
Abstract
Background
Since the release of the combined oral contraceptive pill in 1960, women have shouldered the burden of contraception and family planning. Over 60 years later, this is still the case as the only practical, effective contraceptive options available to men are condoms and vasectomy. However, there are now a variety of promising hormonal and non-hormonal male contraceptive options being studied. The purpose of this narrative review is to provide clinicians and laypeople with focused, up-to-date descriptions of novel strategies and targets for male contraception. We include a cautiously optimistic discussion of benefits and potential drawbacks, highlighting several methods in preclinical and clinical stages of development.
Results
As of June 2023, two hormonal male contraceptive methods are undergoing phase II clinical trials for safety and efficacy. A large-scale, international phase IIb trial investigating efficacy of transdermal segesterone acetate (Nestorone) plus testosterone gel has enrolled over 460 couples with completion estimated for late 2024. A second hormonal method, dimethandrolone undecanoate, is in two clinical trials focusing on safety, pharmacodynamics, suppression of spermatogenesis and hormones; the first of these two is estimated for completion in December 2024. There are also several non-hormonal methods with strong potential in preclinical stages of development.
Conclusions
There exist several hurdles to novel male contraception. Therapeutic development takes decades of time, meticulous work, and financial investment, but with so many strong candidates it is our hope that there will soon be several safe, effective, and reversible contraceptive options available to male patients.
Résumé
Contexte
Depuis la sortie de la pilule contraceptive orale combinée en 1960, les femmes ont assumé le fardeau de la contraception et de la planification familiale. Plus de 60 ans plus tard, c’est toujours le cas, car les seules options contraceptives pratiques et efficaces disponibles pour les hommes sont les préservatifs et la vasectomie. Cependant, il existe maintenant une variété d’options contraceptives masculines hormonales et non hormonales prometteuses qui sont à l’étude. Le but de cette revue narrative est de fournir aux cliniciens et aux profanes des descriptions ciblées et à jour de nouvelles stratégies et cibles pour la contraception masculine. Nous incluons une discussion prudemment optimiste sur les avantages et les inconvénients potentiels, en soulignant plusieurs méthodes aux stades précliniques et cliniques du développement.
Résultats
En juin 2023, deux méthodes contraceptives masculines hormonales faisaient l’objet d’essais cliniques de phase II pour leur innocuité et leur efficacité. Un essai international de phase IIb à grande échelle, portant sur l’efficacité de l’acétate de ségestérone transdermique (Nestorone) et du gel de testostérone, a recruté plus de 460 couples et devrait être achevé pour la fin de 2024. Une seconde méthode hormonale, l’undécanoate de diméthandrolone, fait l’objet de deux essais cliniques axés sur l’innocuité, la pharmacodynamique, la suppression de la spermatogenèse et des hormones; le premier de ces deux essais devrait être achevé en décembre 2024. Il existe également plusieurs méthodes non hormonales à fort potentiel aux stades précliniques de développement.
Conclusions
Il existe plusieurs obstacles à la nouvelle contraception masculine. Le développement thérapeutique nécessite des décennies de temps, un travail méticuleux et un investissement financier ; mais avec autant de candidats solides, nous espérons qu’il y aura bientôt plusieurs options contraceptives sûres, efficaces et réversibles, disponibles pour les hommes.
Similar content being viewed by others


Introduction
In the wake of the Dobbs v. Jackson Women’s Health Organization 2022 decision, the resultant “trigger laws” in 13 U.S. states, and the lingering retraction of reproductive rights in many more [1, 2], the need for novel contraceptive options has gained urgency across the United States. Unfortunately, due to a complex combination of medical challenges and societal beliefs [3,4,5,6], the burden of contraception has fallen almost entirely on women, and the only practical effective options available to males are condoms and vasectomy. Even with ‘perfect use’, the failure rate of condoms is still over 10% [7], and vasectomy is largely irreversible. Further, many of the contraceptive options currently available have high discontinuation rates [8], contributing to high rates of unintended pregnancy in the United States [9, 10]. With that in mind, there is a growing demand for safe, effective, and reversible male contraception that would allow men to share the burden of family planning [11, 12].
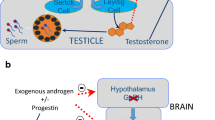
Male fertility is dependent on production of an adequate number of viable, motile sperm capable of moving through the female reproductive tract and fertilizing oocytes. Fertile males generally have seminal sperm concentrations greater than 15 million sperm/mL [13], and adequate sperm suppression for contraception requires sperm levels ≤ 1 million/mL [14]. The process of sperm production is termed spermatogenesis and is controlled by the hypothalamic-pituitary-testicular (HPT) axis (Fig. 1) [15]. Briefly, the hypothalamus produces gonadotropin-releasing hormone (GnRH) in a pulsatile fashion, which stimulates the anterior pituitary to secrete the gonadotrophic hormones luteinizing hormone (LH) and follicle-stimulating hormone (FSH). LH stimulates androgen production by testicular Leydig cells, and FSH, along with high levels of intratesticular T, enables spermatogenesis within the seminiferous tubules [16]. T exerts negative feedback on GnRH release and therefore suppresses LH and FSH secretion; the same effect is seen with exogenous androgens. Similarly, natural and synthetic progesterone, the latter termed progestins, exert negative feedback on the HPT axis to suppress LH and FSH release [16]. These concepts underlie the mechanisms of hormonal contraceptives discussed in this review, which generally target spermatogenesis, sperm motility, or transport through the vas deferens (Fig. 1).

Overview of the hypothalamic-pituitary-testicular (HPT) axis and targets of male contraception. The HPT axis consists of the hypothalamus, pituitary gland, and testes. The hypothalamus releases gonadotropin-releasing hormone (GnRH) in a pulsatile fashion which signals for release of luteinizing hormone (LH) and follicle-stimulating hormone (FSH) from the anterior pituitary. LH and FSH drive testosterone (T) production and spermatogenesis in the testes. T and the hormonal contraceptives exert negative feedback on the hypothalamus to inhibit GnRH, LH, and FSH release, therefore suppressing spermatogenesis. Non-hormonal methods focus on distinct targets to inhibit spermatogenesis, sperm motility, or transit through the vas deferens. Pointed arrows indicate activation; red broad-tipped arrows indicate inhibition. NES/T, Nestorone/testosterone; DMAU, dimethandrolone undecanoate; 11β-MNTDC, 11β-methyl-19-nortestosterone dodecylcarbonate; RARA, retinoic acid receptor alpha; BRDT, bromodomain testis-specific protein; TSSK, testis-specific serine/threonine kinase; sAC, soluble adenylyl cyclase; CatSper, cation channel of sperm; SLO3, slowpoke homolog 3; RISUG, reversible inhibition of sperm under guidance. Figure created by EJL using BioRender.com
There are several promising male contraceptive options in development, and they can be broadly categorized as either hormonal or non-hormonal. The purpose of this review is to provide an overview of the most promising male contraceptive methods under study, including how they work, their current state in research and development, and potential side effects or barriers to marketability. We will also briefly discuss some methods in preclinical stages of development to demonstrate that men may soon have access to a variety of safe, effective, and reversible contraceptive options.
Materials and methods
For this narrative review, authors searched the online databases MEDLINE (via PubMed.gov), Cochrane Reviews, CENTRAL (via CochraneLibrary.com), ClinicalTrials.gov, and the World Health Organization’s International Clinical Trials Registry Platform for publications and ongoing clinical trials through 20 June 2023. Search terms included “contraception”, “male contraception”, “hormonal contraception”, “spermatogenesis inhibition”, “vas deferens occlusion”, and terms related to methods discussed below. Authors considered all identified ongoing studies related to male contraception, but we excluded from discussion those evaluating 7α-Methyl-19-nortestosterone (MENT) [17,18,19] or T combined with GnRH antagonists [20, 21], estradiol [22], or progestins (medroxyprogesterone acetate [23, 24] or norethisterone enanthate [25,26,27,28]) as these treatments ultimately failed to progress in clinical trials. Several past trials evaluating T injection alone [29,30,31,32,33,34,35] were also excluded as ongoing trials use T as a supplemental rather than primary compound. Authors EJL, GFLQ, and KLP completed literature search and assessed methodology of ongoing trials with particular focus on sample size (n), primary and secondary outcomes, and inclusion and exclusion criteria; none of the studies were excluded due to grossly unsound methodology.
Hormonal methods
Three hormonal methods show great promise in male contraception: segesterone acetate (Nestorone; NES), dimethandrolone undecanoate (DMAU), and 11β-methyl-19-nortestosterone dodecylcarbonate (11β-MNTDC). NES and DMAU are currently in phase II clinical trials, and 11β-MNTDC has completed one phase II trial. Each method will be discussed separately below, and the clinical trials investigating these three compounds are summarized in Table 1.
Segesterone acetate + testosterone (NES/T)
Segesterone acetate, most often identified by its trade name Nestorone (NES), is a potent progestin with virtually no affinity for androgen receptors (AR) or estrogen receptors (ER) and minimal glucocorticoid activity [49,50,51]. NES shows low bioavailability when taken orally but is readily absorbed by transdermal application [52]; it has been available with ethinyl estradiol in the ANNOVERA vaginal ring (Mayne Pharma, Raleigh, NC) since 2018 and is a well-tolerated female contraceptive with > 97% efficacy [53,54,55]. NES is now compounded with T in a transdermal gel (NES/T) in a phase II clinical trial evaluating efficacy [36, 37]. T is added to improve suppression of spermatogenesis and minimize potential symptoms of androgen deficiency [56].
Phase I trials of NES/T daily gel (approximately 8.3 mg/62.5 mg) have demonstrated gonadotropin suppression adequate to suppress spermatogenesis in nearly 90% of participants [38, 39], suggesting that NES/T will be an effective form of male birth control [57]. Importantly, in these same studies there were no severe side effects with treatment. The main adverse effects were similar to the combined estrogen-progestin contraceptive pills used by women [58] and included minor mood symptoms, acne, and likely transient gastrointestinal symptoms [38,39,40]. From the most recent Phase I trial and a survey on attitudes towards NES/T, the majority of participants (79% and 56%, respectively) were satisfied or very satisfied with the treatments, and 50–51% reported that they would use NES/T daily gel as a sole form of contraception [38, 59].
A phase IIb trial investigating NES/T efficacy is currently underway at 17 medical centers across 8 U.S. states and 7 other countries [36, 37]; it is sponsored by the Population Council and the Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD). Participants are self-administering the transdermal gel with one of two T doses (both compounded as NES/T; 8 mg/62 mg or 8 mg/74 mg), and participants showing low serum T with symptoms of hypogonadism will be offered additional T [37]. The trial is broken down into phases. There is an initial screening phase, after which participants begin daily NES/T. Within 20 weeks of beginning treatment, participants must show sperm suppression to levels ≤ 1 million sperm/mL before entering the 52-week efficacy phase. The recovery phase is intended to assess sperm production after ceasing NES/T application and continue symptom surveillance of both males and their female partners [37]. Enrollment was completed in November 2022 with 462 couples having started treatment. Completion of the primary endpoint, contraceptive efficacy, is estimated for late 2024, with full study results likely available in early 2025 [36, 37].
Dimethandrolone undecanoate (DMAU)
DMAU is a testosterone-derived pro-drug, metabolized to active form dimethandrolone (DMA), with high affinity for AR and, to a lesser degree, progesterone receptors (PR) [60]. DMAU and DMA are not aromatized and therefore lack estrogenic effects [61], but DMAU is highly lipophilic and experiences first-pass metabolism by the liver [62], requiring study of a variety of formulations to determine the optimal delivery method. In preclinical animal studies including non-human primates, DMAU was shown to effectively, reversibly suppress gonadotropins and spermatogenesis while maintaining physiologic androgenic effects without serious side effects [63,64,65]; importantly, there were no signs of liver toxicity, a well-characterized side effect of many exogenous androgens [66]. DMAU has been studied in several clinical trials for safety, pharmacodynamics, and gonadotropin suppression to evaluate its potential in male contraception, and phase I and phase II clinical trials are currently underway.
Early human trials of DMAU evaluated safety and absorption with doses up to 800 mg. In 2014, the first clinical trial orally dosed DMAU in a powder formulation from 25 to 800 mg, fasting or following high-fat meal (50% calories as fat). With a high-fat meal, authors found considerable, dose-escalating absorption of DMAU and suppression of gonadotropins (12 h later) from 200 mg upwards [45]. In a follow-up 2017 study, authors evaluated daily DMAU absorption at doses up to 400 mg daily and effects on estrogen and T levels [44]. Similarly, they found improved absorption with high-fat meals and suppression of estrogen and T in the absence of any serious side effects [44].
In a placebo-controlled, double-blinded, randomized phase I trial, Thirumalai et al. 2019 [43] investigated safety, tolerability, and adverse events associated with oral DMAU over 28 days of treatment, as well as pharmacokinetics, pharmacodynamics, hormonal changes, and sperm counts. The study found suppression of T at even the lowest dose of DMAU and dose-dependent suppression of LH and FSH, theoretically sufficient to suppress spermatogenesis with treatment for 10 weeks [57]. No serious side effects were observed; several participants reported decreased libido or erectile dysfunction, particularly at the highest tested dose, but participants did not report this affecting their sexual or erectile satisfaction [43]. Of note, DMAU was taken after a meal containing 25–30 g of fat, reflecting a typical Western diet but approximately half the fat content of the Ayoub et al. 2017 study [44]. In a secondary analysis of this trial’s samples and data, Thirumalai et al. 2021 found dose-dependent suppression of T and estrogen as well as an increase in a marker for bone formation over 28 days [67]. In another secondary analysis comparing metabolic effects of DMAU and 11β-MNTDC (discussed below), Yuen et al. 2021 found that DMAU caused a mean weight gain of 1.2 or 2.0 kg with 200 or 400 mg daily dosing, respectively, and mild lipid changes, but there were no serious adverse effects or signs of overt insulin resistance [46]. Collectively, these analyses indicate that orally dosed DMAU is well-tolerated and shows promise as a male contraceptive.
Today, there are two ongoing trials with DMAU, run by Drs. Christina Wang, MD, out of the University of California Los Angeles and Stephanie Page, MD, PhD, out of the University of Washington [41, 42]. Per ClinicalTrials.gov, both are reportedly still recruiting. The first is a phase I trial comparing a single injection of intramuscular (80-800 mg) vs. subcutaneous (50-200 mg) DMAU and is primarily assessing safety, pharmacodynamics, and hormonal suppression in healthy males [41]. Completion is estimated for December 2024. The second is a phase II trial primarily investigating the ability of orally dosed DMAU with or without a low dose of levonorgestrel (a progestin) to suppress spermatogenesis after 12 weeks treatment; secondary outcomes include hormonal suppression, serious adverse events, systemic symptoms, and tolerability [42]. Ideally, these ongoing studies will shed further light on the optimal route and dose of DMAU administration to guide efficacy trials.
11β-methyl-19-nortestosterone dodecylcarbonate (11β-MNTDC)
11β-MNTDC is a testosterone derivative active at both AR and PR; it does not undergo aromatization and therefore lacks estrogenic effects [48, 61, 65]. Like DMAU, 11β-MNTDC is a pro-drug and is converted to 11β-methyl-19-nortestosterone (11β-MNT), which is structurally similar to DMA [68]. However, 11β-MNT’s affinity for AR and PR is more balanced than that of DMA (which favors AR), so side effect profiles may vary [48]. In preclinical animal studies, 11β-MNTDC was shown to effectively suppress serum gonadotropins [65] and exert even less liver toxicity than other androgens, including DMAU [63].
Several clinical trials have investigated 11β-MNTDC. The first major human trial was directed by Drs. Wang and Page and published in 2019 [48]. Twelve healthy adult males were given a single oral dose of 100-800 mg 11β-MNTDC with a high-fat meal or fasting, then assessed for pharmacokinetics, adverse effects, serum gonadotropins, and T levels. Like DMAU, 11β-MNTDC absorption was improved with high-fat meal, treatment was overall well-tolerated, and T was suppressed in a dose-dependent manner from 200 mg upwards [48]. Gonadotropin levels were not significantly reduced with a single dose, but this was addressed in a follow-up study published in 2020 [47]. This randomized, placebo-controlled phase II trial was again directed by Drs. Wang and Page, and participants received a daily oral dose of 200 or 400 mg 11β-MNTDC for 28 consecutive days. 11β-MNTDC was taken after a meal containing 25–30 g of fat [47], a more typical fat content per meal than in the previous trial [48]. Ultimately, 11β-MNTDC was well-tolerated; participants reported no serious adverse events, no one discontinued the trial due to side effects, and all reported side effects were mild or moderate. The most common sides effects were headache, acne, and decreased libido in 16% of participants [47]. Mood symptoms were reported, but they were comparable to those seen with currently available female estrogen-containing contraceptives [69,70,71]. 11β-MNTDC caused dose-dependent suppression of LH and FSH, and more participants in the 400 mg group had suppression to LH and FSH levels < 1.0 IU/L, the threshold at which spermatogenesis will be suppressed in nearly 90% of participants [57].
Efficacy trials are still needed for 11β-MNTDC, but between the two clinical trials and a secondary analysis comparing metabolic effects of DMAU and 11β-MNTDC (DMAU discussed above), 11β-MNTDC demonstrated acceptable safety profiles. Levels of T, estradiol, and sex hormone binding globulin (SHBG) were all suppressed, but these changes did not correlate with side effects or changes in serum chemistries [46,47,48]. 11β-MNTDC slightly increased participant weight and serum low-density lipoprotein (LDL) cholesterol levels, but there were no serious adverse events or signs of overt insulin resistance [46]. Results-to-date warrant clinical trials evaluating efficacy and safety using a larger number of participants.
Non-hormonal methods
Several non-hormonal methods show promise in the field of male contraception, and two are either near human study or recently began human trials. In theory, these methods lack hormonal side effects, such as acne or mood symptoms, as well as the societal stigmas and false beliefs associated with hormonal contraception in the United States [6, 72]. The non-hormonal methods showing the most potential or closest to market, particularly those that inhibit spermatogenesis, motility, or vas deferens passage, will be discussed in greatest depth.
Spermatogenesis
All-trans retinoic acid (RA), also known as tretinoin, is derived from vitamin A and plays global roles in cell growth and development. RA plays essential roles in spermatogenesis and acts through binding the retinoic acid receptor alpha (RARA) located in the testes [73, 74]. The first human trial targeting RARA was conducted over 60 years ago with the non-selective RA biosynthesis inhibitor WIN 18,446 [75]. Sixty men were treated for one year, and spermatogenesis was suppressed in all participants throughout. However, off-target effects including inhibition of aldehyde dehydrogenase 2 in the liver unfortunately lead to a severe disulfiram-like reaction, effectively making the drug unmarketable [75]. Since then, the pharmaceutical company Bristol-Myers Squibb (BMS) designed and, with other labs, demonstrated effective, reversible suppression of spermatogenesis in mice with the pan-antagonist BMS-189,453 [76,77,78]. Theoretically, reversible alpha-selective agents would effectively and safely suppress sperm production without the systemic side effects of pan-antagonists. In other words, this would be an ideal method of contraception. Early attempts, most notably BMS-189,532 and BMS-189,614, lacked the efficacy of the pan-antagonist (WIN 18,446) by oral, intravenous, or intraperitoneal routes [79], but RARA remains a strong potential target for male contraception.
Bromodomains are amino acid segments in proteins that facilitate specific protein-protein interactions and a wide variety of cellular functions [80, 81]. One of these bromodomains, bromodomain testis-specific protein (BRDT), is required for spermatogenesis, and males with BRDT gene mutations are infertile with abnormal sperm morphology and impaired motility [82, 83]. Like RARA inhibition, specific inhibition of BRDT would theoretically suppress sperm production without the systemic effects of pan-inhibitors or hormonal methods. Indeed, inhibition of BRDT has been shown to effectively suppress spermatogenesis in male rodents using the small molecule JQ1 [84]. In this study, JQ1 was safe, reversible, and lacked obvious transgenerational effects, but authors noted potential off-target binding that could be reduced or prevented through design of more specific molecular inhibitors [84]. Progress has been made in the search for more specific BRDT inhibitors [85,86,87,88], but the compounds have yet to be tested in vivo and are therefore far from human trials.
Males express distinct testis-specific serine/threonine kinases (TSSK) that play spermatogenic roles in spermatids [89]. Mice with TSSK1 and TSSK2, TSSK3, or TSSK 6 deletions and human males with TSSK2 mutations are infertile, suggesting potential non-hormonal targets for contraception [90,91,92,93]. Of these, research into TSSK2 has shown the most progress. Since generation of enzymatically active, isolated TSSK2 [94], several inhibitors have demonstrated potent in vitro inhibition of TSSK2 [95]. To our knowledge, these inhibitors have yet to undergo in vivo study.
Motility
In order to reach and fertilize oocytes, sperm must travel through the female reproductive tract. This quality is termed motility, and immotile sperm are a major contributor to male-factor infertility [96]. Theoretically, by targeting enzymes or receptors that play essential roles in motility and are present only in sperm, one may reversibly immobilize sperm without systemic side effects. Eppin is an enzyme made in the testes that binds to the surface of sperm to play essential roles in motility [97]. Both immunization against eppin and molecular inhibition using the inhibitor EP055 has been shown to significantly, transiently reduce sperm motility [98, 99]. Although these studies were both done with small sample sizes and much work is needed before eppin inhibition may see clinical trials, no severe side effects were noted in these animal studies, suggesting that eppin may hold promise as a non-hormonal target [100].
In a similar vein as eppin, soluble adenylyl cyclase (sAC) is an intracellular signaling molecule needed for sperm capacitation, motility, and acrosome formation [101,102,103]. Several compounds have been tested in preclinical in vitro studies and shown to effectively inhibit sAC in mouse and human sperm [101, 104]. Indeed, sAC inhibition stands as a strong candidate for male contraception, and two recent studies have been conducted by Drs. Lonny Levin, PhD, and Jochen Buck, MD, PhD, out of Weill Cornell Medicine.
The first study by the Levin-Buck lab intricately compared capacitation and motility of sperm from sAC null mice and from healthy, wild type mice [105]. In vitro, they demonstrated that sAC plays essential roles in capacitation. In vivo, sAC null mice mated similarly to wild type mice, but their sperm were unable to migrate through the female reproductive tract. Essentially, these sperm were immotile [105]. Improving on the inhibitors mentioned above [102], a recent, well-designed study by the Levin-Buck lab investigated the new compound TDI-11,861; they demonstrated that a single oral or intraperitoneal dose of TDI-11,861 acutely inhibits sAC in mice, impairing capacitation and motility [103]. Importantly, the mice in this study had no changes in behavior, no obvious toxicity, and no pregnancies when treated within 2.5 h of mating [103]. With completion of this proof-of-concept study, authors anticipate additional safety and transgenerational studies to follow.
Calcium plays several signaling roles in sperm, including modulation of motility through activating sAC [96]. Extracellular calcium enters sperm flagella, the organelle that propels sperm, primarily through the cell type-specific cation channel of sperm (CatSper) [106]. Studies nearly 15 years ago demonstrated that immunologic inhibition of CatSper significantly suppresses sperm motility [107]; since, several compounds (RU1968 and HC-056456) have demonstrated effective inhibition of CatSper in vitro [108, 109] and preliminarily in vivo [110]. Several new compounds have been identified, synthesized, and tested on human sperm in vitro with excellent efficacy and safety profiles, at least on a cellular level [111]. Additional in vivo animal studies are anticipated.
Slowpoke homolog 3 (SLO3) is the main potassium channel in sperm and has functions directly related to calcium signaling and the CatSper channel [112, 113]. Like CatSper, SLO3 is specific to sperm and has functions essential for male fertility, making it an ideal target for male contraception [114,115,116]. A highly specific inhibitor of SLO3, VU0546110, has been identified and shown in vitro to inhibit sperm motility and acrosome reactions [117]. Better yet, at least one compound (termed “7 a” by Carlson et al. 2022) has been identified that blocks both SLO3 and CatSper, indicating potential for synergistic inhibition of sperm motility [111].
Vas deferens occlusion
The final target we wish readers to know about is physical obstruction of the vas deferens, termed ‘vas occlusion’, via gel injection to physically disrupt sperm during passage through the vas deferens. The benefits of this approach include fast installation (i.e. a quick injection at an outpatient visit) and relatively fast onset of action. A major barrier has been reversibility, but once overcome this approach may hold strong potential in male contraception. Several distinct polymers have been studied, including two styrene compounds termed “reversible inhibition of sperm under guidance” (RISUG) in India [118,119,120] and Valsalgel in the United States [121,122,123], and silicone and polyurethane compounds in the People’s Republic of China [124, 125]. The most recent trial of RISUG showed high contraceptive efficacy and a favorable safety profile [120], but human trials demonstrating reversibility of RISUG are needed. Despite these setbacks, one newer compound is being investigated in an ongoing clinical trial [126]. This new compound is a proprietary hydrogel, named ADAM by its founding company, Contraline Inc. of Charlottesville, Virginia. The trial started enrolling in late 2022 with a planned 25 total male participants through June 2025; ADAM injections will be done at the Epworth Freemasons Hospital in Melbourne, Australia. The primary outcome is adverse events, and secondary outcomes include percentage of participants achieving azoospermia and any serious adverse events [126].
Limitations of the study
This review is subject to several limitations. The clinical trials discussed above are ongoing, and results have yet to be peer-reviewed and published. This does not yet allow for data-driven conclusions. Although this narrative review focuses on the most recent and ongoing studies of male contraception, authors recognize that it is not comprehensive. As mentioned above in Materials and Methods, several compounds were excluded because they failed to progress to human trials, failed after reaching human trials, or are in early preclinical stages. For these, we advise readers to explore several well-written reviews by Thirumalai and Amory [127], Long et al. [128], or the University of California San Diego urology department [129] that include many of these discontinued approaches.
Conclusions
It is long overdue that male partners share the burden of family planning, and it is the authors’ hope that this will soon be a possibility. Ultimately, we feel that two of the methods discussed above—NES/T and DMAU—show the greatest potential for male contraception in the next decade. However, as clinical trials range from early planning stages to data collection stages, it may be several years before we see the efficacy and safety data needed to apply for FDA approval. In particular, the ongoing phase IIb NES/T trial results will not be published before 2025, and this is the method farthest along ‘the pipeline.’
Despite their many theoretical advantages to hormonal contraception, the non-hormonal targets are further from practical application. Authors recognize that there are many obstacles to reaching human studies, let alone late-stage clinical trials. Clinical trials require years of time, meticulous study, and financial support, and many compounds that perform well in pre-clinical animal studies fall short in human trials. The tools needed to efficiently design and study these non-hormonal targets are relatively young. However, they are already being employed to design and test strong drug candidates. As a society we now possess not only the scientific knowledge, technology, and clinical infrastructure needed to overcome these challenges, but also the social drive. With so many strong candidates, it is our hope that there will soon be several safe, effective, and reversible contraceptive options available to male patients.
Availability of data and materials
Not applicable.
Abbreviations
- 11β-MNT:
-
11β-methyl-19-nortestosterone
- 11β-MNTDC:
-
11β-methyl-19-nortestosterone dodecylcarbonate
- AR:
-
Androgen receptor
- BMS:
-
Bristol-Myers Squibb
- BRDT:
-
Bromodomain testis-specific protein
- CatSper:
-
Cation channel of sperm
- DMA:
-
Dimethandrolone
- DMAU:
-
Dimethandrolone undecanoate
- E2:
-
Estradiol
- FSH:
-
Follicle-stimulating hormone
- GnRH:
-
Gonadotropin-releasing hormone
- HPT:
-
Hypothalamic-pituitary-testicular
- IM:
-
Intramuscular
- LDL:
-
Low-density lipoprotein
- LH:
-
Luteinizing hormone
- LNG:
-
Levonorgestrel
- MENT:
-
7α-Methyl-19-nortestosterone
- NES:
-
Nestorone
- NES/T:
-
Nestorone/testosterone transdermal gel
- NICHD:
-
Eunice Kennedy Shriver National Institute of Child Health and Human Development
- PR:
-
Progesterone receptor
- RA:
-
All-trans retinoic acid
- RARA:
-
Retinoic acid receptor alpha
- RCT:
-
Randomized controlled trial
- sAC:
-
Soluble adenylyl cyclase
- SC:
-
Subcutaneous
- SHBG:
-
Sex hormone binding globulin
- SLO3:
-
Slowpoke homolog 3
- T:
-
Testosterone
- TSSK:
-
Testis-specific serine/threonine kinase
References
Myers C, Jones R, Upadhyay U. Predicted changes in abortion access and incidence in a post-roe world. Contraception. 2019;100(5):367–73. https://doi.org/10.1016/j.contraception.2019.07.139.
Kimport K, Rasidjan MP. Exploring the emotional costs of abortion travel in the United States due to legal restriction. Contraception. 2023;120:109956. https://doi.org/10.1016/j.contraception.2023.109956.
Page ST, Blithe D, Wang C. Hormonal male contraception: getting to market. Front Endocrinol (Lausanne). 2022;13:891589. https://doi.org/10.3389/fendo.2022.891589.
Kimport K. More than a physical burden: women’s mental and emotional work in preventing pregnancy. J Sex Res. 2018;55(9):1096–105. https://doi.org/10.1080/00224499.2017.1311834.
Kimport K. Talking about male body-based contraceptives: the counseling visit and the feminization of contraception. Soc Sci Med. 2018;201:44–50. https://doi.org/10.1016/j.socscimed.2018.01.040.
Alspaugh A, Barroso J, Reibel M, Phillips S. Women’s contraceptive perceptions, beliefs, and attitudes: an integrative review of qualitative research. J Midwifery Womens Health. 2020;65(1):64–84. https://doi.org/10.1111/jmwh.12992.
Sundaram A, Vaughan B, Kost K, Bankole A, Finer L, Singh S, et al. Contraceptive failure in the United States: estimates from the 2006–2010 national survey of family growth. Perspect Sex Reprod Health. 2017;49(1):7–16. https://doi.org/10.1363/psrh.12017.
Vaughan B, Trussell J, Kost K, Singh S, Jones R. Discontinuation and resumption of contraceptive use: results from the 2002 national survey of family growth. Contraception. 2008;78(4):271–83. https://doi.org/10.1016/j.contraception.2008.05.007.
Bearak J, Popinchalk A, Ganatra B, Moller AB, Tunçalp Ö, Beavin C, et al. Unintended pregnancy and abortion by income, region, and the legal status of abortion: estimates from a comprehensive model for 1990–2019. Lancet Glob Health. 2020;8(9):e1152–e61. https://doi.org/10.1016/s2214-109x(20)30315-6.
Finer LB, Zolna MR. Shifts in intended and unintended pregnancies in the United States, 2001–2008. Am J Public Health. 2014;104(Suppl 1):43–8. https://doi.org/10.2105/ajph.2013.301416.
Heinemann K, Saad F, Wiesemes M, White S, Heinemann L. Attitudes toward male fertility control: results of a multinational survey on four continents. Hum Reprod. 2005;20(2):549–56. https://doi.org/10.1093/humrep/deh574.
Eberhardt J, van Wersch A, Meikle N. Attitudes towards the male contraceptive pill in men and women in casual and stable sexual relationships. J Fam Plann Reprod Health Care. 2009;35(3):161–5. https://doi.org/10.1783/147118909788707986.
Krausz C. Male infertility: pathogenesis and clinical diagnosis. Best Pract Res Clin Endocrinol Metab. 2011;25(2):271–85. https://doi.org/10.1016/j.beem.2010.08.006.
Nieschlag E. 10th Summit Meeting consensus: recommendations for regulatory approval for hormonal male contraception. October 22–23, 2006. Contraception. 2007;75(3):166-7. doi: https://doi.org/10.1016/j.contraception.2006.12.001.
Klein CE. The hypothalamic-pituitary-gonadal Axis. In: Kufe DWPR, Weichselbaum RR, et al. editors. Holland-Frei Cancer Medicine. Online: BC Decker Inc; 2003. https://www.ncbi.nlm.nih.gov/books/NBK13386/.
Litwack G. Polypeptide Hormones. In: Human Biochemistry. 2nd ed. Online: Andre Gerhard Wolff, Elsevier Inc. 2022. p. 475–516. doi: https://doi.org/10.1016/B978-0-323-85718-5.00013-3.
von Eckardstein S, Noe G, Brache V, Nieschlag E, Croxatto H, Alvarez F, et al. A clinical trial of 7 alpha-methyl-19-nortestosterone implants for possible use as a long-acting contraceptive for men. J Clin Endocrinol Metab. 2003;88(11):5232–9. https://doi.org/10.1210/jc.2002-022043.
Walton MJ, Kumar N, Baird DT, Ludlow H, Anderson RA. 7alpha-methyl-19-nortestosterone (MENT) vs testosterone in combination with etonogestrel implants for spermatogenic suppression in healthy men. J Androl. 2007;28(5):679–88. https://doi.org/10.2164/jandrol.107.002683.
García-Becerra R, Ordaz-Rosado D, Noé G, Chávez B, Cooney AJ, Larrea F. Comparison of 7α-methyl-19-nortestosterone effectiveness alone or combined with progestins on androgen receptor mediated-transactivation. Reproduction. 2012;143(2):211–9. https://doi.org/10.1530/rep-11-0171.
Pavlou SN, Brewer K, Farley MG, Lindner J, Bastias MC, Rogers BJ, et al. Combined administration of a gonadotropin-releasing hormone antagonist and testosterone in men induces reversible azoospermia without loss of libido. J Clin Endocrinol Metab. 1991;73(6):1360–9. https://doi.org/10.1210/jcem-73-6-1360.
Bagatell CJ, Matsumoto AM, Christensen RB, Rivier JE, Bremner WJ. Comparison of a gonadotropin releasing-hormone antagonist plus testosterone (T) versus T alone as potential male contraceptive regimens. J Clin Endocrinol Metab. 1993;77(2):427–32. https://doi.org/10.1210/jcem.77.2.8345047.
Handelsman DJ, Wishart S, Conway AJ. Oestradiol enhances testosterone-induced suppression of human spermatogenesis. Hum Reprod. 2000;15(3):672–9. https://doi.org/10.1093/humrep/15.3.672.
Turner L, Conway AJ, Jimenez M, Liu PY, Forbes E, McLachlan RI, et al. Contraceptive efficacy of a depot progestin and androgen combination in men. J Clin Endocrinol Metab. 2003;88(10):4659–67. https://doi.org/10.1210/jc.2003-030107.
Handelsman DJ, Conway AJ, Howe CJ, Turner L, Mackey MA. Establishing the minimum effective dose and additive effects of depot progestin in suppression of human spermatogenesis by a testosterone depot. J Clin Endocrinol Metab. 1996;81(11):4113–21. https://doi.org/10.1210/jcem.81.11.8923869.
Kamischke A, Diebäcker J, Nieschlag E. Potential of norethisterone enanthate for male contraception: pharmacokinetics and suppression of pituitary and gonadal function. Clin Endocrinol (Oxf). 2000;53(3):351–8. https://doi.org/10.1046/j.1365-2265.2000.01097.x.
Kamischke A, Heuermann T, Krüger K, von Eckardstein S, Schellschmidt I, Rübig A, et al. An effective hormonal male contraceptive using testosterone undecanoate with oral or injectable norethisterone preparations. J Clin Endocrinol Metab. 2002;87(2):530–9. https://doi.org/10.1210/jcem.87.2.8218.
Meriggiola MC, Costantino A, Saad F, D’Emidio L, Morselli Labate AM, Bertaccini A, et al. Norethisterone enanthate plus testosterone undecanoate for male contraception: effects of various injection intervals on spermatogenesis, reproductive hormones, testis, and prostate. J Clin Endocrinol Metab. 2005;90(4):2005–14. https://doi.org/10.1210/jc.2004-1852.
Behre HM, Zitzmann M, Anderson RA, Handelsman DJ, Lestari SW, McLachlan RI, et al. Efficacy and safety of an injectable combination hormonal contraceptive for men. J Clin Endocrinol Metab. 2016;101(12):4779–88. https://doi.org/10.1210/jc.2016-2141.
WHO Task Force on Methods for the Regulation of Male Fertility. Contraceptive efficacy of testosterone-induced azoospermia in normal men. Lancet. 1990;336(8721):955–9. https://doi.org/10.1016/0140-6736(90)92416-F.
WHO Task Force on Methods for the Regulation of Male Fertility. Contraceptive efficacy of testosterone-induced azoospermia and oligozoospermia in normal men. Fertil Steril. 1996;65(6):821. https://doi.org/10.1016/S0015-0282(16)58221-1.
McLachlan RI, McDonald J, Rushford D, Robertson DM, Garrett C, Baker HWG. Efficacy and acceptability of testosterone implants, alone or in combination with a 5α-reductase inhibitor, for male hormonal contraception. Contraception. 2000;62(2):73–8. https://doi.org/10.1016/S0010-7824(00)00139-6.
Gu Y-Q, Wang X-H, Xu D, Peng L, Cheng L-F, Huang M-K, et al. A multicenter contraceptive efficacy study of injectable testosterone undecanoate in healthy chinese men. J Clin Endocrinol Metab. 2003;88(2):562–8. https://doi.org/10.1210/jc.2002-020447.
Gu Y, Liang X, Wu W, Liu M, Song S, Cheng L, et al. Multicenter contraceptive efficacy trial of injectable testosterone undecanoate in chinese men. J Clin Endocrinol Metab. 2009;94(6):1910–5. https://doi.org/10.1210/jc.2008-1846.
Handelsman DJ, Conway AJ, Boylan LM. Suppression of human spermatogenesis by testosterone implants. J Clin Endocrinol Metab. 1992;75(5):1326–32. https://doi.org/10.1210/jcem.75.5.1430094.
Nieschlag E, Hoogen H, Bölk M, Schuster H, Wickings EJ. Clinical trial with testosterone undecanoate for male fertility control. Contraception. 1978;18(6):607–14. https://doi.org/10.1016/0010-7824(78)90045-8.
Blithe D, Myer K. Study of daily application of Nestorone® (NES) and testosterone (T) combination gel for male contraception. ClinicalTrials.gov: 2023 https://ClinicalTrials.gov/show/NCT03452111.
Amory JK, Blithe DL, Sitruk-Ware R, Swerdloff RS, Bremner WJ, Dart C, et al. Design of an international male contraceptive efficacy trial using a self-administered daily transdermal gel containing testosterone and segesterone acetate (Nestorone). Contraception. 2023:110064. https://doi.org/10.1016/j.contraception.2023.110064.
Anawalt BD, Roth MY, Ceponis J, Surampudi V, Amory JK, Swerdloff RS, et al. Combined nestorone-testosterone gel suppresses serum gonadotropins to concentrations associated with effective hormonal contraception in men. Andrology. 2019;7(6):878–87. https://doi.org/10.1111/andr.12603.
Ilani N, Roth MY, Amory JK, Swerdloff RS, Dart C, Page ST, et al. A new combination of testosterone and nestorone transdermal gels for male hormonal contraception. J Clin Endocrinol Metab. 2012;97(10):3476–86. https://doi.org/10.1210/jc.2012-1384.
Mahabadi V, Amory JK, Swerdloff RS, Bremner WJ, Page ST, Sitruk-Ware R, et al. Combined transdermal testosterone gel and the progestin nestorone suppresses serum gonadotropins in men. J Clin Endocrinol Metab. 2009;94(7):2313–20. https://doi.org/10.1210/jc.2008-2604.
Wang C, Page S. Injectable DMAU for male contraception in healthy male volunteers (CCN015); 2023. https://ClinicalTrials.gov/show/NCT02927210.
Wang C, Page S. Study of spermatogenesis suppression with DMAU alone or with LNG versus placebo alone in normal men. https://ClinicalTrials.gov/show/NCT03455075; 2020.
Thirumalai A, Ceponis J, Amory JK, Swerdloff R, Surampudi V, Liu PY, et al. Effects of 28 days of oral dimethandrolone undecanoate in healthy men: a prototype male pill. J Clin Endocrinol Metab. 2019;104(2):423–32. https://doi.org/10.1210/jc.2018-01452.
Ayoub R, Page ST, Swerdloff RS, Liu PY, Amory JK, Leung A, et al. Comparison of the single dose pharmacokinetics, pharmacodynamics, and safety of two novel oral formulations of dimethandrolone undecanoate (DMAU): a potential oral, male contraceptive. Andrology. 2017;5(2):278–85. https://doi.org/10.1111/andr.12303.
Surampudi P, Page ST, Swerdloff RS, Nya-Ngatchou JJ, Liu PY, Amory JK, et al. Single, escalating dose pharmacokinetics, safety and food effects of a new oral androgen dimethandrolone undecanoate in man: a prototype oral male hormonal contraceptive. Andrology. 2014;2(4):579–87. https://doi.org/10.1111/j.2047-2927.2014.00216.x.
Yuen F, Thirumalai A, Fernando FA, Swerdloff RS, Liu PY, Pak Y, et al. Comparison of metabolic effects of the progestational androgens dimethandrolone undecanoate and 11β-MNTDC in healthy men. Andrology. 2021;9(5):1526–39. https://doi.org/10.1111/andr.13025.
Yuen F, Thirumalai A, Pham C, Swerdloff RS, Anawalt BD, Liu PY, et al. Daily oral administration of the novel androgen 11β-MNTDC markedly suppresses serum gonadotropins in healthy men. J Clin Endocrinol Metab. 2020;105(3):e835–47. https://doi.org/10.1210/clinem/dgaa032.
Wu S, Yuen F, Swerdloff RS, Pak Y, Thirumalai A, Liu PY, et al. Safety and pharmacokinetics of single-dose novel oral androgen 11β-methyl-19-nortestosterone-17β-dodecylcarbonate in men. J Clin Endocrinol Metab. 2019;104(3):629–38. https://doi.org/10.1210/jc.2018-01528.
Kumar N, Koide SS, Tsong Y, Sundaram K. Nestorone: a progestin with a unique pharmacological profile. Steroids. 2000;65(10–11):629–36. https://doi.org/10.1016/s0039-128x(00)00119-7.
Kumar N, Fagart J, Liere P, Mitchell SJ, Knibb AR, Petit-Topin I, et al. Nestorone® as a novel progestin for nonoral contraception: structure-activity relationships and brain metabolism studies. Endocrinology. 2017;158(1):170–82. https://doi.org/10.1210/en.2016-1426.
Sitruk-Ware R, Nath A. The use of newer progestins for contraception. Contraception. 2010;82(5):410–7. https://doi.org/10.1016/j.contraception.2010.04.004.
Fraser IS, Weisberg E, Kumar N, Kumar S, Humberstone AJ, McCrossin L, et al. An initial pharmacokinetic study with a Metered Dose Transdermal System for delivery of the progestogen nestorone as a possible future contraceptive. Contraception. 2007;76(6):432–8. https://doi.org/10.1016/j.contraception.2007.08.006.
FDA News Release. : FDA approves new vaginal ring for one year of birth control. https://www.fda.gov/news-events/press-announcements/fda-approves-new-vaginal-ring-one-year-birth-control (2018). Accessed 2023.
Archer DF, Merkatz RB, Bahamondes L, Westhoff CL, Darney P, Apter D, et al. Efficacy of the 1-year (13-cycle) segesterone acetate and ethinylestradiol contraceptive vaginal system: results of two multicentre, open-label, single-arm, phase 3 trials. Lancet Glob Health. 2019;7(8):e1054–e64. https://doi.org/10.1016/s2214-109x(19)30265-7.
Virro JJ, Besinque K, Carney CE, Gross D, Bernick B, Mirkin S. Long-lasting, patient-controlled, procedure-free contraception: a review of Annovera with a pharmacist perspective. Pharm (Basel). 2020;8(3). https://doi.org/10.3390/pharmacy8030156.
Liu PY, Swerdloff RS, Anawalt BD, Anderson RA, Bremner WJ, Elliesen J, et al. Determinants of the rate and extent of spermatogenic suppression during hormonal male contraception: an integrated analysis. J Clin Endocrinol Metab. 2008;93(5):1774–83. https://doi.org/10.1210/jc.2007-2768.
Roth MY, Ilani N, Wang C, Page ST, Bremner WJ, Swerdloff RS, et al. Characteristics associated with suppression of spermatogenesis in a male hormonal contraceptive trial using testosterone and nestorone(®) gels. Andrology. 2013;1(6):899–905. https://doi.org/10.1111/j.2047-2927.2013.00135.x.
Roe AH, Bartz DA, Douglas PS. Combined estrogen-progestin contraception: Side effects and health concerns. In: Crowley WF, Schreiber CA, editors.Online: UpToDate. 2023. https://www.uptodate.com/contents/combined-estrogen-progestin-contraception-side-effects-and-health-concerns. Accessed 20 April 2023.
Roth MY, Shih G, Ilani N, Wang C, Page ST, Bremner WJ, et al. Acceptability of a transdermal gel-based male hormonal contraceptive in a randomized controlled trial. Contraception. 2014;90(4):407–12. https://doi.org/10.1016/j.contraception.2014.05.013.
Attardi BJ, Hild SA, Reel JR. Dimethandrolone undecanoate: a new potent orally active androgen with progestational activity. Endocrinology. 2006;147(6):3016–26. https://doi.org/10.1210/en.2005-1524.
Attardi BJ, Pham TC, Radler LC, Burgenson J, Hild SA, Reel JR. Dimethandrolone (7alpha,11beta-dimethyl-19-nortestosterone) and 11beta-methyl-19-nortestosterone are not converted to aromatic A-ring products in the presence of recombinant human aromatase. J Steroid Biochem Mol Biol. 2008;110(3–5):214–22. https://doi.org/10.1016/j.jsbmb.2007.11.009.
Sharma S, Ahire D, Basit A, Lajoie M, Wang C, Lee MS, et al. Dimethandrolone, a potential male contraceptive pill, is primarily metabolized by the highly polymorphic UDP-glucuronosyltransferase 2B17 enzyme in human intestine and liver. Drug Metab Dispos. 2022;50(12):1493–500. https://doi.org/10.1124/dmd.122.001041.
Hild SA, Attardi BJ, Koduri S, Till BA, Reel JR. Effects of synthetic androgens on liver function using the rabbit as a model. J Androl. 2010;31(5):472–81. https://doi.org/10.2164/jandrol.109.009365.
Hild SA, Marshall GR, Attardi BJ, Hess RA, Schlatt S, Simorangkir DR, et al. Development of l-CDB-4022 as a nonsteroidal male oral contraceptive: induction and recovery from severe oligospermia in the adult male cynomolgus monkey (Macaca fascicularis). Endocrinology. 2007;148(4):1784–96. https://doi.org/10.1210/en.2006-1487.
Attardi BJ, Marck BT, Matsumoto AM, Koduri S, Hild SA. Long-term effects of dimethandrolone 17β-undecanoate and 11β-methyl-19-nortestosterone 17β-dodecylcarbonate on body composition, bone mineral density, serum gonadotropins, and androgenic/anabolic activity in castrated male rats. J Androl. 2011;32(2):183–92. https://doi.org/10.2164/jandrol.110.010371.
NIDDK. Androgenic Steroids. In: LiverTox: Clinical and Research Information on Drug-Induced Liver Injury [Internet]. Bethesda, MD: National Institute of Diabetes and Digestive and Kidney Diseases. 2020. https://www.ncbi.nlm.nih.gov/books/NBK548931/.
Thirumalai A, Yuen F, Amory JK, Hoofnagle AN, Swerdloff RS, Liu PY, et al. Dimethandrolone undecanoate, a novel, nonaromatizable androgen, increases P1NP in healthy men over 28 days. J Clin Endocrinol Metab. 2021;106(1):e171–e81. https://doi.org/10.1210/clinem/dgaa761.
Attardi BJ, Hild SA, Koduri S, Pham T, Pessaint L, Engbring J, et al. The potent synthetic androgens, dimethandrolone (7α,11β-dimethyl-19-nortestosterone) and 11β-methyl-19-nortestosterone, do not require 5α-reduction to exert their maximal androgenic effects. J Steroid Biochem Mol Biol. 2010;122(4):212–8. https://doi.org/10.1016/j.jsbmb.2010.06.009.
Mu E, Kulkarni J. Hormonal contraception and mood disorders. Aust Prescr. 2022;45(3):75–9. https://doi.org/10.18773/austprescr.2022.025.
de Wit AE, Booij SH, Giltay EJ, Joffe H, Schoevers RA, Oldehinkel AJ. Association of use of oral contraceptives with depressive symptoms among adolescents and young women. JAMA Psychiatry. 2020;77(1):52–9. https://doi.org/10.1001/jamapsychiatry.2019.2838.
Skovlund CW, Mørch LS, Kessing LV, Lidegaard Ø. Association of hormonal contraception with depression. JAMA Psychiatry. 2016;73(11):1154–62. https://doi.org/10.1001/jamapsychiatry.2016.2387.
Frost JJ, Lindberg LD, Finer LB. Young adults’ contraceptive knowledge, norms and attitudes: associations with risk of unintended pregnancy. Perspect Sex Reprod Health. 2012;44(2):107–16. https://doi.org/10.1363/4410712.
Noman MAA, Kyzer JL, Chung SSW, Wolgemuth DJ, Georg GI. Retinoic acid receptor antagonists for male contraception: current status†. Biol Reprod. 2020;103(2):390–9. https://doi.org/10.1093/biolre/ioaa122.
Schleif MC, Havel SL, Griswold MD. Function of retinoic acid in development of male and female gametes. Nutrients. 2022;14(6). https://doi.org/10.3390/nu14061293.
Heller CG, Moore DJ, Paulsen CA. Suppression of spermatogenesis and chronic toxicity in men by a new series of bis(dichloroacetyl) diamines. Toxicol Appl Pharmacol. 1961;3:1–11. https://doi.org/10.1016/0041-008x(61)90002-3.
Chung SS, Wang X, Roberts SS, Griffey SM, Reczek PR, Wolgemuth DJ. Oral administration of a retinoic acid receptor antagonist reversibly inhibits spermatogenesis in mice. Endocrinology. 2011;152(6):2492–502. https://doi.org/10.1210/en.2010-0941.
Chung SS, Wang X, Wolgemuth DJ. Prolonged oral administration of a pan-retinoic acid receptor antagonist inhibits spermatogenesis in mice with a rapid recovery and changes in the expression of influx and efflux transporters. Endocrinology. 2016;157(4):1601–12. https://doi.org/10.1210/en.2015-1675.
Amory JK, Muller CH, Shimshoni JA, Isoherranen N, Paik J, Moreb JS, et al. Suppression of spermatogenesis by bisdichloroacetyldiamines is mediated by inhibition of testicular retinoic acid biosynthesis. J Androl. 2011;32(1):111–9. https://doi.org/10.2164/jandrol.110.010751.
Chung SS, Cuellar RA, Wang X, Reczek PR, Georg GI, Wolgemuth DJ. Pharmacological activity of retinoic acid receptor alpha-selective antagonists in vitro and in vivo. ACS Med Chem Lett. 2013;4(5):446–50. https://doi.org/10.1021/ml300365k.
Cochran AG, Conery AR, Sims RJ 3. Bromodomains: a new target class for drug development. Nat Rev Drug Discov. 2019;18(8):609–28. https://doi.org/10.1038/s41573-019-0030-7.
Fujisawa T, Filippakopoulos P. Functions of bromodomain-containing proteins and their roles in homeostasis and cancer. Nat Rev Mol Cell Biol. 2017;18(4):246–62. https://doi.org/10.1038/nrm.2016.143.
Wisniewski A, Georg GI. BET proteins: investigating BRDT as a potential target for male contraception. Bioorg Med Chem Lett. 2020;30(6):126958. https://doi.org/10.1016/j.bmcl.2020.126958.
Shang E, Nickerson HD, Wen D, Wang X, Wolgemuth DJ. The first bromodomain of Brdt, a testis-specific member of the BET sub-family of double-bromodomain-containing proteins, is essential for male germ cell differentiation. Development. 2007;134(19):3507–15. https://doi.org/10.1242/dev.004481.
Matzuk MM, McKeown MR, Filippakopoulos P, Li Q, Ma L, Agno JE, et al. Small-molecule inhibition of BRDT for male contraception. Cell. 2012;150(4):673–84. https://doi.org/10.1016/j.cell.2012.06.045.
Ayoub AM, Hawk LML, Herzig RJ, Jiang J, Wisniewski AJ, Gee CT, et al. BET bromodomain inhibitors with one-step synthesis discovered from virtual screen. J Med Chem. 2017;60(12):4805–17. https://doi.org/10.1021/acs.jmedchem.6b01336.
Law RP, Atkinson SJ, Bamborough P, Chung CW, Demont EH, Gordon LJ, et al. Discovery of tetrahydroquinoxalines as bromodomain and extra-terminal domain (BET) inhibitors with selectivity for the second bromodomain. J Med Chem. 2018;61(10):4317–34. https://doi.org/10.1021/acs.jmedchem.7b01666.
Yu Z, Ku AF, Anglin JL, Sharma R, Ucisik MN, Faver JC, et al. Discovery and characterization of bromodomain 2-specific inhibitors of BRDT. Proc Natl Acad Sci U S A. 2021;118(9). https://doi.org/10.1073/pnas.2021102118.
Guan X, Cheryala N, Karim RM, Chan A, Berndt N, Qi J, et al. Bivalent BET bromodomain inhibitors Confer increased potency and selectivity for BRDT via protein conformational plasticity. J Med Chem. 2022;65(15):10441–58. https://doi.org/10.1021/acs.jmedchem.2c00453.
Salicioni AM, Gervasi MG, Sosnik J, Tourzani DA, Nayyab S, Caraballo DA, et al. Testis-specific serine kinase protein family in male fertility and as targets for non-hormonal male contraception†. Biol Reprod. 2020;103(2):264–74. https://doi.org/10.1093/biolre/ioaa064.
Xu B, Hao Z, Jha KN, Zhang Z, Urekar C, Digilio L, et al. Targeted deletion of Tssk1 and 2 causes male infertility due to haploinsufficiency. Dev Biol. 2008;319(2):211–22. https://doi.org/10.1016/j.ydbio.2008.03.047.
Nayyab S, Gervasi MG, Tourzani DA, Caraballo DA, Jha KN, Teves ME, et al. TSSK3, a novel target for male contraception, is required for spermiogenesis. Mol Reprod Dev. 2021;88(11):718–30. https://doi.org/10.1002/mrd.23539.
Spiridonov NA, Wong L, Zerfas PM, Starost MF, Pack SD, Paweletz CP, et al. Identification and characterization of SSTK, a serine/threonine protein kinase essential for male fertility. Mol Cell Biol. 2005;25(10):4250–61. https://doi.org/10.1128/mcb.25.10.4250-4261.2005.
Zhang H, Su D, Yang Y, Zhang W, Liu Y, Bai G, et al. Some single-nucleotide polymorphisms of the TSSK2 gene may be associated with human spermatogenesis impairment. J Androl. 2010;31(4):388–92. https://doi.org/10.2164/jandrol.109.008466.
Shetty J, Sinville R, Shumilin IA, Minor W, Zhang J, Hawkinson JE, et al. Recombinant production of enzymatically active male contraceptive drug target hTSSK2 - localization of the TSKS domain phosphorylated by TSSK2. Protein Expr Purif. 2016;121:88–96. https://doi.org/10.1016/j.pep.2016.01.009.
Hawkinson JE, Sinville R, Mudaliar D, Shetty J, Ward T, Herr JC, et al. Potent pyrimidine and pyrrolopyrimidine inhibitors of testis-specific serine/threonine kinase†|2 (TSSK2). ChemMedChem. 2017;12(22):1857–65. https://doi.org/10.1002/cmdc.201700503.
Dcunha R, Hussein RS, Ananda H, Kumari S, Adiga SK, Kannan N, et al. Current insights and latest updates in sperm motility and associated applications in assisted reproduction. Reprod Sci. 2022;29(1):7–25. https://doi.org/10.1007/s43032-020-00408-y.
O’Rand MG, Widgren EE, Hamil KG, Silva EJ, Richardson RT. Functional studies of eppin. Biochem Soc Trans. 2011;39(5):1447–9. https://doi.org/10.1042/bst0391447.
O’Rand MG, Widgren EE, Sivashanmugam P, Richardson RT, Hall SH, French FS, et al. Reversible immunocontraception in male monkeys immunized with eppin. Science. 2004;306(5699):1189–90. https://doi.org/10.1126/science.1099743.
O’Rand MG, Hamil KG, Adevai T, Zelinski M. Inhibition of sperm motility in male macaques with EP055, a potential non-hormonal male contraceptive. PLoS ONE. 2018;13(4):e0195953. https://doi.org/10.1371/journal.pone.0195953.
Silva AAS, Raimundo TRF, Mariani NAP, Kushima H, Avellar MCW, Buffone MG, et al. Dissecting EPPIN protease inhibitor domains in sperm motility and fertilizing ability: repercussions for male contraceptive development. Mol Hum Reprod. 2021;27(12). https://doi.org/10.1093/molehr/gaab066.
Balbach M, Ghanem L, Rossetti T, Kaur N, Ritagliati C, Ferreira J, et al. Soluble adenylyl cyclase inhibition prevents human sperm functions essential for fertilization. Mol Hum Reprod. 2021;27(9). https://doi.org/10.1093/molehr/gaab054.
Miller M, Rossetti T, Ferreira J, Ghanem L, Balbach M, Kaur N, et al. Design, synthesis, and pharmacological evaluation of second-generation soluble adenylyl cyclase (sAC, ADCY10) inhibitors with slow dissociation rates. J Med Chem. 2022;65(22):15208–26. https://doi.org/10.1021/acs.jmedchem.2c01133.
Balbach M, Rossetti T, Ferreira J, Ghanem L, Ritagliati C, Myers RW, et al. On-demand male contraception via acute inhibition of soluble adenylyl cyclase. Nat Commun. 2023;14(1):637. https://doi.org/10.1038/s41467-023-36119-6.
Ramos-Espiritu L, Kleinboelting S, Navarrete FA, Alvau A, Visconti PE, Valsecchi F, et al. Discovery of LRE1 as a specific and allosteric inhibitor of soluble adenylyl cyclase. Nat Chem Biol. 2016;12(10):838–44. https://doi.org/10.1038/nchembio.2151.
Ritagliati C, Ayoub S, Balbach M, Buck J, Levin LR. In vivo characterization of sAC null sperm. Front Cell Dev Biol. 2023;11:1134051. https://doi.org/10.3389/fcell.2023.1134051.
Rahban R, Nef S, CatSper. The complex main gate of calcium entry in mammalian spermatozoa. Mol Cell Endocrinol. 2020;518:110951. https://doi.org/10.1016/j.mce.2020.110951.
Li H, Ding X, Guan H, Xiong C. Inhibition of human sperm function and mouse fertilization in vitro by an antibody against cation channel of sperm 1: the contraceptive potential of its transmembrane domains and pore region. Fertil Steril. 2009;92(3):1141–6. https://doi.org/10.1016/j.fertnstert.2008.07.1751.
Rennhack A, Schiffer C, Brenker C, Fridman D, Nitao ET, Cheng YM, et al. A novel cross-species inhibitor to study the function of CatSper ca(2+) channels in sperm. Br J Pharmacol. 2018;175(15):3144–61. https://doi.org/10.1111/bph.14355.
Carlson AE, Burnett LA, del Camino D, Quill TA, Hille B, Chong JA, et al. Pharmacological targeting of native CatSper channels reveals a required role in maintenance of sperm hyperactivation. PLoS ONE. 2009;4(8):e6844. https://doi.org/10.1371/journal.pone.0006844.
Curci L, Carvajal G, Sulzyk V, Gonzalez SN, Cuasnicú PS. Pharmacological inactivation of CatSper blocks sperm fertilizing ability independently of the capacitation status of the cells: implications for non-hormonal contraception. Front Cell Dev Biol. 2021;9:686461. https://doi.org/10.3389/fcell.2021.686461.
Carlson EJ, Francis R, Liu Y, Li P, Lyon M, Santi CM, et al. Discovery and characterization of multiple classes of human CatSper blockers. ChemMedChem. 2022;17(15):e202000499. https://doi.org/10.1002/cmdc.202000499.
Chávez JC, Ferreira JJ, Butler A, De La Vega Beltrán JL, Treviño CL, Darszon A, et al. SLO3 K + channels control calcium entry through CATSPER channels in sperm. J Biol Chem. 2014;289(46):32266–75. https://doi.org/10.1074/jbc.M114.607556.
Tan Z, Garcia TX. SLO3 in the fast lane: the latest male contraceptive target with a promising small-molecule inhibitor. Proc Natl Acad Sci U S A. 2023;120(8):e2221758120. https://doi.org/10.1073/pnas.2221758120.
Schreiber M, Wei A, Yuan A, Gaut J, Saito M, Salkoff L. Slo3, a novel pH-sensitive K + channel from mammalian spermatocytes. J Biol Chem. 1998;273(6):3509–16. https://doi.org/10.1074/jbc.273.6.3509.
Santi CM, Martínez-López P, de la Vega-Beltrán JL, Butler A, Alisio A, Darszon A, et al. The SLO3 sperm-specific potassium channel plays a vital role in male fertility. FEBS Lett. 2010;584(5):1041–6. https://doi.org/10.1016/j.febslet.2010.02.005.
Zeng XH, Yang C, Kim ST, Lingle CJ, Xia XM. Deletion of the Slo3 gene abolishes alkalization-activated K + current in mouse spermatozoa. Proc Natl Acad Sci U S A. 2011;108(14):5879–84. https://doi.org/10.1073/pnas.1100240108.
Lyon M, Li P, Ferreira JJ, Lazarenko RM, Kharade SV, Kramer M, et al. A selective inhibitor of the sperm-specific potassium channel SLO3 impairs human sperm function. Proc Natl Acad Sci U S A. 2023;120(4):e2212338120. https://doi.org/10.1073/pnas.2212338120.
Guha SK, Singh G, Ansari S, Kumar S, Srivastava A, Koul V, et al. Phase II clinical trial of a vas deferens injectable contraceptive for the male. Contraception. 1997;56(4):245–50. https://doi.org/10.1016/s0010-7824(97)00142-x.
Sharma RS, Mathur AK, Singh R, Das HC, Singh GJ, Toor DPS, et al. Safety & efficacy of an intravasal, one-time injectable & non-hormonal male contraceptive (RISUG): a clinical experience. Indian J Med Res. 2019;150(1):81–6. https://doi.org/10.4103/ijmr.IJMR_635_18.
Lohiya NK, Ansari AS, Sadasukhi TC, Pachera S, Khilwani B, Dhaked RK. RISUG® offers early contraception: an experience during phase III clinical trials. J Reprod Healthc Med. 2022;3:11. https://doi.org/10.25259/JRHM_8_2022.
Waller D, Bolick D, Lissner E, Premanandan C, Gamerman G. Azoospermia in rabbits following an intravas injection of Vasalgel ™. Basic Clin Androl. 2016;26:6. https://doi.org/10.1186/s12610-016-0033-8.
Waller D, Bolick D, Lissner E, Premanandan C, Gamerman G. Reversibility of Vasalgel™ male contraceptive in a rabbit model. Basic Clin Androl. 2017;27:8. https://doi.org/10.1186/s12610-017-0051-1.
Colagross-Schouten A, Lemoy MJ, Keesler RI, Lissner E, VandeVoort CA. The contraceptive efficacy of intravas injection of Vasalgel™ for adult male rhesus monkeys. Basic Clin Androl. 2017;27:4. https://doi.org/10.1186/s12610-017-0048-9.
Zhao SC, Zhang SP, Yu RC. Intravasal injection of formed-in-place silicone rubber as a method of vas occlusion. Int J Androl. 1992;15(6):460–4. https://doi.org/10.1111/j.1365-2605.1992.tb01138.x.
Zhao SC, Lian YH, Yu RC, Zhang SP. Recovery of fertility after removal of polyurethane plugs from the human vas deferens occluded for up to 5 years. Int J Androl. 1992;15(6):465–7. https://doi.org/10.1111/j.1365-2605.1992.tb01139.x.
Eisenfrats K, Lawrentschuk N, Contraline. Safety evaluation of the ADAM system. https://www.clinicaltrials.gov/ct2/show/NCT05134428; 2023.
Thirumalai A, Amory JK. Emerging approaches to male contraception. Fertil Steril. 2021;115(6):1369–76. https://doi.org/10.1016/j.fertnstert.2021.03.047.
Long JE, Lee MS, Blithe DL. Update on novel hormonal and nonhormonal male contraceptive development. J Clin Endocrinol Metab. 2021;106(6):e2381–e92. https://doi.org/10.1210/clinem/dgab034.
Service CA, Puri D, Hsieh TC, Patel DP. Emerging concepts in male contraception: a narrative review of novel, hormonal and non-hormonal options. Ther Adv Reprod Health. 2023;17:26334941221138323. https://doi.org/10.1177/26334941221138323.
Acknowledgements
Not applicable.
Funding
This manuscript was supported by the University of South Dakota Sanford School of Medicine Department of Obstetrics and Gynecology.
Author information
Authors and Affiliations
Contributions
First author EJL and senior author KAH conceived and wrote this review paper. EJL created Fig. 1 using BioRender.com. GFLQ and KLP assisted with literature search and interpretation. All authors approved the final version of this manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Louwagie, E.J., Quinn, G.F., Pond, K.L. et al. Male contraception: narrative review of ongoing research. Basic Clin. Androl. 33, 30 (2023). https://doi.org/10.1186/s12610-023-00204-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12610-023-00204-z
Keywords
- Male contraception
- Reproductive health
- Endocrinology
- Androgens
- Progestin
- Spermatogenesis
- Sperm motility
- Vas deferens