrated that PEI–IONPs can inhibit the migration and invasion of the murine Pan02 pancreatic cell line, consistently disrupting invadosome dynamics (Mulens-Arias et al. 2015a). Since MNK1 has also been implicated in tumor cell migration (Wang et al. 2018), we studied how apMNKQ2@PEI–IONPs might affect MDA-MB-231 cell migration. In a transwell migration assay, exposure to apMNKQ2 did not alter the migration rate relative to untreated cells, even at the highest aptamer concentrations: 100% cell migration when exposed to 2 μM apMNKQ2 and 91.20% at 4 μM. PEI–IONPs partially inhibited MDA-MB-231 cell migration, although stronger inhibition of migration was observed in the cells exposed to the lower PEI–IONP concentration: 76.38% cell migration at Eq. 2 μM and 91.26% for Eq. 4 μM PEI–IONP (Fig. 3a, b). Notably, when cells were incubated with the apMNKQ2@PEI–IONP nanoconjugate, we detected a synergism in the inhibition of cell migration that was slightly higher when cells were exposed to 4 μM apMNKQ2@PEI–IONP: 59.96% cell migration at 2 μM and 53.64% at 4 μM apMNKQ2@PEI–IONP (Fig. 3a, b). Thus, apMNKQ2@PEI–IONPs potentiate the negative effect of both the apMNKQ2 and the PEI–IONP on MDA-MB-231 cell migration.

ApMNKQ2@PEI–IONPs potentiate the apMNKQ2– and PEI–IONP-dependent inhibition of MDA-MB-231 cell migration. a Transwell migration assay in which cells were incubated on 8 μM pore inserts in cell culture medium containing 2% FBS alone, or the apMNKQ2, the PEI–IONPs or the nanoconjugates. The inserts were then maintained in culture medium with 10% FBS for 24 h. The figure shows representative microphotographs of stained transmigrated MDA-MB-231 cells. b Quantification of the migrating MDA-MB-231 cells in transwell migration assays: two-tailed Mann–Whitney test, **p < 0.01, ***p < 0.001. c SGK1 and phospho(Thr346)-NDRG1 in MDA-MB-231 cells in a representative western blot. d Summary of SGK1 and phospho(Thr346)-NDRG1 expression in MDA-MB-231 cell lysates treated for 24 h with the apMNKQ2@PEI–IONPs, equivalent to apMNKQ2 or PEI–IONPs. The graphs represent three independent experiments. e MT1-MMP and TIMP-2 mRNA expression in MDA-MB-231 cell lysates treated for 24 h with apMNKQ2@PEI–IONPs, equivalent to apMNKQ2 or PEI–IONPs. f TIMP-1 quantification in the supernatant of MDA-MB-231 cells treated for 24 h with apMNKQ2@PEI–IONPs, equivalent to apMNKQ2 or PEI–IONPs. The graphs represent three independent experiments
MNK1 regulates the NDRG1 suppressor of metastasis in MDA-MB-231 cells (Tian et al. 2017), which is also modulated by SGK1, thereby regulating the migratory behavior of the tumor cells (Chekmarev et al. 2021). Altogether, the axis MNK1–SGK1–NDRG1 is tightly interconnected in regulating the migration ability of breast tumor cells. The analysis of the expression/activation level of these two proteins might indicate whether the MNK1 targeting has some effects on them. Therefore, we determined the effect of apMNKQ2@PEI–IONPs on these migration/invasion-related factors. PEI–IONPs, apMNKQ2 or apMNKQ2@PEI–IONPs induced the activating Thr346 phosphorylation of NDRG1 (Fig. 3c). Exposure to 2 μM or 4 μM apMNKQ2 increased the phospho(Thr346)-NDRG1 levels by 6.33-fold and 3.12-fold, respectively. Curiously, PEI–IONP treatment also augmented the phosphorylation of NDRG1 2.83-fold and 1.88-fold at the equivalent iron concentrations, respectively. Hence, both the MNK1-specific aptamer and the PEI–IONPs activate NDRG1 signaling. However, we found that the apMNKQ2@PEI–IONP nanoconjugate induced a stronger increase in phospho(Thr346)-NDRG1 (4.37-fold) at a 4 μM apMNKQ2 equivalent, suggesting a synergistic effect. This effect, however, was not observed at 2 µM. Importantly, such synergism was also evident in the increase in SGK1 at the two apMNKQ2 concentrations tested. While apMNKQ2 and the equivalent PEI–IONPs enhanced the expression of SGK1 to a similar extent (5.45- and 5.46-fold for 2 μM apMNKQ2; and 5.6- and 5.73-fold for 4 μM apMNKQ2), apMNKQ2@PEI–IONP treatment augmented the level of SGK1 further to 7.34- and 8.41-fold, respectively (Fig. 3c). Therefore, these nanoconjugates potentiate the induction of SGK1 and its target phospho(Thr346)-NDRG1 by the MNK1 specific aptamer or that of PEI–IONPs alone, which might in turn be involved in the inhibition of the MDA-MB-231 cell migration.
To better understand the mechanism by which apMNKQ2@PEI–IONPs inhibit migration, we evaluated the expression of some genes associated with tumor cell migration/invasion: the membrane-associated metalloproteinase MT1-MMP (Niland et al. 2021) and the tissue inhibitor of metalloproteinase-2 TIMP-2(Groblewska et al. 2012). These factors can reflect the migration status of MDA-MB-231 cells as it has been demonstrated their involvement in MDA-MB-231 cell migration. Indeed, the membrane-associated metalloproteinase MT1-MMP is upregulated in MDA-MB-231 cells upon migration-induced factors, such as TNF-α, and mediate cell migration (Wolczyk et al. 2016). On the contrary, the tissue inhibitor of metalloproteinases-1 and -2 (TIMP-1 and TIMP-2) have been associated with the inhibition of the metalloproteinase-dependent and -independent cell migration (Akahane et al. 2004). Interestingly, while treatment with apMNKQ2 increased the level of MT1-MMP mRNA (2.18-, 1.72-, 1.81- and 1.06-fold with 1, 2, 3 and 4 μM apMNKQ2, respectively), a similar trend was evident for TIMP-2 mRNA expression (1.55-, 1.49-, 1.52- and 0.92-fold increase for 1, 2, 3 and 4 μM apMNKQ2, respectively), suggesting the existence of some compensatory mechanism (Fig. 3e). Importantly, this effect on both these gene transcripts appeared to be inversely correlated with the amount of aptamer, such that no significant increase was detected at 4 μM, indicating an activation of the different signaling pathways as the aptamer concentration increased. While treatment with the apMNKQ2@PEI–IONP nanoconjugate also tended to increase the expression of both these transcripts, we did not observe any obvious correlation with the doses of the aptamer applied (Fig. 3e). Indeed, the nanoconjugate increased MT1-MMP mRNA at 1 µM (1.88-fold change), 3 µM (1.48-fold change) and 4 µM (1.99-fold change) and TIMP-2 mRNA at 1 μM (1.6-fold change) and 3 μM (1.31-fold change). However, the increase in MT1-MMP and TIMP-2 mRNA seemed to be induced by the apMNKQ2 as no changes in these transcripts were seen when the cells were treated with equivalent doses of the PEI–IONPs alone.
We then assessed the release of TIMP-1by MDA-MB-231 cells, another essential MMP inhibitor (Warner et al. 2020). Like TIMP-2 mRNA, cells released more TIMP-1 into the supernatant following a 24 h treatment with 2 μM (7.33 ng/mL) and 4 μM apMNKQ2 (7.9 ng/mL), as opposed to that released by untreated cells (6.12 ng/mL: Fig. 3f). Like the mRNA transcripts, treatment with apMNKQ2@PEI–IONPs did not further enhance TIMP-1 secretion, and since an equivalent of 4 μM PEI–IONPs did not affect TIMP-1 secretion (5.89 ng/mL), the effect on TIMP-1 was attributed to apMNKQ2. Together, the analyses of MT1-MMP, TIMP-1 and TIMP-2 does not explain the synergistic effect of the apMNKQ2@PEI–IONP on MDA-MB-231 cell migration per se, leaving the increase in SGK1/phospho(Thr346)-NDRG1 signaling as the most likely cause. Nonetheless, the trend towards triggering a smaller increment as the dose of apMNKQ2 or apMNKQ2@PEI–IONPs increased was also evident for phospho(Thr346)-NDRG1, suggesting the activation of more than one signaling pathway (Additional file 1: Fig. S3).
It was recently shown that apMNKQ2 induces the expression of Occludin-1 mRNA and protein in vitro (Pinto-Díez et al. 2022), a protein involved in tight-junction stability and epithelial barrier integrity in conjunction with Claudin-3. Indeed, a decrease in occludin-1 is implicated in BC cell metastasis to bone (Martin et al. 2016) and its loss correlates positively with BC progression (Martin et al. 2010). Therefore, an increase in Occludin-1 might imply enhanced cell-to-cell contact, impairing tumor cell migration, consistent with our results.
The conjugated apMNKQ2 reverts the antitumor effect of PEI–IONPs in the MDA-MB-231 tumor model in vivo
Having demonstrated that the apMNKQ2@PEI–IONP nanoconjugate has a synergistic effect on MDA-MB-231 cells relative to its individual components (apMNKQ2 and PEI–IONPs), we assessed whether such effects were translated to an in vivo model. We generated an MDA-MB-231-based tumor model in vivo by injecting 1 × 106 of these cells into the right flank of nude mice. Once the tumors that formed reached a volume of ~ 200 mm3, the animals were injected intravenously through the retro-orbital sinuses with 100 μL of the corresponding formulation twice weekly, delivering a maximum of 5 doses (apMNKQ2 doses, 1 mg/kg; PEI–IONP equivalent, ~ 0.3 mg/kg: Additional file 1: Fig. S5). When magnetic targeting was required, the animals were anesthetized and a magnet was situated above the tumor for 1 h. The animals were sacrificed 4 days after the last dose administered, and their organs and the tumors were preserved for further analysis.
MDA-MB-231 tumors grew in a sustained manner over 18 days, increasing their volume ~ 4.47-fold (± 0.95) and while apMNKQ2 marginally slowed down tumor growth, which increased only ~ 3.35-fold (± 0.37), this did not differ significantly from the control (Fig. 4a). The PEI–IONPs did significantly dampen tumor growth, which augmented ~ 2.76-fold (± 0.60) with magnetic targeting or ~ 2.12-fold (± 0.49) without. However, when the nanoconjugate was administered, no such beneficial effects were evident and the tumors grew ~ 4.39-fold (± 0.53: Fig. 4a). Therefore, apMNKQ2 conjugation with the PEI–IONPs blocks the intrinsic antitumor activity of the latter in the in vivo model of MDA-MB-231 tumors. We did observe a significant inhibition of tumor growth when the animals administered the apMNKQ2@PEI–IONP nanoconjugate underwent magnetic targeting (~ 2.29 ± 0.29), suggesting that the accumulation of PEI–IONPs in the tumor might be an important feature of the IONP driven antitumor effect (Fig. 4a). Indeed, PEI–IONP administration produced an increase in the tumor iron content, rising from 38.93 (± 14.31) to 55.43 (± 11.12) μg/g based on ICP–OES measurements (Fig. 4b). This further increased to 129.1 (± 78.52) μg/g when magnetic targeting was employed, although the administration of PEI–IONP alone was sufficient to induce a local antitumor effect, suggesting that the 16.5 μg/g increase in tumor iron content might be sufficient. Alternatively, PEI–IONPs may produce a systemic effect and indeed, PEI appears to be a potent adjuvant in various scenarios, such as when administered subcutaneously with the viral HIV-1 gp140 glycoprotein (Sheppard et al. 2014) or intranasally along with the influenza hemagglutinin or herpes simplex virus type-2 (HSV2) glycoprotein d (Wegmann et al. 2012). The intrinsic adjuvant property of PEI is associated with the ability of the polycation to activate the innate immune response by engaging toll-like receptors, triggering the secretion of several cytokines that attract and modulate leukocytes or antigen-presenting cells at the site of administration or accumulation (Mulens-Arias et al. 2015b; Wegmann et al. 2012; Cubillos-Ruiz et al. 2009).

Therapeutic effect of the apMNKQ2@PEI–IONP formulation. a MDA-MB-231 cells were inoculated into the right flank of athymic nude mice and once the tumors reached ~ 200 mm3, the mice were randomized and treated with five doses of the corresponding formulation. Tumor growth curve over 18 days from the beginning of the treatment, normalizing the tumor volumes to the initial volume. The arrows indicate the days of administration: two-way ANOVA, **p < 0.01. b Tumor iron content from animals sacrificed 18 days after the first treatment determined by ICP and normalized to the corresponding tumor weight: two-tailed Mann–Whitney test, *p < 0.05, n = 3 per group. c Hepatic enzyme levels in the serum from treated mice. d Blood cell counts in treated animals: red blood cells (RBC), white blood cells (WBC), and platelets. e Subpopulations of the white blood cells in the treated mice
Curiously, apMNKQ2@PEI–IONP administration did not increase the tumor's iron content (34.82 ± 6.3 μg/g) relative to the saline control (38.93 ± 14.71 μg/g), which might be related to the formation of more serum protein/IONP aggregates in the presence of mouse serum, as demonstrated in vitro (Additional file 1: Fig. S4). ApMNKQ2@PEI–IONP forms aggregates of ~ 5289 nm when in contact with mouse serum as opposed to those of a diameter of ~ 2577 nm formed in saline [20 mmol/L Tris–HCl (pH 7.4), 1 mmol/L MgCl2, 150 mmol/L NaCl, and 5 mmol/L KCl]. By contrast, the aggregates of PEI–IONPs formed in mouse serum were ~ 3168 nm in diameter, > 2000 nm smaller than the apMNKQ2@PEI–IONP aggregates formed in mouse serum (Fig. 1c). This increase in the size of the nanoconjugate aggregates might impede IONPs from reaching tumor microenvironment by passive targeting alone. When a magnetic field was applied to attract the NPs to the tumor, this targeting could overcome the passive infiltration of tumor tissues by NPs and facilitate PEI–IONP accumulation. Indeed, magnetic targeting increased the iron content in animals treated with apMNKQ2@PEI–IONPs from 34.82 (± 6.39) to 50.52 (± 17.82) μg/g, similar to the content of PEI–IONP treated animals (Fig. 4b). Therefore, magnetic targeting overcomes the more limited PEI–IONP uptake by the tumor when conjugated to apMNKQ2, possibly enhancing the antitumor effects of these nanoconjugates.
Significant toxic repercussions of the IONP/aptamer administration were not evident when measured through cellular hepatic enzymes (alanine aminotransferase, ALT; aspartate aminotransferase, AST; alkaline phosphatase, ALP; and lactate dehydrogenase, LDH), blood components (white blood cells, WBCs; red blood cells, RBCs; and platelets), and body weight changes (Additional file 1: Fig. S6). The hepatic enzyme in the blood that differed most notably was LDH. Administering apMNKQ2@PEI–IONPs provoked a mild increase in LDH from 1056 Units/L in saline-treated animals to 1367 and 1345 Units/L, although these differences were not significant (Fig. 4c). The variation in RBCs was negligible among the treatments, varying from 7.6% (saline) to 8.6% (apMNK1@PEI–IONPs), and similar behavior was observed for WBCs, although magnetic targeting produced a slight increase in the WBCs following PEI–IONP administration (10.25% vs. 8.6% saline-treated: Fig. 4d). No significant variation was found for the percentage of specific WBCs (Fig. 4e), indicating that on the whole, all the treatments were safe. Noteworthy, due to the large size of nanocomplex and PEI–IONPs when in contact with serum proteins, it is plausible that the nanoparticles are rapidly eliminated from circulation by phagocytosis thus leading to a reduced passive accumulation in the site-of-interest. However, it appears that these events do not induce systemic toxicity.
Close inspection of the MDA-MB-231 tumors in vivo revealed that apMNKQ2 did not affect Occludin-1 mRNA expression as occurred in vitro (Pinto-Díez et al. 2022). However, the systemic administration of PEI–IONPs tended to increase the Occludin-1 mRNA in tumors, which increased further upon external application of a magnetic field (Additional file 1: Fig. S6). This increase in Occludin-1 might be associated with the intrinsic effects of PEI–IONPs on migration/invasion seen here (Fig. 3) and elsewhere (Mulens-Arias et al. 2015a). Conjugation with apMNKQ2 abrogated the effect of PEI–IONPs on Occludin-1 mRNA in vivo (Additional file 1: Fig. S6), confirming the loss-of-activity of the apMNKQ2–PEI–IONP complex in vivo (Fig. 4a). Importantly, the increase in Occludin-1 mRNA observed when apMNKQ2–PEI–IONP administration was combined with an external magnetic field, suggesting that the accumulation of PEI–IONPs in the tumor niche was sufficient to trigger Occludin-1 mRNA expression (Additional file 1: Fig. S7).
Conjugated apMNKQ2 synergizes with PEI–IONPs to impair the MDA-MB-231 tumor vasculature but it antagonizes the increase of tumor-infiltrating macrophages in vivo
NPs must comply with several conditions to achieve high concentrations in neoplastic tissues due to enhanced permeability and retention (EPR) phenomena, often linked to passive targeting. When injected systemically, NPs must evade the reticuloendothelial system (RES) and renal clearance, which ultimately leads to more prolonged circulation (> 6 h) (Greish 2007). Moreover, since the EPR depends on molecular weight, it would be expected that the larger the NPs, the longer they would remain in circulation. Indeed, the reversion of the anti-tumor effect of PEI–IONPs on MDA-MB-231 tumor when conjugated with the aptamer apMNKQ2 can be explained by the inability of the nanoconjugate to infiltrate the tumor microenvironment, i.e., an impaired EPR, precluding the PEI–IONPs from exerting their effects. Indeed, we observed a reduction of iron content from 55.42 µg Fe/g in PEI–IONPs-treated tumors to 34.82 µg Fe/g in apMNKQ2@PEI–IONPs-treated tumors (Fig. 4b). Such inference arises from the increase in the hydrodynamic diameter of the nanoconjugates compared to the PEI–IONPs in aptamer buffer (> 2000 times), which is further increased upon contact with serum proteins. However, not only must the NPs circulate longer but they must also be able to infiltrate the neoplastic vasculature and matrix. Therefore, if the tumor vasculature is affected, the nanoconjugates might encounter resistance to their penetration of the tumor matrix.
We analyzed the vasculature density of tumors from mice treated with different formulations by CD31 staining. Although we previously demonstrated that administration of the PEI–IONPs studied here exerted an antiangiogenic effect on MDA-MB-231 tumors in vivo, whereby PEI–IONPs decreased tumor vessel number and promoted M0, M1, and M2 macrophage infiltration, with or without magnetic targeting (Mulens-Arias et al. 2019), we did not observe a significant reduction in tumor vessel density (6.95 ± 3.15% CD31) relative to untreated tumors (6.16 ± 2.08% CD31: Fig. 5a). When magnetic targeting was employed, we detected only a marginal, non-significant reduction in the tumor vessel density (5.97 ± 4.26% CD31). Such differences in the effects of PEI–IONPs on angiogenesis between the earlier study and the data presented here can be explained by the IONP dose administered: while the previous study 1.2 mg/kg of PEI–IONPs was used, here only 0.3 mg/kg was administered, fourfold less IONPs. This difference in doses might lead to less iron content in tumor during the current study due the passive infiltration, and, thus, less PEI–IONP-dependent effect on angiogenesis. By contrast, apMNKQ2 provoked a significant decrease in tumor vascular density (5.28 ± 3.19% CD31) compared to untreated mice (6.16 ± 2.08% CD31), indicating an antiangiogenic effect of MNK1 inhibition (Fig. 5a). Indeed, this corroborates earlier data, where the inhibition of MNK1 by the antiangiogenic cercosporasmide suppresses pro-angiogenic proteins, such as VEGF and hypoxia-inducible factor (HIF), by targeting the MNK1/eIF4E pathway (Chen et al. 2020). In that study, cercosporasmide facilitates the response of renal cell carcinoma to the antiangiogenic tyrosine kinase and mTOR inhibitors. Here, the apMNKQ2@PEI–IONP nanoconjugate further reduced the tumor vascular density (4.58 ± 2.57% CD31), suggesting that PEI–IONPs might facilitate the antiangiogenic effect of apMNKQ2 in MDA-MB-231 tumors or they may have a synergistic effect with apMNKQ2 in disrupting the tumor vasculature.

Immunohistochemical analysis of the tumor tissues. a Blood vessel density was determined by CD31 staining of snap frozen MDA-MB-231 tumors from the animals treated with the different formulations (Top) and quantified as shown in the graph (Bottom). b Tumor-infiltrating macrophages were determined by F4/80 staining of snap frozen MDA-MB-231 tumors from animals treated with the different formulations (Top) and quantified as shown in the graph (Bottom): two-tailed Mann–Whitney test, *p < 0.05, **p < 0.01, ***p < 0.001. ‘#’ indicates differences between the two groups at each edge of the line; ‘*’ indicates differences between saline group and the indicated one
The nanoconjugates did not increase the tumor iron content and the application of a magnetic field to the tumors did not further restrict the tumor vasculature (4.66 ± 3.04% CD31) in response to apMNKQ2@PEI–IONPs (4.58 ± 2.57% CD31). Therefore, the antiangiogenic effect of apMNKQ2 is independent of IONP accumulation in the tumor (Fig. 5a). However, the same reduction in vessel density when tumors were treated with apMNKQ2@PEI–IONPs can obstruct passive retention of the nanoconjugates in the tumor microenvironment, thereby abolishing the antitumor effect of PEI–IONPs.
As previously demonstrated (Mulens-Arias et al. 2019), there were more tumor-infiltrating macrophages in tumors from animals administered PEI–IONPs and subjected to a magnetic field, as seen by the relative area of the tumor that was F4/80+ (15.19 ± 9.16% vs. 7.94 ± 6.62%: Fig. 5b). However, no other treatment triggered a significant increase in tumor-infiltrating macrophages and thus, this alone cannot explain the antitumor effect.
Together, the decrease in tumor vasculature density induced by apMNKQ2 and the formation of large protein corona-nanoconjugate aggregates might partly explain why the electrostatic conjugation of the aptamer with the PEI–IONP abrogates the intrinsic antitumor activity of the latter. While the increase in aggregate size in the bloodstream can preclude the accumulation of the IONPs due to an EPR effect, the more limited tumor vasculature can further impair NP accumulation in the tumor microenvironment. Application of an external magnetic field overcomes this impediment and it enhances the accumulation of magnetic IONPs in the tumor.
Conclusions
We assessed whether electrostatic complexing of the functional MNK1b-specific aptamer with PEI–IONPs facilitates apMNKQ2 delivery in vitro and in vivo as a therapeutic approach to treat TNBC. We demonstrate that the apMNKQ2@PEI–IONP nanoconjugates delivered three times more apMNKQ2 intracellularly than the aptamer alone. This enhanced intracellular delivery of the aptamer has consequences for MNK1 signaling, reducing MNK1 and its target phospho(Ser209)-eIF4E. We also observed a synergistic effect of the apMNKQ2 and PEI–IONPs on the inhibition of MDA-MB-231 cell migration, most likely associated with the increase in SGK1 and phospho(Thr346)-NDRG1. Despite the effect of the apMNKQ2 on MDA-MB-231 cell proliferation, migration and MNK1 signaling, the intravenous administration of apMNKQ2 alone did not significantly retarded tumor growth in vivo. By contrast, we detected significant inhibition of tumor growth when the PEI–IONPs are administered alone. However, since we did not observe an anti-tumor effect of the nanoconjugate apMNKQ2@PEI–IONP alone in vivo, and the effects observed in terms of tumor growth retardation of the nanocomplex plus magnet was like that of the treatment with PEI–IONPs alone or PEI–IONPs plus magnet, is its plausible that the anti-tumor mechanisms are only related to the molecular signaling triggered by PEI–IONPs per se. Although we expected that the aptamer complex with the IONPs would facilitate its delivery in vivo, exerting an enhanced antitumoral effect, administration of the apMNKQ2@PEI–IONP nanocomplex did not dampen tumor growth, probably by impeding the accumulation of the IONPs in the tumor. However, this apMNKQ2-induced reversion of the intrinsic antitumor effect of the PEI–IONPs was abolished when an external magnetic field was applied to the tumor site, promoting the accumulation of the IONPs. Thus, electrostatic conjugation of the apMNKQ2 aptamer to the PEI–IONPs appears to impede the accumulation of the PEI–IONPs in the tumor, which would be necessary for these NPs to exert their antitumor activity. Two mechanisms might explain this effect on NP accumulation phenomenon: (1) an enlargement of the protein corona-NP aggregates, impeding the passive accumulation of the NPs in the tumor; and (2) a reduction in the tumor vascular density induced by apMNKQ2. This study highlights the need to better comprehend the nanosystem intended for a therapeutic application, as the combination of two or more components could alter the intrinsic effects of each part alone.
Availability of data and materials
All data generated or analyzed during this study are included in this published article and its additional files.
References
Adesso L, Calabretta S, Barbagallo F, Capurso G, Pilozzi E, Geremia R et al (2013) Gemcitabine triggers a pro-survival response in pancreatic cancer cells through activation of the MNK2/eIF4E pathway. Oncogene 32:2848–2857
Akahane T, Akahane M, Shah A, Connor CM, Thorgeirsson UP (2004) TIMP-1 inhibits microvascular endothelial cell migration by MMP-dependent and MMP-independent mechanisms. Exp Cell Res 301:158–167
Al-Deen FMN, Xiang SD, Ma C, Wilson K, Coppel RL, Selomulya C et al (2017) Magnetic nanovectors for the development of DNA blood-stage malaria vaccines. Nanomaterials 7:30
Banerjee K, Resat H (2016) Constitutive activation of STAT3 in breast cancer cells: a review. Int J Cancer 138:2570–2578
Beggs JE, Tian S, Jones GG, Xie J, Iadevaia V, Jenei V et al (2015) The MAP kinase-interacting kinases regulate cell migration, vimentin expression and eIF4E/CYFIP1 binding. Biochem J 467:63–76
Brock EJ, Jackson RM, Boerner JL, Li Q, Tennis MA, Sloane BF et al (2021) Sprouty4 negatively regulates ERK/MAPK signaling and the transition from in situ to invasive breast ductal carcinoma. PLoS ONE 16:e0252314–e0252314
Cao Y, Zhang S, Ma M, Zhang Y (2022) Fluorinated PEG-PEI coated magnetic nanoparticles for siRNA delivery and CXCR4 knockdown. Nanomaterials 12:1692
Chekmarev J, Azad MG, Richardson DR (2021) The oncogenic signaling disruptor, NDRG1: molecular and cellular mechanisms of activity. Cells 10:2382
Chen Y, Lian G, Liao C, Wang W, Zeng L, Qian C et al (2012) Characterization of polyethylene glycol-grafted polyethylenimine and superparamagnetic iron oxide nanoparticles (PEG-g-PEI-SPION) as an MRI-visible vector for siRNA delivery in gastric cancer in vitro and in vivo. J Gastroenterol 48:809–821. https://doi.org/10.1007/s00535-012-0713-x
Chen S, Cui L, Hu Q, Shen Y, Jiang Y, Zhao J (2020) Preclinical evidence that MNK/eIF4E inhibition by cercosporamide enhances the response to antiangiogenic TKI and mTOR inhibitor in renal cell carcinoma. Biochem Biophys Res Commun 530:142–148
Cheng J, Zheng Z, Tang W, Shao J, Jiang H, Lin H (2022) A new strategy for stem cells therapy for erectile dysfunction: adipose-derived stem cells transfect Neuregulin-1 gene through superparamagnetic iron oxide nanoparticles. Investig Clin Urol 63:359–367
Cruz-Hernández CD, Rodríguez-Martínez G, Cortés-Ramírez SA, Morales-Pacheco M, Cruz-Burgos M, Losada-García A et al (2022) Aptamers as theragnostic tools in prostate cancer. Biomolecules 12:1056
Cubillos-Ruiz JR, Engle X, Scarlett UK, Martinez D, Barber A, Elgueta R et al (2009) Polyethylenimine-based siRNA nanocomplexes reprogram tumor-associated dendritic cells via TLR5 to elicit therapeutic antitumor immunity. J Clin Invest 119:2231–2344
Culjkovic B, Topisirovic I, Skrabanek L, Ruiz-Gutierrez M, Borden KLB (2005) eIF4E promotes nuclear export of cyclin D1 mRNAs via an element in the 3′UTR. J Cell Biol 169:245–256
Culjkovic-Kraljacic B, Baguet A, Volpon L, Amri A, Borden KLB (2012) The oncogene eIF4E reprograms the nuclear pore complex to promote mRNA export and oncogenic transformation. Cell Rep 2:207–215
Dillekås H, Rogers MS, Straume O (2019) Are 90% of deaths from cancer caused by metastases? Cancer Med 8:5574–5776
Fan W, Wang W, Mao X, Chu S, Feng J, Xiao D et al (2017) Elevated levels of p-Mnk1, p-eIF4E and p-p70S6K proteins are associated with tumor recurrence and poor prognosis in astrocytomas. J Neurooncol 131:485–493
Frisk G, Svensson T, Bäcklund LM, Lidbrink E, Blomqvist P, Smedby KE (2012) Incidence and time trends of brain metastases admissions among breast cancer patients in Sweden. Br J Cancer 106:1850–1853
Furic L, Rong L, Larsson O, Koumakpayi IH, Yoshida K, Brueschke A et al (2010) eIF4E phosphorylation promotes tumorigenesis and is associated with prostate cancer progression. Proce Natl Acad Sci 107:14134–14139
Gao F, Yin J, Chen Y, Guo C, Hu H, Su J (2022) Recent advances in aptamer-based targeted drug delivery systems for cancer therapy. Front Bioeng Biotechnol 10:972933–972933
García-Recio EM, Pinto-Díez C, Pérez-Morgado MI, García-Hernández M, Fernández G, Martín ME et al (2016) Characterization of MNK1b DNA aptamers that inhibit proliferation in MDA-MB231 breast cancer cells. Mol Ther Nucleic Acids 5:e275–e275
Geter PA, Ernlund AW, Bakogianni S, Alard A, Arju R, Giashuddin S et al (2017) Hyperactive mTOR and MNK1 phosphorylation of eIF4E confer tamoxifen resistance and estrogen independence through selective mRNA translation reprogramming. Genes Dev 31:2235–2249
Godbole M, Togar T, Patel K, Dharavath B, Yadav N, Janjuha S et al (2018) Up-regulation of the kinase gene SGK1 by progesterone activates the AP-1-NDRG1 axis in both PR-positive and -negative breast cancer cells. J Biol Chem 293:19263–19276
Gong M, Liu H, Sun N, Xie Y, Yan F, Cai L (2020) Polyethylenimine-dextran-coated magnetic nanoparticles loaded with miR-302b suppress osteosarcoma in vitro and in vivo. Nanomedicine 15:711–723
Gou Q, Liu Z, Xie Y, Deng Y, Ma J, Li J et al (2022) Systematic evaluation of tumor microenvironment and construction of a machine learning model to predict prognosis and immunotherapy efficacy in triple-negative breast cancer based on data mining and sequencing validation. Front Pharmacol. https://doi.org/10.3389/fphar.2022.995555
Greish K (2007) Enhanced permeability and retention of macromolecular drugs in solid tumors: a royal gate for targeted anticancer nanomedicines. J Drug Target 15:457–464
Groblewska M, Siewko M, Mroczko B, Szmitkowski M (2012) The role of matrix metalloproteinases (MMPs) and their inhibitors (TIMPs) in the development of esophageal cancer. Folia Histochem Cytobiol 50:12–19
He Y, Cheng G, Xie L, Nie Y, He B, Gu Z (2012) Polyethyleneimine/DNA polyplexes with reduction-sensitive hyaluronic acid derivatives shielding for targeted gene delivery. Biomaterials 34:1235–1245
Hoang M-D, Lee H-J, Lee H-J, Jung S-H, Choi N-R, Vo M-C et al (2015) Branched polyethylenimine-superparamagnetic iron oxide nanoparticles (bPEI-SPIONs) improve the immunogenicity of tumor antigens and enhance th1 polarization of dendritic cells. J Immunol Res 2015:1–9
Hola K, Markova Z, Zoppellaro G, Tucek J, Zboril R (2015) Tailored functionalization of iron oxide nanoparticles for MRI, drug delivery, magnetic separation and immobilization of biosubstances. Biotechnol Adv 33:1162–1176
Hou S, Du P, Wang P, Wang C, Liu P, Liu H (2017) Significance of MNK1 in prognostic prediction and chemotherapy development of epithelial ovarian cancer. Clin Transl Oncol 19:1107–1116
Huang F-W, Wang H-Y, Li C, Wang H-F, Sun Y-X, Feng J et al (2010) PEGylated PEI-based biodegradable polymers as non-viral gene vectors. Acta Biomater 6:4285–4295
Jin H, Gui R, Gong J, Huang W (2017) Aptamer and 5-fluorouracil dual-loading Ag2S quantum dots used as a sensitive label-free probe for near-infrared photoluminescence turn-on detection of CA125 antigen. Biosens Bioelectron 92:378–384
Joshi S, Platanias LC (2012) Mnk kinases in cytokine signaling and regulation of cytokine responses. Biomol Concepts 3:127–139
Korneeva NL, Soung YH, Kim HI, Giordano A, Rhoads RE, Gram H et al (2010) Mnk mediates integrin α6β4–dependent eIF4E phosphorylation and translation of VEGF mRNA. Mol Cancer Res 8:1571–1578
Koromilas AE, Lazaris-Karatzas A, Sonenberg N (1992) mRNAs containing extensive secondary structure in their 5′ non-coding region translate efficiently in cells overexpressing initiation factor eIF-4E. EMBO J 11:4153–4158
Lian Z, Chang T, Ma S, Li J, Zhang H, Wang X et al (2022) MiR-96-5p induced NDRG1 deficiency promotes prostate cancer migration and invasion through regulating the NF-κB signaling pathway. Cancer Biomark 35:83–98
Liu R, Chen Y, Liu G, Li C, Song Y, Cao Z et al (2020) PI3K/AKT pathway as a key link modulates the multidrug resistance of cancers. Cell Death Dis 11:797–797
Liu L, Yang Z, Liu C, Wang M, Chen X (2022) Preparation of PEI-modified nanoparticles by dopamine self-polymerization for efficient DNA delivery. Biotechnol Appl Biochem. https://doi.org/10.1002/bab.2402
Łukasiewicz S, Czeczelewski M, Forma A, Baj J, Sitarz R, Stanisławek A (2021) Breast cancer-epidemiology, risk factors, classification, prognostic markers, and current treatment strategies-an updated Review. Cancers 13:4287
Luther DC, Huang R, Jeon T, Zhang X, Lee Y-W, Nagaraj H et al (2020) Delivery of drugs, proteins, and nucleic acids using inorganic nanoparticles. Adv Drug Deliv Rev 156:188–213
Ma L, Sun N, Meng Y, Tu C, Cao X, Wei Y et al (2018) Harnessing the affinity of magnetic nanoparticles toward dye-labeled DNA and developing it as an universal aptasensor revealed by lipopolysaccharide detection. Anal Chim Acta 1036:107–114
Manore SG, Doheny DL, Wong GL, Lo H-W (2022) IL-6/JAK/STAT3 signaling in breast cancer metastasis: biology and treatment. Front Oncol 12:866014–866014
Martin TA, Mansel RE, Jiang WG (2010) Loss of occludin leads to the progression of human breast cancer. Int J Mol Med 26:723–734
Martin TA, Jordan N, Davies EL, Jiang WG (2016) Metastasis to bone in human cancer is associated with loss of occludin expression. Anticancer Res 36:1287
Mohammad Gholinia Sarpoli L, Zare-Karizi S, Heidari E, Hasanzadeh A, Bayandori M, Azedi F et al (2022) Co-delivery of curcumin and Bcl-2 siRNA to enhance therapeutic effect against breast cancer cells using PEI-functionalized PLGA nanoparticles. Pharma Dev Technol 27:785–793. https://doi.org/10.1080/10837450.2022.2120003
Mulens-Arias V, Rojas JM, Pérez-Yagüe S, del Morales PM, Barber DF (2015a) Polyethylenimine-coated SPION exhibits potential intrinsic anti-metastatic properties inhibiting migration and invasion of pancreatic tumor cells. J Controll Releas 216:78–92
Mulens-Arias V, Rojas JM, Pérez-Yagüe S, Morales MP, Barber DF (2015b) Polyethylenimine-coated SPIONs trigger macrophage activation through TLR-4 signaling and ROS production and modulate podosome dynamics. Biomaterials 52:494–506. https://doi.org/10.1016/j.biomaterials.2015.02.068
Mulens-Arias V, Rojas JM, Sanz-Ortega L, Portilla Y, Pérez-Yagüe S, Barber DF (2019) Polyethylenimine-coated superparamagnetic iron oxide nanoparticles impair in vitro and in vivo angiogenesis. Nanomedicine 21:102063
Niland S, Riscanevo AX, Eble JA (2021) Matrix metalloproteinases shape the tumor microenvironment in cancer progression. Int J Mol Sci 23:146
O’Loghlen A, González VM, Piñeiro D, Pérez-Morgado MI, Salinas M, Martı́n ME (2004) Identification and molecular characterization of Mnk1b, a splice variant of human MAP kinase-interacting kinase Mnk1. Exp Cell Res 299:343–355
Ortega MA, Fraile-Martínez O, Asúnsolo Á, Buján J, García-Honduvilla N, Coca S (2020) signal transduction pathways in breast cancer: the important role of PI3K/Akt/mTOR. J Oncol 2020:9258396
Pelletier EM, Shim B, Goodman S, Amonkar MM (2008) Epidemiology and economic burden of brain metastases among patients with primary breast cancer: results from a US claims data analysis. Breast Cancer Res Treat 108:297–305
Perou CM, Sørlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA et al (2000) Molecular portraits of human breast tumours. Nature 406:747–752
Pinto-Díez C, García-Recio EM, Pérez-Morgado MI, García-Hernández M, Sanz-Criado L, Sacristán S et al (2018) Increased expression of MNK1b, the spliced isoform of MNK1, predicts poor prognosis and is associated with triple-negative breast cancer. Oncotarget 9:13501–13516
Pinto-Díez C, Ferreras-Martín R, Carrión-Marchante R, Klett-Mingo JI, García-Hernández M, Pérez-Morgado MI et al (2022) An optimized MNK1b aptamer, apMNKQ2, and its potential use as a therapeutic agent in breast cancer. Mol Ther Nucleic Acids 30:553–568
Pugazhenthi S, Nesterova A, Sable C, Heidenreich KA, Boxer LM, Heasley LE et al (2000) Akt/Protein Kinase B Up-regulates Bcl-2 Expression through cAMP-response element-binding protein. J Biol Chem 275:10761–10766
Scheper GC, Proud CG (2002) Does phosphorylation of the cap-binding protein eIF4E play a role in translation initiation? Eur J Biochem 269:5350–5359
Scheper GC, Morrice NA, Kleijn M, Proud CG (2001) The mitogen-activated protein kinase signal-integrating kinase Mnk2 is a eukaryotic initiation factor 4E kinase with high levels of basal activity in mammalian cells. Mol Cell Biol 21:743–754
Scheper GC, Parra JL, Wilson M, Van Kollenburg B, Vertegaal ACO, Han Z-G et al (2003) The N and C termini of the splice variants of the human mitogen-activated protein kinase-interacting kinase Mnk2 determine activity and localization. Mol Cell Biol 23:5692–5705
Schmid P, Rugo HS, Adams S, Schneeweiss A, Barrios CH, Iwata H et al (2020) Atezolizumab plus nab-paclitaxel as first-line treatment for unresectable, locally advanced or metastatic triple-negative breast cancer (IMpassion130): updated efficacy results from a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol 21:44–59
Sheppard NC, Brinckmann SA, Gartlan KH, Puthia M, Svanborg C, Krashias G et al (2014) Polyethyleneimine is a potent systemic adjuvant for glycoprotein antigens. Int Immunol 26:531–538
Sizikov AA, Nikitin PI, Nikitin MP (2021) Magnetofection in vivo by nanomagnetic carriers systemically administered into the bloodstream. Pharmaceutics 13:1927
Szymczyk A, Drozd M, Kamińska A, Matczuk M, Trzaskowski M, Mazurkiewicz-Pawlicka M et al (2022) Comparative evaluation of different surface coatings of Fe(3)O(4)-based magnetic nano sorbent for applications in the nucleic acids extraction. Int J Mol Sci 23:8860
Thomas M, Klibanov AM (2003) Conjugation to gold nanoparticles enhances polyethylenimine’s transfer of plasmid DNA into mammalian cells. Proc Natl Acad Sci USA 100:9138–9143
Tian S, Wang X, Proud CG (2017) Oncogenic MNK signalling regulates the metastasis suppressor NDRG1. Oncotarget 8:46121–46135
Ueda T, Watanabe-Fukunaga R, Fukuyama H, Nagata S, Fukunaga R (2004) Mnk2 and Mnk1 are essential for constitutive and inducible phosphorylation of eukaryotic initiation factor 4E but not for cell growth or development. Mol Cell Biol 24:6539–6549
Wang X, Wang Y, Zhang Q, Zhuang H, Chen B (2018) MAP Kinase-interacting kinase 1 promotes proliferation and invasion of hepatocellular carcinoma and is an unfavorable prognostic biomarker. Med Sci Monit 24:1759–1767
Wang X, Pei K, Sun H, Wang Q (2021) A magnetic relaxation switch sensor for determination of 17β-estradiol in milk and eggs based on aptamer-functionalized Fe3O4@Au nanoparticles. J Sci Food Agric 101:5697–5706
Warner RB, Najy AJ, Jung YS, Fridman R, Kim S, Kim H-RC (2020) Establishment of structure-function relationship of tissue inhibitor of metalloproteinase-1 for its interaction with CD63: implication for cancer therapy. Sci Rep 10:2099
Waskiewicz AJ, Flynn A, Proud CG, Cooper JA (1997) Mitogen-activated protein kinases activate the serine/threonine kinases Mnk1 and Mnk2. EMBO J 16:1909–1920
Wee P, Wang Z (2017) Epidermal growth factor receptor cell proliferation signaling pathways. Cancers 9:52
Wegmann F, Gartlan KH, Harandi AM, Brinckmann SA, Coccia M, Hillson WR et al (2012) Polyethyleneimine is a potent mucosal adjuvant for viral glycoprotein antigens. Nat Biotechnol 30:883–888
Wei M, Zhang Y, Yang X, Ma P, Li Y, Wu Y et al (2021) Claudin-2 promotes colorectal cancer growth and metastasis by suppressing NDRG1 transcription. Clin Transl Med 11:e667–e667
Wendel H-G, Silva RLA, Malina A, Mills JR, Zhu H, Ueda T et al (2007) Dissecting eIF4E action in tumorigenesis. Genes Dev 21:3232–3237
Wheater MJ, Johnson PW, Blaydes JP (2010) The role of MNK proteins and eIF4E phosphorylation in breast cancer cell proliferation and survival. Cancer Biol Ther 10:728–735
Wolczyk D, Zaremba-Czogalla M, Hryniewicz-Jankowska A, Tabola R, Grabowski K, Sikorski AF et al (2016) TNF-α promotes breast cancer cell migration and enhances the concentration of membrane-associated proteases in lipid rafts. Cell Oncol 39:353–363
Xiao X, Li H, Zhao L, Zhang Y, Liu Z (2021) Oligonucleotide aptamers: recent advances in their screening, molecular conformation and therapeutic applications. Biomed Pharmacother 143:112232
Xu W, Kannan S, Verma CS, Nacro K (2022) Update on the development of MNK inhibitors as therapeutic agents. J Med Chem 65:983–1007
Zhan Y, Guo J, Yang W, Goncalves C, Rzymski T, Dreas A et al (2017) MNK1/2 inhibition limits oncogenicity and metastasis of KIT-mutant melanoma. J Clin Invest 127:4179–4192
Zhao Y, Wang J, Cai X, Ding P, Lv H, Pei R (2020) Metal-organic frameworks with enhanced photodynamic therapy: synthesis, erythrocyte membrane camouflage, and aptamer-targeted aggregation. ACS Appl Mater Interfaces 12:23697–23706
Zheng J, Li J, Xu L, Xie G, Wen Q, Luo J et al (2014) Phosphorylated Mnk1 and eIF4E are associated with lymph node metastasis and poor prognosis of nasopharyngeal carcinoma. PLoS ONE 9:e89220–e89220
Acknowledgements
The authors acknowledge the Scientific and Technical Assistance of the Transmission Electron Microscopy, Flow cytometry, Histology and Animal services at the Centro Nacional de Biotecnología (CNB, CSIC). ICP–OES analysis was carried out in the support laboratories at the Instituto de Ciencia de Materiales de Madrid (ICMM, CSIC). The authors are also grateful to Dr M. Sefton for author editing of the manuscript.
Funding
This study was supported by the DTS18/00029 Grant (to VM Gonzalez and DF Barber) from the Instituto de Salud Carlos III (Plan Estatal de I + D + I 2017–2020), co-financed by the European Development Regional Fund ‘‘A way to achieve Europe’’ (ERDF), and by the PID2020-112685RB-100 Grant (to DF Barber) funded by MCIN/AEI/10.13039/501100011033. V. Mulens-Arias was a postdoctoral scholar working under a Juan de La Cierva-Incorporación Contract (IJCI-2017-31447, funded by MCIN/AEI/10.13039/501100011033) and Y. Portilla received a predoctoral FPU Grant (FPU15/06170 funded by MCIN/AEI/10.13039/501100011033 and the ESF Investing in your future). This research work was performed in the framework of the Nanomedicine CSIC HUB and Cancer CSIC HUB.
Author information
Authors and Affiliations
Contributions
DFB and VMG designed and supervised the study, and they contributed to the writing, review and revision of the manuscript. VMA and YP participated in the design of the study and performed the statistical analysis, conducted the experimental work, data analysis, as well as preparing the figures and helped draft the manuscript. SPY assisted with the experimental work. RFM and MEM assisted with the experimental work, contributed to the writing and revision of the manuscript, and helped with the analysis and interpretation of the results. All the authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
All animal studies were approved by the Ethics in Animal Experimentation Committee at the National Center for Biotechnology (CEEA–CNB), the Spanish Scientific Research Council (CSIC) Ethics Committee, and by the Division of Animal Protection of the Comunidad Autómoma de Madrid (CAM, PROEX 185/19), and they were carried out in compliance with national and European Union legislation (Directive 2010/63/EU).
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1
: Table S1. Primer sequences used for the RT-PCR assays. Fig. S1. Iron oxide nanoparticle internalization as measured by ICP–OES. A MDA-MB-231, B RAW264.7, and C SVEC4-10 cells were incubated with 1 mM apMNK1@PEI–IONPs or the equivalent of PEI–IONPs for 24 h, and the cells were then harvested and digested to determine their iron content by ICP–OES. Fig. S2. Cytotoxicity of apMNK1 and its formulations in MDA-MB-231 cells over 48 h. Fig. S3. SGK1 and phospho-NDRG1 expression in MDA-MB-231 cells exposed to increasing aptamer concentrations. A Representative western blot. B–D A summary of SGK1, B phospho-NDRG1 and C NDRG1 expression in MDA-MB-231 cell lysates exposed for 24 h to apMNK1@PEI–IONPs, or the equivalent apMNK1 or PEI–IONPs.phospho-NDRG1/NDRG1 ratio. The graphs reflect the results of at least three independent replicates. Fig. S4. Variation in hydrodynamic diameter of the PEI–IONPs and the apMNK1@PEI–IONP complex over time in a standard buffer with 10 % mouse serum. Fig. S5. In vivo therapeutic treatment of mice bearing MDA-MB-231 tumors with apMNKQ2@PEI–IONPs. Fig. S6. Body weight variation following treatment. Fig. S7. In vivo expression of Occludin mRNA. The tumors were excised and processed for RNA extraction, quantifying Occludin-1 mRNA by semi-quantitative RT-PCR.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Mulens-Arias, V., Portilla, Y., Pérez-Yagüe, S. et al. An electrostatically conjugated-functional MNK1 aptamer reverts the intrinsic antitumor effect of polyethyleneimine-coated iron oxide nanoparticles in vivo in a human triple-negative cancer xenograft. Cancer Nano 14, 64 (2023). https://doi.org/10.1186/s12645-023-00204-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12645-023-00204-8