
Abstract
Cryptorchidism occurs frequently in children with cystic fibrosis. Among boys with cryptorchidism and abrogated mini-puberty, the development of the epididymis and the vas deferens is frequently impaired. This finding suggests that a common cause underlies the abnormal development of Ad spermatogonia and the epididymis. The cystic fibrosis transmembrane conductance regulator (CFTR) is an ATP-binding cassette transporter protein that acts as a chloride channel. The CFTR gene has been associated with spermatogenesis and male fertility. In boys with cryptorchidism, prepubertal hypogonadotropic hypogonadism induces suboptimal expression of the ankyrin-like protein gene, ASZ1, the P-element induced wimpy testis-like gene, PIWIL, and CFTR. The abrogated expression of these gene leads to transposon reactivation, and ultimately, infertility. Curative gonadotropin-releasing hormone agonist (GnRHa) treatment stimulates the expression of CFTR and PIWIL3, which play important roles in the development of Ad spermatogonia and fertility. Furthermore, GnRHa stimulates the expression of the epididymal androgen-sensitive genes, CRISP1, WFDC8, SPINK13, and PAX2, which thereby promotes epididymal development. This review focuses on molecular evidence that favors a role for CFTR in cryptorchidism-induced infertility. Based on information available in the literature, we interpreted our RNA-Seq expression data obtained from samples before and after randomized GnRHa treatment in boys with bilateral cryptorchidism. We propose that, in boys with cryptorchidism, CFTR expression is controlled by luteinizing hormone and testosterone. Moreover, CFTR regulates the activities of genes that are important for fertility and Wolffian duct differentiation.
Résumé
La cryptorchidie survient fréquemment chez les enfants atteints de mucoviscidose et une altération du développement de l’épididyme et du canal déférent a été observée chez les garçons cryptorchides avec une mini-puberté abrogée. Cela suggère que le développement anormal des spermatogonies Ad et de l’épididyme ont une cause commune. CFTR est. une protéine de transport de cassette de liaison à l’ATP qui agit comme un canal chlorure. Plus précisément, le CFTR a été associé à la spermatogenèse et à la fertilité masculine. Chez les garçons cryptorchides, l’hypogonadisme hypogonadotrope prépubère induit une expression sous-optimale d’ASZ1, de quatre gènes PIWIL et de CFTR, entraînant la réactivation des transposons et l’infertilité. Le traitement curatif à la GnRHa stimule l’expression des gènes CFTR et PIWIL3 qui sont importants pour le développement des spermatogonies Ad et la fertilité subséquente. En outre. Le traitement à la GnRHa stimule les gènes épididymaires sensibles aux androgènes CRISP1, WFDC8, SPINK13, PAX2 favorisant le développement de l’épididyme. Cette revue se concentre sur les preuves moléculaires en faveur du rôle du CFTR dans l’infertilité causée par la cryptorchidie. Nous avons interprété les données d’expression de RNAseq obtenues avec des échantillons avant et après un traitement randomisé à la GnRHa chez des garçons cryptorchides bilatéraux dans le contexte des informations disponibles dans la littérature. Nous proposons que chez les garçons cryptorchides, le CFTR est. contrôlé par l’hormone lutéinisante (LH) et la testostérone et influence les activités des gènes qui sont importants pour la fertilité et la différenciation du canal de Wolff.
Similar content being viewed by others


Introduction
The cystic fibrosis transmembrane conductance regulator (CFTR) belongs to the family of ATP-binding cassette (ABC) transporter proteins, and it functions as a chloride channel. CFTR undergoes various post-translational modifications, including phosphorylation, SUMOylation, and mono-methylation (Fig. 1A; from www.phosphosite.org). Studies on the structure of CFTR revealed a helix-loop transition in transmembrane helix 8, which is likely critical for the protein’s channel function. This structure constitutes a unique feature not found in other ABC transporters (Fig. 1B) [1]. CFTR is critical for the secretion of ions and the transport of water molecules in epithelial tissues. Point mutations in the CTFR gene have been shown to cause cystic fibrosis. Among the best-studied mutations causes the deletion of a phenylalanine at position 508, which interferes with the folding and localization of the mutant protein [2].

CFTR post-translational modifications and protein structure. A A lollipop plot (from www.phosphosite.org) shows the primary sequence, including functional domains (y-axis), plotted against the number of observations of post-translational modifications (x-axis) described in the literature. Phosphorylation is shown in blue, SUMOylation and tri-methylation are shown in green. B The 3D structure of CFTR, from www.rcsb.org (5UAK), is reproduced with permission from Liu et al. [1]
CFTR is also important for fetal development, epithelial differentiation and regeneration, and the regulation of the epithelial-to-mesenchymal transition [3]. In addition, CFTR is involved in the excitability of neurons, the proper functioning of skeletal-, cardiac-, and smooth muscles, the regulation of cell volume, the transepithelial transport of salt ions, and the acidification of intra- and extracellular compartments [3].
It has been suggested that CFTR plays a role in spermiogenesis [4,5,6]. During this process, spermatids differentiate into spermatozoa by undergoing extensive remodeling, including chromatin condensation, acrosome formation, elongation, cell volume reduction, and flagellum formation [4]. Genetic data are in keeping with single-cell RNA sequencing (scRNA-Seq) expression data that have revealed that a sub-population of male germ cells, at different developmental stages, expressing the CTFR gene above the threshold level of detection (Fig. 2A). These expression data were confirmed at the protein level by mass spectrometry-based proteome profiling and immunohistochemical analysis of testicular sections (Fig. 2B) [5]. Furthermore, CFTR is also expressed in the human epididymis [6]. Human genetic studies have shown that point mutations in the CFTR gene were associated with non-obstructive azoospermia and impaired spermatogenesis [7, 8]. Indeed, males with cystic fibrosis caused by mutations in CFTR have exhibited a wide range of testicular histology, from normal to severely pathological [9, 10]. It has been suggested that defects in CFTR may result in insufficient activation of follicle stimulating hormone (FSH)-induced signal transduction and gene expression, which could lead to impaired spermatogenesis [11]. Infertile, but otherwise healthy males have CFTR mutations at significantly higher frequencies than the expected frequencies in the general population [12]. This observation implies that, although certain CFTR mutations give rise to clinical cystic fibrosis, with debilitating lung and pancreatic problems and congenital bilateral absence of the vas deferens, other CFTR mutations might occur in healthy men, whose only known clinical condition is reduced sperm quality [12].

CFTR mRNA and protein expression. A Single-cell RNA-Seq data are from Wang et al. [13]. The images were retrieved from the Reproductive Genomics Viewer (RGV; https://rgv.genouest.org). B Heat map shows protein profiling data for the indicated samples; data were reported by Kim et al. [14]. The image was retrieved from www.humanproteomemap.org. The color-scale indicates the protein levels
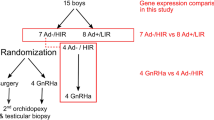
A literature review of the PubMed database was performed using CFTR and cryptorchidism as query terms alone and in combination. Additional publications were identified via the reference lists in the articles found in the PubMed search. Patients, the biopsy samples, histological analyses, and the RNA-Sequencing protocol were described in detail in previous work [15,16,17]. The aim of this review article is to highlight the potential causative role of CTFR in adult male infertility and abnormal Wolffian duct development.
Molecular processes underlying azoospermia induced by cryptorchidism
The pathogenesis of azoospermia in males with mutated CFTR genes might be explained by a mechanism that involves the cAMP-response element binding protein (CREB) pathway [11]. CREB protein levels are reduced in human azoospermia testes, which is consistent with the protein’s down-regulation in cystic fibrosis mouse models and in CFTR-inhibited cultured Sertoli cells [11]. In contrast, among patients with cryptorchidism and abrogated mini-puberty, who are at high risk of developing azoospermia, we observed a weak increase in CREB1 and ATF2 (CREB2) expression levels, compared to the low infertility risk group (LIR), and the CREB3 and CREB5 expression levels remained unaltered (Table 1). Moreover, all CREB genes were downregulated after a curative GnRHa treatment (Table 1). Thus, in cryptorchidism-induced azoospermia, the CREB pathway appears to be less important than it is in patients diagnosed with nonobstructive azoospermia due to conditions unrelated to undescended testes [11]. CFTR is a major hub protein that physically interacts with 831 proteins (see https://thebiogrid.org), including protein involved in targeted proteolysis, epigenetics, and temperature stress response. It was claimed that CFTR was a temperature-sensitive protein. Low temperature favored the proper folding and maturation of CFTR and promoted its insertion into the cell membrane. Conversely, elevated temperatures inhibited these processes [18]. Consequently, it was proposed that the elevated testicular temperature in cryptorchidism might cause a spermatogenic defect by impairing CFTR function, which in turn, might up-regulate the COX20 pathway and disrupt the blood-testis barrier [19]. Moreover, a key part of the heat shock response is the strong upregulation of heat shock proteins (HSPs), which primarily depends on heat shock transcription factors (HSFs). Importantly, HSF genes, endoplasmic reticulum stress genes, and six out of seven HSP genes showed no increase in expression when exposed to elevated body temperatures [20] (Table 2). Furthermore, in contrast to the adult testis, prepubertal cryptorchid testes lack increases in COX20 expression before and after treatment [log2–0.67;FDR0.005], which argues against a major effect of temperature stress.
It was reported that Finish, but not Danish, 3-month-old boys with cryptorchidism had elevated luteinizing hormone (LH) levels, compared to healthy controls [25]. The divergent results were most likely the consequence of diagnostic failure because cryptorchidism was not confirmed with histological analyses of testicular biopsies. Nevertheless, the observed high LH levels were thought to compensate for mild Leydig cell dysfunction, which suggested that cryptorchidism resulted from primary testicular failure [26]. This statement contrasted with at least 10 different reports on LH-RH stimulation tests that demonstrated abnormally low LH responses in boys with cryptorchidism (for references see [27]). Thus, the cause of the low testosterone response is induced at the level of the hypothalamus, and the result is insufficient Leydig cell stimulation. Therefore, most published studies do not support the hypothesis, proposed by Toppari et al., which postulates that that mild Leydig cell dysfunction results from end-organ failure in cryptorchidism [26].
The foundation of the hypogonadotropic hypogonadism hypothesis was laid out in 1976, in Stresa (Italy), when we reported that hormonal values must be analyzed in the context of the presence or absence of Ad spermatogonia [28]. Thus, we grouped patients with cryptorchidism into two categories [28]. The first group comprised patients at high infertility risk (HIR), with testes that lacked Ad spermatogonia (indicating abnormal mini-puberty) and showed pathologically low LH levels, basal and upon stimulation. The other group comprised patients at low infertility risk (LIR), with testes that contained Ad spermatogonia and displayed normal plasma LH values [28]. Ad spermatogonia have a characteristic nuclear feature that distinguishes them from the other germ cells (e.g., fetal, transient, and pale-type (Ap) spermatogonia) of developmental stages [21]. This is a major transformation of gonocytes into Ad spermatogonia and is not simply another step in a succession it represents the switch from a fetal reservoir of stem cells (gonocytes) to an adult reservoir of stem cells (Ad spermatogonia), from which all future germ cells are generated [21]. Insufficient testosterone levels fail to direct gonocytes into the differentiation process in boys with defective mini-puberty, resulting in both abrogated Ad spermatogonia development and infertility [21].
To improve our understanding of cryptorchidism-induced azoospermia, we interpreted testicular GeneChip and RNA-Seq expression data on genes involved in regulating CFTR expression [15, 17, 21, 22, 29]. One of these regulators, FOXA1, facilitates transcription-factor binding to chromatin; for example, it facilitates the binding of androgen and estrogen receptors to chromatin [30]. FOXA1 expression was reduced in samples from an HIR group, and expression was upregulated with a curative GnRHa treatment. Thus, FOXA1 could stimulate CFTR expression in response to GnRHa treatment (Table 2). Another regulator of CFTR, ASZ1, plays a central role during spermatogenesis by repressing transposable elements, which is essential for genome integrity in the germline [31]. ASZ1 is evolutionarily conserved, and it is expressed in germ cells. The ASZ1 protein acts by metabolizing Piwi-interacting RNA (piRNA). piRNA mediates the repression of transposable elements during meiosis by forming complexes composed of piRNAs and PIWI proteins; these complexes govern the methylation and subsequent repression of transposons. ASZ1 expression is down-regulated in patients with HIR (Table 3). PIWIL biogenesis is regulated by ASZ1, HENMT1, FKBP6, and HSP90AA1. These genes were all down-regulated in HIR samples (Table 2). FKBP6 acts as a co-chaperone and represses transposable elements via its interaction with HSP90AA1. piRNA processing is also critically dependent on poly A+-specific RNase-like domain containing 1 (PNLDC1). Men with dysfunctional PNLDC1 and nonobstructive azoospermia showed a concomitant loss of PIWIL1 expression [32]. While PNLDC1 expression in HIR testes is downregulated, no increase after GnRHa treatment was observed, which is consistent with the notion that this gene is not directly involved in the development of azoospermia in cryptorchid patients (Table 2).
Previously, we provided evidence that supported the notion that infertility in cryptorchidism is a consequence of hypogonadotropic hypogonadism-induced alterations in the PIWIL-pathway that undermine transposon repression [23, 24]. It was also shown that testosterone altered testicular function by regulating the expression of Piwi-interacting RNAs [33]. Mutant mice with insufficient testosterone secretion expressed lower levels of Miwi [34]. Moreover, mice deficient in each of the genes essential for silencing the L1 retrotransposons were sterile [35]. We found that individuals with HIR showed little or no expression of the PIWIL4 protein or seven out of the 12 TDRD genes that are important for spermatogenesis [23, 24]. This deficiency was accompanied by low expression of the RNA-helicases, DDX4 and DDX25, which are dependent on gonadotropin and testosterone stimulation [23, 24].
Abnormal gametogenesis results from disturbed PIWIL biogenesis (which involves four PIWIL genes) and insufficient ASZ1, FOXA1, and CFTR functions (Table 2). Importantly, curative GnRHa treatment stimulates the expression of several genes involved in pituitary development and differentiation, neuronal development, and testosterone synthesis pathways. GnRHa treatment also rescues fertility by increasing the expression of CFTR, DMRTC2, PAX7, BRACHYURY/T, TERT, and PIWIL3.
CFTR in abnormal development of the epididymis and vas deferens in cryptorchidism
It was reported that among children with cystic fibrosis the incidence of undescended testis is five to 12 times more common than that observed in a control population [36]. Furthermore, Fedder et al. found the CFTR intron variant IVS8-5 T to be associated with cryptorchidism requiring orchidopexy. However the patient cohort was not classified into HIR and LIR groups [37], Importantly, defective development of the epididymis and the vas deferens occurs more often in boys with cryptorchidism and abrogated mini-puberty, which suggests that testicular and epididymal pathologies share a common cause [38].
Mutations in CFTR are thought to be responsible for the absence of the vas deferens and the distal half of the epididymis. It was suggested that these structural anomalies were caused by abnormal fluid transport in the Wolffian duct, which would then fail to differentiate into the epididymis and vas deferens during post-natal stages of development [12, 39, 40]. In mice, impaired expression of Slc9a3 (a Na/H exchanger), reduces the levels of CFTR and causes obstructive azoospermia [41]. Among patients in the HIR group, expression of SLC9A3’s regulatory cofactor SLC9A3R1 is downregulated (Table 3). Given that GnRHa treatment upregulates SLC9A2, SLC9A4, SLC9AS1, and CFTR and that Slc9a3 mutant mice develop epididymal obstruction, we propose that these genes contribute to the development of the epididymis (Table 3).
Other genes that are important for epididymal development, such as SCNN1A and SCNN1G, are also downregulated in patients with HIR patients (Table 4). SCNN1A encodes a sodium channel that enables cells to generate and transmit electrical signals [42]. An increasing number of studies show that CFTR plays a role in fundamental cellular processes such as fetal development, epithelial differentiation/polarization and regeneration, and the epithelial–mesenchymal transition [43].
Androgens are the primary regulators of epididymal development and function. However, a large body of evidence has suggested that growth factors also play important roles in these processes. Among others, fibroblast growth factor (FGF) is involved in the development and normal functioning of male reproductive organs, including the testis and epididymis [44]. In 2010, we reported that boys with unilateral cryptorchidism showed impaired FGFR1 expression in the undescended testis [45]. Moreover, reduced FGFR1 protein levels were observed in cryptorchid epididymis samples from both humans and rodents [46]. These findings suggested that FGFR1 regulates the development of the epididymal mesenchyme. It appears likely that, in humans and rodents with cryptorchidism, the impairment in FGFR1 protein secretion in the abnormal mesenchyme contributes to epididymis malformation and the lack of epididymis-testes descent. Interestingly, FGFR signaling regulates specific chaperones that control CFTR maturation [39].
LH is necessary for epididymis-testicular descent. LH-receptor knockout mice exhibit bilateral cryptorchidism that can be corrected with testosterone replacement therapy. Specifically, this therapy reverses morphological alterations and changes in gene expression in the knockout mice, except those related to insulin-like factor 3. This finding suggests that testosterone, rather than INSL3, facilitates the completion of testicular descent [47]. Furthermore, in 66% of naturally cryptorchid mice, treatment with LH-releasing hormone induces epididymis-testicular descent, increases testosterone secretion, and normalizes the altered morphology of the cryptorchid epididymis [48]. In boys with cryptorchidism, GnRHa treatment increases testosterone secretion, stimulates epididymis development, and induces completion of the epididymis-testicular descent [49]. Most patients who fail to respond to hormonal treatment have small, irregular epididymides [49].
The presence of GnRH and GnRH receptor mRNAs in normal human non-reproductive tissues suggests that, in addition to regulating gonadotropin secretion from the anterior pituitary, these proteins play an important role in regulating cellular functions in an autocrine or paracrine manner [50]. The hypogonadotropic manifestations of cystic fibrosis may be partly explained by abnormal neuropeptide-vesicle trafficking, due to CFTR mutations [51]. Indeed, a six-month treatment with GnRHa normalized both pituitary function and CFTR expression. In addition, GnRHa treatment promoted chloride-channel function in F508del-CFTR cells, by increasing the stability of CFTR in the membrane [52]. Thus, it is possible that GnRHa directly regulates GnRH-dependent chloride transport in cystic fibrosis (Fig. 3). Consequently, it was suggested that both topical and intra-nasal applications of GnRHa may be potentially beneficial for treating cystic fibrosis [53].

A model of CFTR and cryptorchidism-induced infertility. A schematic diagram outlines the relationships between CFTR and biological processes. A disturbance in any of the steps results in infertility, epididymal maldevelopment, and cryptorchidism
GnRHa treatment also stimulates LH release, which in turn, stimulates testosterone release. Testosterone stimulates the expression of CFTR (Fig. 3) and other androgen-sensitive epidydimal genes, such as CRISP1, WFDC8, SPINK13, PAX2 (Table 4), and the epithelial sodium channel subunits, SCNN1A and SCNN1G (Table 4). Thus, GnRHa therapy contributes to rescuing fertility and improving the morphology and function of the epididymis through numerous pathways (Fig. 3).
In conclusion, the high incidence of abnormal epididymides in patients with HIR may stem from a combination of a degree of prepubertal hypogonadotropic hypogonadism, insufficient CFTR activity, and FGFR1 deficiency. In addition, given that the bronchial system expresses GnRH-R [53], Buserelin could potentially be useful for treating cystic fibrosis in general and for treating boys with cryptorchidism combined with cystic fibrosis, in particular.
Availability of data and materials
Not applicable.
Abbreviations
- ASZ1 :
-
Ankyrin Repeat
SAM And Basic Leucine Zipper Domain Containing 1
- BRACHYURY :
-
T-Box Transcription Factor T
- CFTR :
-
Cystic Fibrosis Transmembrane Conductance Regulator
- COX2 :
-
Cytochrome C Oxidase Assembly Factor COX20
- CREB1 :
-
cAMP Responsive Element Binding Protein 1
- CREB2(ATF2):
-
cAMP Responsive Element Binding Protein 2
- CREB3 :
-
cAMP Responsive Element Binding Protein 3
- CREB5 :
-
cAMP Responsive Element Binding Protein 5
- CRISP1 :
-
Cysteine Rich Secretory Protein 1
- CTTNBP2 :
-
Cortactin Binding Protein 2
- DDX4/25 :
-
DEAD-Box Helicase 4/25
- DMRTC2 :
-
DMRT-Like Family C2
- FGFR1 :
-
Fibroblast Growth Factor Receptor 1
- FKBP6 KBP :
-
Prolyl Isomerase Family Member 6 (Inactive)
- FOXA1 :
-
Fork-head Box A1
- GnRHa:
-
Gonadotropin releasing hormone agonist
- HENMT1 :
-
HEN Methyltransferase 1
- HIR:
-
High infertility risk group
- HSF :
-
Heat Shock Transcription Factor
- HSP90AA1:
-
Heat Shock Transcription Factor 1
- INSL3 :
-
Insulin-Like 3
- LIR:
-
Low infertility risk group
- LH:
-
Luteinizing hormone
- PAX2/7 :
-
Paired Box 2/7
- PIWIL 1–4:
-
Piwi-Like RNA-Mediated Gene Silencing 1–4
- PNLDC1 :
-
PARN-Like Ribonuclease Domain Containing Exonuclease 1
- SCNN1A :
-
Sodium Channel Epithelial 1 Subunit Alpha
- SCNN1G :
-
Sodium Channel Epithelial 1 Subunit Gamma
- SLC9A3 :
-
Solute Carrier Family 9 Member A3
- SPINK13 :
-
Serine Peptidase Inhibitor Kazal Type 13
- TERT :
-
Telomerase Reverse Transcriptase
- WFDC8 :
-
WAP Four-Disulfide Core Domain 8
References
Liu F, Zhang Z, Csanády L, Gadsby DC, Chen J. Molecular structure of the human CFTR. Ion Channel. Cell. 2017;169(1):85–95. e8. https://doi.org/10.1016/j.cell.2017.02.024 PMID: 28340353.
Lukacs GL, Verkman AS. CFTR: folding, misfolding and correcting the ΔF508 conformational defect. Trends Mol Med. 2012;18(2):81–91. https://doi.org/10.1016/j.molmed.2011.10.003 Epub 2011 Dec 3. PMID: 22138491; PMCID: PMC3643519.
Quaresma MC, Pankonien I, Clarke LA, Sousa LS, Silva IAL, Railean V, et al. Mutant CFTR Drives TWIST1 mediated epithelial-mesenchymal transition. Cell Death Dis. 2020;11(10):920. https://doi.org/10.1038/s41419-020-03119-z PMID: 33106471; PMCID: PMC7588414.
Gong XD, Li JC, Cheung KH, Leung GP, Chew SB, Wong PY. Expression of the cystic fibrosis transmembrane conductance regulator in rat spermatids: implication for the site of action of antispermatogenic agents. Mol Hum Reprod. 2001;7(8):705–13. https://doi.org/10.1093/molehr/7.8.705 PMID: 114708575.
Teixeira S, Sá R, Grangeia A, Silva J, Oliveira C, Ferráz L, et al. Immunohystochemical analysis of CFTR in normal and disrupted spermatogenesis. Syst Biol Reprod Med. 2013;59(1):53–9. https://doi.org/10.3109/19396368.2012.718851 Epub 2012 Sep 18. PMID: 22989055.
Leir SH, Yin S, Kerschner JL, Cosme W, Harris A. An atlas of human proximal epididymis reveals cell-specific functions and distinct roles for CFTR. Life Sci Alliance. 2020;3(11):e202000744. https://doi.org/10.26508/lsa.202000744 PMID: 32855272; PMCID: PMC7471510.
Sharma H, Mavuduru RS, Singh SK, Prasad R. Increased frequency of CFTR gene mutations identified in Indian infertile men with non-CBAVD obstructive azoospermia and spermatogenic failure. Gene. 2014;548(1):43–7. https://doi.org/10.1016/j.gene.2014.07.005 Epub 2014 Jul 7.
Levkova M, Chervenkov T, Hachmeriyan M, Angelova L. CFTR gene variants as a reason for impaired spermatogenesis: a pilot study and a Meta-analysis of published data. Hum Fertil (Camb). 2021;15:1–10. https://doi.org/10.1080/14647273.2021.1900608 Epub ahead of print. PMID: 33719834.
Tuerlings JH, Mol B, Kremer JA, Looman M, Meuleman EJ, te Meerman GJ, et al. Mutation frequency of cystic fibrosis transmembrane regulator is not increased in oligozoospermic male candidates for intracytoplasmic sperm injection. Fertil Steril. 1998;69(5):899–903.
Larriba S, Bassas L, Gimenez J, Ramos MD, Segura A, Nunes V, et al. Testicular CFTR splice variants in patients with congenital absence of the vas deferens. Hum Mol Genet. 1998;7(11):1739–43. https://doi.org/10.1093/hmg/7.11.1739 PMID: 9736775.
Xu WM, Chen J, Chen H, Diao RY, Fok KL, Dong JD, et al. Defective CFTR-dependent CREB activation results in impaired spermatogenesis and azoospermia. PLoS One. 2011;6(5):e19120.
Wong PYD. CFTR gene and male fertility. Mol Hum Reprod. 1998;4:107–10.
Wang M, Liu X, Chang G, Chen Y, An G, Yan L, et al. Single-Cell RNA Sequencing Analysis Reveals Sequential Cell Fate Transition during Human Spermatogenesis. Cell Stem Cell. 2018;23(4):599–614.e4. https://doi.org/10.1016/j.stem.2018.08.007 Epub 2018 Aug 30. PMID: 30174296.
Kim MS, Pinto SM, Getnet D, Nirujogi RS, Manda SS, Chaerkady R, et al. A draft map of the human proteome. Nature. 2014;509(7502):575–81. https://doi.org/10.1038/nature13302 PMID: 24870542; PMCID: PMC4403737.
Vincel B, Verkauskas G, Bilius V, Dasevicius D, Malcius D, Jones B, et al. Gonadotropin-releasing hormone agonist corrects defective Mini-puberty in boys with cryptorchidism: a prospective randomized study. Biomed Res Int. 2018;2018:4651218. https://doi.org/10.1155/2018/4651218 PMID: 30065939; PMCID: PMC6051297.
Hadziselimovic F, Hoecht B. Testicular histology related to fertility outcome and postpubertal hormone status in cryptorchidism. Klin Padiatr. 2008;220(5):302–7. https://doi.org/10.1055/s-2007-993194 Epub 2008 Apr 9. PMID: 18401814.
Hadziselimovic F, Hadziselimovic NO, Demougin P, Krey G, Hoecht B, Oakeley EJ. EGR4 is a master gene responsible for fertility in cryptorchidism. Sex Dev. 2009;3(5):253–63. https://doi.org/10.1159/000249147 Epub 2009 Oct 14. PMID: 19828938.
Denning GM, Anderson MP, Amara JF, Marshall J, Smith AE, Welsh MJ. Processing of mutant cystic fibrosis transmembrane conductance regulator is temperature sensitive. Nature. 1992;358:761–4.
Chen H, Ruan YC, Xu WM, Chen J, Chan HC. Regulation of male fertility by CFTR and implications in male infertility. Hum Reprod Update. 2012;18(6):703–13. https://doi.org/10.1093/humupd/dms027 Epub 2012 Jun 17. Erratum in: Hum Reprod Update. 2012;18(6):715. PMID: 22709980.
Hadziselimovic F. Temperature is not a major factor in the differentiation of gonocytes into ad spermatogonia and fertility outcome in congenitally cryptorchid boys. Basic Clin Androl. 2022;32(1):2. https://doi.org/10.1186/s12610-021-00152-6 PMID: 35000579; PMCID: PMC8744351.
Gegenschatz-Schmid K, Verkauskas G, Demougin P, Bilius V, Dasevicius D, Stadler MB, et al. DMRTC2, PAX7, BRACHYURY/T and TERT are implicated in male germ cell development following curative hormone treatment for cryptorchidism-induced infertility. Genes (Basel). 2017;8(10):267. https://doi.org/10.3390/genes8100267 PMID: 29019938; PMCID: PMC5664117.
Hadziselimovic F, Gegenschatz-Schmid K, Verkauskas G, Demougin P, Bilius V, Dasevicius D, et al. GnRHa treatment of Cryptorchid boys affects genes involved in hormonal control of the HPG Axis and fertility. Sex Dev. 2017;11(3):126–36. https://doi.org/10.1159/000471937 Epub 2017 May 16. PMID: 28505621.
Hadziselimovic F, Hadziselimovic NO, Demougin P, Krey G, Oakeley EJ. Deficient expression of genes involved in the endogenous defense system against transposons in cryptorchid boys with impaired mini-puberty. Sex Dev. 2011;5:287–93.
Hadziselimovic F, Hadziselimovic NO, Demougin P, Krey G, Oakeley E. Piwi-pathway alteration induces LINE-1 transposon derepression and infertility development in cryptorchidism. Sex Dev. 2015;9:98–104. https://doi.org/10.1159/000375351 Epub 2015 Mar 13. PMID: 25791297.
Suomi A-M, Main KM, Kaleva M, Schmidt IM, Chellakooty M, Virtanen HE, et al. Hormonal changes in 3-month-old cryptorchid boys. J Clin Endocrinol Metab. 2006;91:953e8.
Toppari J, Kaleva M, Virtanen HE, Main KM. Skakkebaek NE Luteinizing hormone in testicular descent. Mol Cell Endocrinol. 2007;269:34e7.
Hadziselimovic F. On the descent of the epididymo-testicular unit, cryptorchidism, and prevention of infertility. Basic Clin Androl. 2017;27:21. https://doi.org/10.1186/s12610-017-0065-8 PMID: 29163975; PMCID: PMC5686796.
Hadziselimovic F, Herzog B, Girard J. Lack of germ cells and endocrinology in cryptorchid boys from one to six years of life. In: Biereich JR, Giarola A, editors. Cryptorchidism. London: Academic Press; 1979. p. 129–34.
Hadziselimovic F, Hadziselimovic NO, Demougin P, Oakeley EJ. Testicular gene expression in cryptorchid boys at risk of azoospermia. Sex Dev. 2011;5:49–59.
Gao S, Chen S, Han D, Wang Z, Li M, Han W, et al. Chromatin binding of FOXA1 is promoted by LSD1-mediated demethylation in prostate cancer. Nat Genet. 2020;52(10):1011–7. https://doi.org/10.1038/s41588-020-0681-7.
Ott CJ, Blackledge NP, Kerschner JL, Leir SH, Crawford GE, Cotton CU, et al. Intronic enhancers coordinate epithelial-specific looping of the active CFTR locus. Proc Natl Acad Sci U S A. 2009;106(47):19934–9. https://doi.org/10.1073/pnas.0900946106 Epub 2009 Nov 6. PMID:19897727; PMCID: PMC2785270.
Nagirnaja L, Mørup N, Nielsen JE, Stakaitis R, Golubickaite I, Oud MS, et al. Variant PNLDC1, defective piRNA processing, and Azoospermia. N Engl J Med. 2021;385(8):707–19.
Kang HJ, Moon MJ, Lee HY, Han SW. Testosterone alters testis function through regulation of piRNA expression in rats. Mol Biol Rep. 2014;41(10):6729–35. https://doi.org/10.1007/s11033-014-3558-y Epub 2014 Jul 6. PMID: 24997694.
Chuma S, Nakano T. piRNA and spermatogenesis in mice. Phil Trans R Soc Lond B Biol Sci. 2012;368(1609):20110338. https://doi.org/10.1098/rstb.2011.0338 PMID: 23166399; PMCID: PMC3539364.
Bortvin A. Piwi-interacting RNAs (piRNAs) mouse testis perspective. Biochemistry (Mosc). 2013;78:592–602.
Holsclaw DS, Perlmutter AD, Jockin H, Shwachman H. Genital abnormalities in male patients with cystic fibrosis. J Urol. 1971;106(4):568–74. https://doi.org/10.1016/s0022-5347(17)61343-0 PMID: 4399160.
Fedder J, Jørgensen MW, Engvad B. Prevalence of CBAVD in azoospermic men carrying pathogenic CFTR mutations - evaluated in a cohort of 639 non-vasectomized azoospermic men. Andrology. 2021;9:588–98. https://doi.org/10.1111/andr.12925.
Hadziselimovic F, Herzog B. Hodenerkrankungen im Kindesalter. Bibliothek für Kinderheilkunde. Stuttgart: Hippokrates Verlag; 1990. p. 3-7773-0929-X.
Trzcińska-Daneluti AM, Chen A, Nguyen L, Murchie R, Jiang C, Moffat J, et al. RNA interference screen to identify kinases that suppress rescue of ΔF508-CFTR. Mol Cell Proteomics. 2015;14(6):1569–83.
Tizzano EF, Buchwald M. Recent advances in cystic fibrosis research. J Pediatr. 1993;122(6):985–8. https://doi.org/10.1016/s0022-3476(09)90033-6 PMID: 8501582.
Wang YY, Lin YH, Wu YN, Chen YL, Lin YC, Cheng CY, et al. Loss of SLC9A3 decreases CFTR protein and causes obstructed azoospermia in mice. PLoS Genet. 2017;13(4):e1006715. https://doi.org/10.1371/journal.pgen.1006715 PMID: 28384194; PMCID: PMC5398719.
Pierandrei S, Truglio G, Ceci F, Del Porto P, Bruno SM, Castellani S, et al. DNA methylation patterns correlate with the expression of SCNN1A, SCNN1B, and SCNN1G (epithelial Sodium Channel, ENaC) genes. Int J Mol Sci. 2021;22(7):3754. https://doi.org/10.3390/ijms22073754.
Farinha CM, Gentzsch M. Revisiting CFTR interactions: old partners and new players. Int J Mol Sci. 2021;22(24):13196. https://doi.org/10.3390/ijms222413196 PMID: 34947992; PMCID: PMC8703571.
Cotton LM, O’Bryan MK, Hinton BT. Cellular signaling by fibroblast growth factors (FGFs) and their receptors (FGFRs) in male reproduction. Endocr Rev. 2008;29:193–216.
Hadziselimovic NO, de Geyter C, Demougin P, Oakeley EJ, Hadziselimovic F. Decreased expression of FGFR1, SOS1, RAF1 genes in cryptorchidism. Urol Int. 2010;84:353–61.
Hadziselimovic F. Involvement of Fibroblast Growth Factors and Their Receptors in Epididymo-Testicular Descent and Maldescent. Mol Syndromol. 2016;6(6):261–7. https://doi.org/10.1159/000444033 Epub 2016 Feb 2. PMID: 27022326; PMCID: PMC4802980.
Yuan FP, Lin DX, Rao CV, Lei ZM. Cryptorchidism in LhrKO animals and the effect of testosterone- replacement therapy. Hum Reprod. 2006;21:936–42.
Hadziselimović F. Funktionelle Morphologie und Pathologie der Nebenhoden und ihr Einfluss auf den Descensus Testiculorum. [Functional morphology and pathology of the epididymis and influence on testicular descent]. Morphol Med. 1981;1:31–42.
Bica DTG, Hadziselimovic F. The behavior of epididymis, processus vaginalis, and testicular descent in cryptorchid boys treated with buserelin. Eur J Pediatr. 1993;152(Suppl 2):S38–42.
Kakar SS, Jennes L. Expression of gonadotropin-releasing hormone and gonadotropin-releasing hormone receptor mRNAs in various non-reproductive human tissues. Cancer Lett. 1995;98(1):57–62.
Liposits Z, Merchenthaler I, Wetsel WC, Reid JJ, Mellon PL, Weiner RI, et al. Morphological characterization of immortalized hypothalamic neurons synthesizing luteinizing hormone-releasing hormone. Endocrinology. 1991;129(3):1575–83. https://doi.org/10.1210/endo-129-3-1575 PMID: 1874189.
Benz N, Le Hir S, Norez C, Kerbiriou M, Calvez M-L, Becq F, et al. Improvement of chloride transport defect by gonadotropin-releasing hormone (GnRH) in cystic fibrosis epithelial cells. PLoS One. 2014;9(2):e88964.
Calvez ML, Benz N, Huguet F, Saint-Pierre A, Rouillé E, Coraux C, et al. Buserelin alleviates chloride transport defect in human cystic fibrosis nasal epithelial cells. PLoS One. 2017;12(11):e0187774. https://doi.org/10.1371/journal.pone.0187774 PMID: 29145426; PMCID: PMC5690610.
Acknowledgements
We gratefully acknowledge the excellent services provided by the PubMed literature database and the PDB, Phosphosite and BioGrid protein databases.
Funding
none.
Author information
Authors and Affiliations
Contributions
FH interpreted the data and wrote the manuscript. GV and MS contributed to the manuscript. All author(s) read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no conflicts of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Hadziselimovic, F., Verkauskas, G. & Stadler, M. A novel role for CFTR interaction with LH and FGF in azoospermia and epididymal maldevelopment caused by cryptorchidism. Basic Clin. Androl. 32, 10 (2022). https://doi.org/10.1186/s12610-022-00160-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12610-022-00160-0