
Abstract
Many hormones act on the hypothalamus to control hunger and satiety through various pathways closely associated with several factors. When food is present in the gastro intestinal (GI) tract, enteroendocrine cells (EECs) emit satiety signals such as cholecystokinin (CCK), glucagon like peptide-1 (GLP-1) and peptide YY (PYY), which can then communicate with the vagus nerve to control food intake. More specifically, satiety has been shown to be particularly affected by the GLP-1 hormone and its receptor agonists that have lately been acknowledged as a promising way to reduce weight. In addition, there is increasing evidence that normal flora is also involved in the peripheral, central, and reward system that impact satiety. Moreover, neurologic pathways control satiety through neurotransmitters. In this review, we discuss the different roles of each of the GLP-1 hormone and its agonist, gut microbiomes, as well as neurotransmitters and their interconnected relation in the regulation of body’s satiety homeostasis.
Similar content being viewed by others
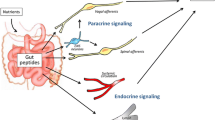


Introduction
Hormones involved in satiety
The hypothalamus is a brain structure that plays a major part in the complex neural network that controls the homeostatic regulation of energy balance [1]. It is the center for all hormones involved in the satiety mechanism regulation. Of these hormones are peripheral anorexigenic hormones that improve satiety such as glucagon like peptide -1 (GLP-1), peptide YY (PYY), insulin, cholecystokinin (CCK) and leptin. Even ghrelin, which is a hormone that stimulates hunger, acts on hypothalamus [2]. It is known that central Pro-opiomelanocortin (POMC) and cocaine–amphetamine-regulated transcript-containing (CART) neurons in the hypothalamus increase satiety, while neurons carrying neuropeptide Y (NPY) and agouti-related peptide (AgRP) trigger the desire to eat [3]. The hypothalamus also transmits signals to the mesolimbic rewards circuit, which is crucial for addiction, impulsive behavior, and food reward [4]. Signaling of the peripheral hormones mentioned earlier have been even connected to impulsivity [5] and addiction [6]. This emphasizes further the role of physiological and/or psychological factors in eating disorders [7].
Microbiota role in metabolism
Over the last decade, there has been an increasing interest in the role of the gut microbiota in the physiology of both health and disease [8]. These multitudes of intestinal inhabitants collaborate with the host in a crucial evolutionary relationship to preserve homeostasis [9]. A growing body of evidence demonstrates that the gut microbiota has a substantial impact on the bidirectional connection between the GI tract and the brain, known as the microbiota–gut–brain axis [10]. Particularly, it is becoming more recognized that the host's microbiome affects how efficiently it uses its own energy, which can lead to metabolic and eating disorders [11]. For instance, changing gut microbiota composition has been linked to anorexia nervosa [12] and obesity [13].
Neuroscience in relation to satiety
The arcuate nucleus of the hypothalamus is the region that seems to be most crucial in the integration of signals about energy flux. It gets signals related to immediate satisfaction (satiety related to the early stages of digestion, particularly in the stomach and initial parts of the digestive system) that interact with signals derived from adiposity. POMC, a precursor for a number of peptides like α-melanocyte stimulating hormone (α-MSH), endorphins, and adrenocorticotropic hormone (ACTH), is expressed by melanocortin system neurons in the arcuate nucleus [14]. Melanocortin 4 receptor (MC4R), the main receptor for α -MSH, is found in the arcuate nucleus as well as various other parts of the brain [15]. When α-MSH or an agonist binds to the MC4R, catabolic pathways are triggered, resulting in hypophagia, thermogenesis, and weight loss [16], whereas MC4R antagonists cause weight gain and hyperphagia [17].
Body
Hormones and satiety-GLP-1
GLP-1 and satiety
GLP-1 is an incretin hormone made by the L cells of the intestine. GLP-1 receptors are abundant in the arcuate nucleus as well as the hypothalamus which contains projections to the hunger centers [18, 19]. By its peripheral and central activities, GLP-1 decreases calorie intake, boosts feelings of satiety, and encourages weight loss [20]. It was shown that food intake is inhibited by acute intracerebroventricular GLP-1 injection, and food intake is increased—even in satiated rats—by antagonists to the GLP-1 receptor [21]. The strong evidence that the paraventricular nucleus is the principal site for brain-derived GLP-1 satiety comes from the direct delivery of GLP-1 into this region of the brain. GLP-1 exerts its effects by acting directly on the paraventricular nucleus. Yet, because POMC neurons express GLP-1 receptors, GLP-1 also has anorexigenic actions in the arcuate nucleus [22]. Neuronal circuits are activated, and food intake declines as satiety signals like CCK and GLP-1 are created during food ingestion, signaling the conclusion of the meal. Finally, GLP-1 injections into the body over an extended period of time decrease weight gain and promote weight loss [23]. This is also supported by the observation that obese people have lower GLP-1 levels than lean people [24].
GLP-1 agonist effect on microbiota
The GLP-1 receptor agonist liraglutide has lately been acknowledged as a promising anti-obesity medication in obese and/or diabetic people [25]. The literature demonstrated that alterations in gut microbiota also significantly impacted satiety, lipid metabolism, and ectopic fat deposition, through the effect of GLP-1or its agonists. For instance, liraglutide, may thereby prevent weight gain via modifying the composition of the gut's microbial population [26]. More specifically, a previous study showed that liraglutide can, in fact, alter the makeup of the gut microbiota by boosting the lean-related profile, which is consistent with its ability to reduce body weight in mice with streptozotocin-induced transient hyperglycemia [27]. Similarly, liraglutide was found to reduce weight gain in both diabetic and nondiabetic obese patients by altering the composition of the gut flora [28]. According to another study, liraglutide causes gut microbial structural alterations in diet-induced obese (DIO) mice, with the distribution of Proteobacteria and Verrucomicrobia phylotypes changing the most, while Firmicutes remain relatively unaffected [29]. The reduction in Proteobacteria lead to a drop in total body mass and the adiposity index, which were indicators of decreased food intake and feeding effectiveness [29]. Since Verrucomicrobia support the human gut’s glucose balance, its reduction will disrupt glucose homeostasis and therefore satiety [30]. Interestingly, it has been suggested that Firmicutes, a phylum that produces a significant amount of short chain fatty acids (SCFAs), particularly butyrate, may contribute to host obesity by enabling weight gain mechanism such as increase nutrition processing and energy extraction [31]. This can explain why GLP-1 does not stimulate Firmicutes as they both have opposite outcomes. Moreover, the abundance of Akkermansia muciniphila, a species known to degrade mucin and produce SCFAs, was found to be positively correlated with indicators of gut inflammation and significantly associated with body weight loss when its proportion increase due to liraglutide administration [32]. Another study suggested that the changes in the microbiome may be related to the GLP-1 and receptor signaling's convergent physiologic effects on calorie intake, glucose metabolism, and lipid management [33].
Microbiota in relationship to satiety
Microbiome and peripheral satiety mechanism
The story of the interaction between the gut microbiome and the neural circuits to the brain starts when food is ingested where CCK, GLP-1 and PYY are secreted by enteroendocrine cells (EECs) in order to send satiety signals via the vagus nerve in the purpose to control food intake [34]. There are many studies that reveal a “gut–brain” communication which is modulated by the gastrointestinal (GI) bacterial composition as we will mention in the following:
Microbiota and CCK
A person's gut microbiota may modify the expression and release of GI satiety peptides, which in turn may impact how much they eat. This idea was demonstrated in rodents. For instance, studies show that when compared to typical mice of the same weight, germ-free (GF) mice (that lack microbiota) exhibit reduced intestine expression of the CCK peptide [35]. Although the role of food receptors in the increased caloric intake seen in GF mice is unknown, it is suggested that the activation of nutrient responsive receptors triggers the release of satiety peptides from the intestinal tract, including CCK [36]. This indicates that these receptors play a role through the regulation of satiety peptides availability. For example, in cases of fructose malabsorption, there are higher relative abundances of Actinobacteria, Bacteroidetes, and Lactobacillaceae (especially Lactobacillus johnsonii). This indicates that fructose malabsorption induces CCK expression into the intestine by changing microbiota composition and metabolism. These adjustments are followed by a large rise in the number of CCK-positive enteroendocrine cells (EECs), proving that fructose malabsorption-induced changes in CCK release require the microbiota [37]. Moreover, increased CCK release is seen in the murine EEC line STC-1 when specific fatty acid metabolites generated by colonized lactic acid bacteria are applied [38]. All these data suggest that the relation between CCK and microbiota is thought to be due to microbial-derived products like lipopolysaccharide (LPS) and metabolites such SCFA [39] which act on enteroendocrine cells to release CCK.
Microbiota and GLP-1
The impact of the microbiome on gut satiety peptides extends beyond CCK. GLP-1 is secreted from intestinal L-cells and reduces appetite through a vagal-mediated mechanism [40]. It was found that intestinal bacteria ferment the prebiotic fiber beta-glucan to generate propionate, one of the important SCFAs [41]. Evidence suggests that a healthy microbiome's production of SCFAs affects the release of GLP-1. In a study conducted by Tolhurst and his colleagues, the free fatty acid receptor (FFAR 2), a nutrient-sensing G-protein coupled receptor, is activated when SCFAs—acetate, propionate, and butyrate—are applied to mouse colonic cell cultures. The activation of (FFAR 2) receptor resulted in an increase in GLP-1 production. Hence, the interaction of bacterial metabolites like propionate with intestinal L-cells can directly control GLP-1 production [42]. Moreover, according to recent research, acute inulin-propionate ester supplementation elevated plasma GLP-1 and PYY levels. It was linked to lower food consumption at meals after supplementation in humans. This demonstrates the immediate impact of propionate on meal consumption [43], and shows how microbial product help in physiologic control of satiety hormonal release. Also, it is interesting to note that prebiotic supplementation increases colon mass in mice when compared to non-supplemented controls [44], which can be partially attributed to an increase in the number of secretory cells [45]. Another way that the microbiota may affect GLP-1 release is through metabolites. For example, levels of acyl-glycerols in the gut are restored in obese mice when Akkermansia muciniphila is administered as a probiotic [46] as mentioned in Table 1. Acylglycerols, which are byproducts of fat digestion, activate a G-protein-coupled receptor, which in turn prompts L cells to release gut peptides including GLP-1 [47].
Microbiota and central satiety mechanism: satiety and neurological inflammation
As mentioned above, the hypothalamus is home to important anorexigenic and orexigenic neuronal populations that control hunger and energy expenditure. Leptin in particular can alter the expression and release of neuropeptides in the hypothalamus to control energy homeostasis.
The hypothalamus and the nucleus of tractus solitarius (NTS) have been related to inflammation which lead to loss of function as a result of bacterial inflammatory agents generated by the obese-type microbiome [48]. Leptin sensitivity in neurons is compromised by inflammation and cytokine signaling [49]. Particularly, leptin can alter the expression and release of neuropeptides in the hypothalamus to control energy homeostasis. Thus, affecting leptin sensitivity can disturb energy homeostasis in the hypothalamus. In diet-induced obesity (DIO) mice, taking a probiotic supplement containing Lactobacillus rhamnosus, Lactobacillus acidophilus, and Bifidobacterium bifidum reduces body weight and food intake. It also normalizes and restores leptin-induced phosphorylated signal transducer and activator of transcription 3 (pSTAT3) expression (Table 1) [50]. Lactobacillus rhamnosus supplementation alone has resulted in identical preservation of leptin signaling, proving that hypothalamic leptin signaling is affected by the presence of specific bacteria [51].
On the other hand, a study In diet-induced obesity (DIO) mice, taking a probiotic supplement containing Lactobacillus rhamnosus, Lactobacillus acidophilus, and Bifidobacterium bifidum reduces body weight and food intake. It also normalizes and restores leptin-induced pSTAT3 expression [50]. Lactobacillus rhamnosus supplementation alone has resulted in identical preservation of leptin signaling, proving that hypothalamic leptin signaling is affected by the presence of specific bacteria [51].
In brief, the microbiome plays an important role in controlling central satiety metabolism and this role is mediated by the release of different cytokines.
Microbiota and reward system
In addition to the role of neurologic inflammation as central mechanism, brain reward system can also alter satiety.
Food accessibility, social and environmental cues, and flavor are a few examples of external influences that might interfere with the body’s natural intake control [40]. The dorsal striatum's dopamine (DA) levels are sufficiently raised by optogenetic activation of vagal afferent neurons (VANs) that innervate the upper GI tract to promote reward-related behaviors such self-stimulation, location preference, and flavor conditioning [34].
Antibiotic-free and germ-free mice show changes in dopaminergic reward pathways [54]. Contrary to conventional mice, GF mice had greater desire for even low concentrations of intralipid [35]. GF mice showed enhanced DA turnover in the striatum and reduced expression of D1 receptor mRNA in the striatum and nucleus accumbens (NAc) [55], two areas implicated in food-seeking behavior [56]. This shows that in situations where dopamine is high, people tend to have higher desire to eat. Antimicrobial therapy elevated L-3,4-dihydroxyphenylalanine (L-DOPA) in young mice’s amygdala and lowered DA turnover in rats' amygdala and striatum, indicating that the microbiome regulates DA neurochemistry [57]. Adolescent rats with periodic daily access to the high fat/high sucrose (HFHS) diet have higher total energy expenditure and changed monoamine gene expression in the hippocampus and prefrontal cortex. One of the changes observed is related to the monoamine oxidase A (MAO-A) that is an enzyme involved in removing the neurotransmitters norepinephrine, serotonin and dopamine from the brain. These changes are correlated with bacterial distribution and abundance. In particular, MAO-A expression in the hippocampus is linked to several other bacterial genus including unspecified Bifidobacteriales, Bifidobacteriaceae, and an unspecified genus of the Lachnospiraceae family. In contrast, MAO-A expression in the prefrontal cortex is positively linked to an unspecified genus of the Lachnospiraceae family [58].
Food preferences could be influenced by microbiota exposed to particular conditions. For instance, mice under social stress show increased preference for sucrose, and this preference is eliminated by SCFA supplementation, suggesting that the microbiota controls stress-induced sucrose preference through the synthesis of SCFA [59].
Also, artificial sweeteners with low or no calories are another topic of concern in terms of how they affect intake and satisfaction. This is because it has been shown that some sweeteners, like stevia, are digested by gut flora [60]. Although the consumption of artificial sweeteners does not appear to trigger compensatory overeating in humans in short-term or long-term trials, it has been shown to modify reward circuits in both rodents and humans [61]. For instance, tyrosine hydroxylase and dopamine transporter (DAT) mRNA expression in the NAc is reduced in rats exposed to a chronic low dosage of the stevia glycoside rebaudioside A (RebA), which can be reversed by supplementing with the prebiotic oligo-fructose [62]. These findings imply that the metabolism of artificial sweeteners by bacteria may change reward signaling and that the rewarding qualities of food might override the basic satiety signals produced by homeostatic regions [63].
In conclusion, the presence of microbiota leads to the activation of the reward system which thus increases food desire, whereas their absence can depress reward system and reduce food desire.
Neuroscience in relation to satiety
Many studies have shown that the homeostatic regulator of food intake interacts with the dopamine reward system leading to a boosting effect on food intake. This interaction is based on the involvement of the dopamine reward system in the behavior of food seeking [64]. For instance, it has been shown that ghrelin stimulates ventral tegmental area (VTA) dopamine neurons whereas leptin and insulin inhibit them [65]. According to research by Hommel et al., leptin receptors are expressed on VTA dopamine neurons and inhibit their activity. Food intake was observed to decrease when leptin was administered to the VTA, but it increased when leptin receptors were knocked down in the VTA, along with activity levels and hedonic feeding [66].
Neurotransmitter effect on satiety
In the parabrachial nucleus, a region located in the pons, the neurotransmitter gamma amino benzoic acid (GABA) produced by NPY and AgRP neurons, maintains energy balance [67]. The dorsal raphe nucleus (DRN) contains a population of heat-activated GABAergic neurons that control energy expenditure via altering motility and thermogenesis [68]. The increase in motility and thermogenesis lead to increased desire to eat and replenish energy sources. Its significance in obesity is clear from the fact that eliminating the vesicular transporter for GABA in AgRP neurons causes resistance to obesity brought on by a high-fat diet, regardless of changes in food intake [69]. Serotonin receptor, which is found in certain arcuate POMC neurons, is another neurotransmitter that controls how much food is consumed and how much energy is expended [70]. Independently of changes in energy expenditure, these POMC serotonin receptors are engaged in controlling energy homeostasis through changes in eating behavior [71] as shown in Table 2. Through the MC4R sympathetic preganglionic neurons, POMC neurons that project to the spinal cord are also engaged in maintaining homeostasis of energy by promoting adaptive thermogenesis in brown adipose tissue [72]. As a result of energy expenditure, the desire to eat will also expand. Moreover, due to its role in maintaining energy homeostasis, oxytocin, a centrally acting neurotransmitter and hormone, is receiving more attention as a potential anti-obesity target [73]. Obesity was shown to be a characteristic of mice lacking either oxytocin or oxytocin receptors [74]. Also, in diet-induced obesity and genetically obese mouse models, long-term peripheral or central administration of oxytocin causes an inhibition of food intake, an increase in energy expenditure, and weight loss as seen in Table 2 [75].
Conclusion
The gut microbiota, which is the term for the whole microbial community inhabiting the digestive system, has been shown in several studies to be influenced by GLP-1. By encouraging the development of specific advantageous bacteria in the gut, GLP-1 may make it easier to produce satiety related microbial products. On the other hand, GLP-1 shortage or resistance may cause dysbiosis, or an imbalance of harmful and helpful microbes, which can exacerbate metabolic diseases like obesity and insulin resistance.
In conclusion, each of the hormonal, microbial and neurotransmitters are important for controlling satiety and glucose metabolism. Moreover, ongoing research on the connection between GLP-1 and microbiota could yield new insights and treatments for metabolic disorders.
Data availability
Lebanese Internation University (our instituation) funds publishing this article.
References
Waterson MJ, Horvath TL (2015) Neuronal regulation of energy homeostasis: beyond the hypothalamus and feeding. Cell Metab 22(6):962–970
Schellekens H, Finger BC, Dinan TG, Cryan JF (2012) Ghrelin signalling and obesity: at the interface of stress, mood and food reward. Pharmacol Ther 135(3):316–326
Wardlaw SL (2011) Hypothalamic proopiomelanocortin processing and the regulation of energy balance. Eur J Pharmacol 660(1):213–219
Murray S, Tulloch A, Gold MS, Avena NM (2014) Hormonal and neural mechanisms of food reward, eating behaviour and obesity. Nat Rev Endocrinol 10(9):540–552
Anderberg RH, Hansson C, Fenander M, Richard JE, Dickson SL, Nissbrandt H et al (2016) The stomach-derived hormone ghrelin increases impulsive behavior. Neuropsychopharmacology 41(5):1199–1209
Engel JA, Jerlhag E (2014) Role of appetite-regulating peptides in the pathophysiology of addiction: implications for pharmacotherapy. CNS Drugs 28(10):875–886
Cardi V, Leppanen J, Treasure J (2015) The effects of negative and positive mood induction on eating behaviour: a meta-analysis of laboratory studies in the healthy population and eating and weight disorders. Neurosci Biobehav Rev 57:299–309
Marchesi JR, Adams DH, Fava F, Hermes GDA, Hirschfield GM, Hold G et al (2016) The gut microbiota and host health: a new clinical frontier. Gut 65(2):330–339
Ley RE, Lozupone CA, Hamady M, Knight R, Gordon JI (2008) Worlds within worlds: evolution of the vertebrate gut microbiota. Nat Rev Microbiol 6(10):776–788
Cryan JF, Dinan TG (2012) Mind-altering microorganisms: the impact of the gut microbiota on brain and behaviour. Nat Rev Neurosci 13(10):701–712
Arora T, Bäckhed F (2016) The gut microbiota and metabolic disease: current understanding and future perspectives. J Intern Med 280(4):339–349
Mack I, Cuntz U, Grämer C, Niedermaier S, Pohl C, Schwiertz A et al (2016) Weight gain in anorexia nervosa does not ameliorate the faecal microbiota, branched chain fatty acid profiles and gastrointestinal complaints. Sci Rep 6(1):26752
Le Chatelier E, Nielsen T, Qin J, Prifti E, Hildebrand F, Falony G et al (2013) Richness of human gut microbiome correlates with metabolic markers. Nature 500(7464):541–546
Park JH, Lee MJ, Song MY, Bose S, Shin BC, Kim HJ (2012) Efficacy and safety of mixed oriental herbal medicines for treating human obesity: a systematic review of randomized clinical trials. J Med Food 15(7):589–597
Mountjoy KG (2010) Functions for pro-opiomelanocortin-derived peptides in obesity and diabetes. Biochem J 428(3):305–324
Williams KW, Scott MM, Elmquist JK (2011) Modulation of the central melanocortin system by leptin, insulin, and serotonin: co-ordinated actions in a dispersed neuronal network. Eur J Pharmacol 660(1):2–12
Tao YX (2010) The melanocortin-4 receptor: physiology, pharmacology, and pathophysiology. Endocr Rev 31(4):506–543
Secher A, Jelsing J, Baquero AF, Hecksher-Sørensen J, Cowley MA, Dalbøge LS et al (2014) The arcuate nucleus mediates GLP-1 receptor agonist liraglutide-dependent weight loss. J Clin Investig 124(10):4473–4488
Jin SLC, Han VKM, Simmons JG, Towle AC, Lauder JM, Lund PK (1988) Distribution of glucagonlike peptide I (GLP-I), glucagon, and glicentin in the rat brain: an immunocytochemical study. J Comp Neurol 271(4):519–532
Barrera JG, Sandoval DA, D’Alessio DA, Seeley RJ (2011) GLP-1 and energy balance: an integrated model of short-term and long-term control. Nat Rev Endocrinol 7(9):507–516
Hansotia T, Maida A, Flock G, Yamada Y, Tsukiyama K, Seino Y et al (2007) Extrapancreatic incretin receptors modulate glucose homeostasis, body weight, and energy expenditure. J Clin Investig 117(1):143–152
Sandoval DA, Bagnol D, Woods SC, D’Alessio DA, Seeley RJ (2008) Arcuate glucagon-like peptide 1 receptors regulate glucose homeostasis but not food intake. Diabetes 57(8):2046–2054
Tilinca MC, Tiuca RA, Burlacu A, Varga A (2021) A 2021 update on the use of liraglutide in the modern treatment of ‘diabesity’: a narrative review. Medicina (B Aires) 57(7):669
Anandhakrishnan A, Korbonits M (2016) Glucagon-like peptide 1 in the pathophysiology and pharmacotherapy of clinical obesity. World J Diabetes 7(20):572
Astrup A, Rössner S, Van Gaal L, Rissanen A, Niskanen L, Al Hakim M et al (2009) Effects of liraglutide in the treatment of obesity: a randomised, double-blind, placebo-controlled study. Lancet 374(9701):1606–1616
Basso N, Soricelli E, Castagneto-Gissey L, Casella G, Albanese D, Fava F et al (2016) Insulin resistance, microbiota, and fat distribution changes by a new model of vertical sleeve gastrectomy in obese rats. Diabetes 65(10):2990–3001
Wang L, Li P, Tang Z, Yan X, Feng B (2016) Structural modulation of the gut microbiota and the relationship with body weight: compared evaluation of liraglutide and saxagliptin treatment. Sci Rep 6(1):33251
Zhao L, Chen Y, Xia F, Abudukerimu B, Zhang W, Guo Y et al (2018) A Glucagon-like peptide-1 receptor agonist lowers weight by modulating the structure of gut microbiota. Front Endocrinol (Lausanne) 17:9
Moreira G, Azevedo F, Ribeiro L, Santos A, Guadagnini D, Gama P et al (2018) Liraglutide modulates gut microbiota and reduces NAFLD in obese mice. J Nutr Biochem 62:143–154
Fujio-Vejar S, Vasquez Y, Morales P, Magne F, Vera-Wolf P, Ugalde JA et al (2017) The gut microbiota of healthy chilean subjects reveals a high abundance of the phylum Verrucomicrobia. Front Microbiol 30:8
Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE et al (2009) A core gut microbiome in obese and lean twins. Nature 457(7228):480–484
Derrien M, Belzer C, de Vos WM (2017) Akkermansia muciniphila and its role in regulating host functions. Microb Pathog 106:171–181
Madsen MSA, Holm JB, Pallejà A, Wismann P, Fabricius K, Rigbolt K et al (2019) Metabolic and gut microbiome changes following GLP-1 or dual GLP-1/GLP-2 receptor agonist treatment in diet-induced obese mice. Sci Rep 9(1):15582
Han W, Tellez LA, Perkins MH, Perez IO, Qu T, Ferreira J et al (2018) A neural circuit for gut-induced reward. Cell 175(3):665-678.e23
Duca FA, Swartz TD, Sakar Y, Covasa M (2012) Increased oral detection, but decreased intestinal signaling for fats in mice lacking gut microbiota. PLoS ONE 7(6):e39748
Samuel BS, Shaito A, Motoike T, Rey FE, Backhed F, Manchester JK et al (2008) Effects of the gut microbiota on host adiposity are modulated by the short-chain fatty-acid binding G protein-coupled receptor, Gpr41. Proc Natl Acad Sci 105(43):16767–16772
Zhang X, Grosfeld A, Williams E, Vasiliauskas D, Barretto S, Smith L et al (2019) Fructose malabsorption induces cholecystokinin expression in the ileum and cecum by changing microbiota composition and metabolism. FASEB J 33(6):7126–7142
Hira T, Ogasawara S, Yahagi A, Kamachi M, Li J, Nishimura S et al (2018) Novel mechanism of fatty acid sensing in enteroendocrine cells: specific structures in oxo-fatty acids produced by gut bacteria are responsible for CCK secretion in STC-1 cells via GPR40. Mol Nutr Food Res 62(19):1800146
Sun LJ, Li JN, Nie YZ (2020) Gut hormones in microbiota–gut–brain cross-talk. Chin Med J (Engl) 133(7):826–833
Riediger T (2012) The receptive function of hypothalamic and brainstem centres to hormonal and nutrient signals affecting energy balance. Proc Nutr Soc 71(4):463–477
Carlson J, Erickson J, Hess J, Gould T, Slavin J (2017) Prebiotic dietary fiber and gut health: comparing the in vitro fermentations of beta-glucan, inulin and xylooligosaccharide. Nutrients 9(12):1361
Tolhurst G, Heffron H, Lam YS, Parker HE, Habib AM, Diakogiannaki E et al (2012) Short-chain fatty acids stimulate glucagon-like peptide-1 secretion via the G-protein–coupled receptor FFAR2. Diabetes 61(2):364–371
Chambers ES, Viardot A, Psichas A, Morrison DJ, Murphy KG, Zac-Varghese SEK et al (2015) Effects of targeted delivery of propionate to the human colon on appetite regulation, body weight maintenance and adiposity in overweight adults. Gut 64(11):1744–1754
Chassaing B, Miles-Brown J, Pellizzon M, Ulman E, Ricci M, Zhang L et al (2015) Lack of soluble fiber drives diet-induced adiposity in mice. Am J Physiol Gastrointest Liver Physiol 309(7):528–541
Delzenne NM, Cani PD, Daubioul C, Neyrinck AM (2005) Impact of inulin and oligofructose on gastrointestinal peptides. Br J Nutr 93(S1):S157–S161
Everard A, Belzer C, Geurts L, Ouwerkerk JP, Druart C, Bindels LB et al (2013) Cross-talk between Akkermansia muciniphila and intestinal epithelium controls diet-induced obesity. Proc Natl Acad Sci 110(22):9066–9071
Hansen KB, Rosenkilde MM, Knop FK, Wellner N, Diep TA, Rehfeld JF et al (2011) 2-oleoyl glycerol Is a GPR119 agonist and signals GLP-1 release in humans. J Clin Endocrinol Metab 96(9):E1409–E1417
Maldonado-Ruiz R, Cárdenas-Tueme M, Montalvo-Martínez L, Vidaltamayo R, Garza-Ocañas L, Reséndez-Perez D et al (2019) Priming of hypothalamic ghrelin signaling and microglia activation exacerbate feeding in rats’ offspring following maternal overnutrition. Nutrients 11(6):1241
de Git KCG, Adan RAH (2015) Leptin resistance in diet-induced obesity: the role of hypothalamic inflammation. Obes Rev 16(3):207–224
Bagarolli RA, Tobar N, Oliveira AG, Araújo TG, Carvalho BM, Rocha GZ et al (2017) Probiotics modulate gut microbiota and improve insulin sensitivity in DIO mice. J Nutr Biochem 50:16–25
Cheng YC, Liu JR (2020) Effect of Lactobacillus rhamnosus GG on energy metabolism, leptin resistance, and gut microbiota in mice with diet-induced obesity. Nutrients 12(9):2557
Dudele A, Fischer CW, Elfving B, Wegener G, Wang T, Lund S (2015) Chronic exposure to low doses of lipopolysaccharide and high-fat feeding increases body mass without affecting glucose tolerance in female rats. Physiol Rep 3(11):e12584
Vaughn AC, Cooper EM, DiLorenzo PM, O’Loughlin LJ, Konkel ME, Peters JH et al (2017) Energy-dense diet triggers changes in gut microbiota, reorganization of gut–brain vagal communication and increases body fat accumulation. Acta Neurobiol Exp (Wars). 77(1):18–30
González-Arancibia C, Urrutia-Piñones J, Illanes-González J, Martinez-Pinto J, Sotomayor-Zárate R, Julio-Pieper M et al (2019) Do your gut microbes affect your brain dopamine? Psychopharmacology 236(5):1611–1622
Heijtz RD, Wang S, Anuar F, Qian Y, Björkholm B, Samuelsson A et al (2011) Normal gut microbiota modulates brain development and behavior. Proc Natl Acad Sci 108(7):3047–3052
Roitman MF, Stuber GD, Phillips PEM, Wightman RM, Carelli RM (2004) Dopamine operates as a subsecond modulator of food seeking. J Neurosci 24(6):1265–1271
Desbonnet L, Clarke G, Traplin A, O’Sullivan O, Crispie F, Moloney RD et al (2015) Gut microbiota depletion from early adolescence in mice: Implications for brain and behaviour. Brain Behav Immun 48:165–173
Reichelt AC, Loughman A, Bernard A, Raipuria M, Abbott KN, Dachtler J et al (2020) An intermittent hypercaloric diet alters gut microbiota, prefrontal cortical gene expression and social behaviours in rats. Nutr Neurosci 23(8):613–627
van de Wouw M, Boehme M, Lyte JM, Wiley N, Strain C, O’Sullivan O et al (2018) Short-chain fatty acids: microbial metabolites that alleviate stress-induced brain–gut axis alterations. J Physiol 596(20):4923–4944
Magnuson BA, Carakostas MC, Moore NH, Poulos SP, Renwick AG (2016) Biological fate of low-calorie sweeteners. Nutr Rev 74(11):670–689
Pang MD, Goossens GH, Blaak EE (2021) The impact of artificial sweeteners on body weight control and glucose homeostasis. Front Nutr 7:7
Nettleton JE, Klancic T, Schick A, Choo AC, Shearer J, Borgland SL et al (2019) Low-dose stevia (Rebaudioside A) consumption perturbs gut microbiota and the mesolimbic dopamine reward system. Nutrients 11(6):1248
Alonso-Alonso M, Woods SC, Pelchat M, Grigson PS, Stice E, Farooqi S et al (2015) Food reward system: current perspectives and future research needs. Nutr Rev 73(5):296–307
Baik JH (2021) Dopaminergic control of the feeding circuit. Endocrinol Metab 36(2):229–239
Cone JJ, McCutcheon JE, Roitman MF (2014) Ghrelin acts as an interface between physiological state and phasic dopamine signaling. J Neurosci 34(14):4905–4913
Hommel JD, Trinko R, Sears RM, Georgescu D, Liu ZW, Gao XB et al (2006) Leptin receptor signaling in midbrain dopamine neurons regulates feeding. Neuron 51(6):801–810
Wu Q, Palmiter RD (2011) GABAergic signaling by AgRP neurons prevents anorexia via a melanocortin-independent mechanism. Eur J Pharmacol 660(1):21–27
Schneeberger M, Parolari L, Das Banerjee T, Bhave V, Wang P, Patel B et al (2019) Regulation of energy expenditure by brainstem GABA neurons. Cell 178(3):672-685.e12
Aponte Y, Atasoy D, Sternson SM (2011) AGRP neurons are sufficient to orchestrate feeding behavior rapidly and without training. Nat Neurosci 14(3):351–355
Xu Y, Berglund ED, Sohn JW, Holland WL, Chuang JC, Fukuda M et al (2010) 5-HT2CRs expressed by pro-opiomelanocortin neurons regulate insulin sensitivity in liver. Nat Neurosci 13(12):1457–1459
Ho AJ, Raji CA, Becker JT, Lopez OL, Kuller LH, Hua X et al (2010) Obesity is linked with lower brain volume in 700 AD and MCI patients. Neurobiol Aging 31(8):1326–1339
Xu B, Xie X (2016) Neurotrophic factor control of satiety and body weight. Nat Rev Neurosci 17(5):282–292
Morton GJ, Thatcher BS, Reidelberger RD, Ogimoto K, Wolden-Hanson T, Baskin DG et al (2012) Peripheral oxytocin suppresses food intake and causes weight loss in diet-induced obese rats. Am J Physiol Endocrinol Metab 302(1):E134–E144
Camerino C (2009) Low sympathetic tone and obese phenotype in oxytocin-deficient mice. Obesity 17(5):980–984
Blevins JE, Thompson BW, Anekonda VT, Ho JM, Graham JL, Roberts ZS et al (2016) Chronic CNS oxytocin signaling preferentially induces fat loss in high-fat diet-fed rats by enhancing satiety responses and increasing lipid utilization. Am J Physiol Regul Integr Comp Physiol 310(7):R640–R658
Funding
Author(s) received no financial support for the research, authorship, and/or publication of this article.
Author information
Authors and Affiliations
Contributions
All authors contributed equally for publication in this journal.
Corresponding author
Ethics declarations
Competing interests
The authors declare that they have no competing interests related to the manuscript.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Barakat, G.M., Ramadan, W., Assi, G. et al. Satiety: a gut–brain–relationship. J Physiol Sci 74, 11 (2024). https://doi.org/10.1186/s12576-024-00904-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12576-024-00904-9