
Abstract
Clonidine is a first-generation central antihypertensive that reduces sympathetic nerve activity (SNA). Although clonidine also exerts peripheral vasoconstriction, the extent to which this vasoconstriction offsets the centrally mediated arterial pressure (AP)-lowering effect remains unknown. In anesthetized rats (n = 8), we examined SNA and AP responses to stepwise changes in carotid sinus pressure under control conditions and after intravenous low-dose (2 μg/kg) and high-dose clonidine (5 μg/kg). In the baroreflex equilibrium diagram analysis, the operating-point AP under the control condition was 115.2 (108.5–127.7) mmHg [median (25th–75th percentile range)]. While the operating-point AP after low-dose clonidine was not significantly different with or without the peripheral effect, the operating-point AP after high-dose clonidine was higher with the peripheral effect than without [81.3 (76.2–98.2) mmHg vs. 70.7 (57.7–96.9), P < 0.05]. The vasoconstrictive effect of clonidine partly offset the centrally mediated AP-lowering effect after high-dose administration.
Similar content being viewed by others

Introduction
Clonidine is a first-generation antihypertensive agent that suppresses sympathetic outflow from the central nervous system by acting on α2-adrenergic and I1-imidazoline receptors [1,2,3]. After clonidine, several central antihypertensive agents with different properties have been developed. Rilmenidine and moxonidine preferentially act on I1-imidazoline receptors [1,2,3,4], thereby reducing side effects associated with central α2-adrenergic stimulation, such as dry mouth and sedation. Guanfacine is selective for α2A, a subtype mainly responsible for central sympathetic suppression [5].
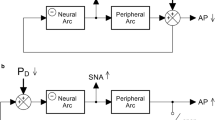
Despite classified as a central antihypertensive agent, the effect of a given drug on arterial pressure (AP) is not determined by its central action alone when the drug is administered systemically. In our previous study, intravenously administered moxonidine increased AP at any given sympathetic nerve activity (SNA), suggesting that a peripheral effect of moxonidine significantly offset the centrally mediated AP-lowering effect [6]. Intravenously administered guanfacine also exhibited a peripheral effect that nearly canceled the reduction of AP at the operating point of the arterial baroreflex [7]. By contrast, intravenously administered rilmenidine showed a weak vasoconstrictive effect that was unmasked by opening the baroreflex negative feedback loop [8].
Regarding clonidine, how exactly the central and peripheral effects interact to determine AP remains unknown. Since clonidine may serve as a reference drug to evaluate other central antihypertensive agents, we quantified the effect of clonidine on the baroreflex-mediated sympathetic AP regulation in the present study. Intravenously administered clonidine typically induces an initial AP elevation through peripheral vasoconstriction but eventually decreases AP through central sympathetic suppression. We hypothesized that the peripheral vasoconstrictive effect of clonidine persists even during the hypotensive phase and partly offsets the centrally mediated AP-lowering effect.
Materials and methods
Surgical preparation
Animal care was provided in strict accordance with the Guiding Principles for the Care and Use of Animals in the Field of Physiological Sciences, which has been approved by the Physiological Society of Japan. The experimental protocols were reviewed and approved by the Animal Subject Committee of the National Cerebral and Cardiovascular Center (No. 20008, 21009).
Eight male Wistar-Kyoto rats (304–395 g) were anesthetized with an intraperitoneal injection (2 mL/kg) of a mixture of urethane (250 mg/mL) and α-chloralose (40 mg/mL). The anesthetic mixture was diluted 18-fold and administered continuously (2 mL·kg−1 h−1) from the right femoral vein. The rats were mechanically ventilated with oxygen-supplied air. AP was measured from the right femoral artery, and the heart rate (HR) was detected from a body surface electrocardiogram. A pair of stainless-steel wire electrodes (AS633, Cooner Wire, CA, USA) was attached to a postganglionic branch of the left splanchnic sympathetic nerve and fixed with silicone glue (Kwik-Sil, World Precision Instruments, FL, USA). The nerve activity was amplified using a biosignal amplifier (AB-610J, Nihon Kohden, Japan) with a bandpass filter setting between 150 and 1000 Hz. The amplified signal was then full-wave rectified and low-pass filtered at a cut-off frequency of 30 Hz to quantify SNA. Bilateral carotid sinus baroreceptor regions were isolated from the systemic circulation [9, 10], and the carotid sinus pressure (CSP) was controlled with a servo-pump system (ET-126, Labworks, Costa Mesa, CA, USA). Bilateral vagal and aortic depressor nerves were sectioned at the neck to minimize reflex effects from the cardiopulmonary region and aortic arch.
Protocol
After completing the surgical preparation, we waited for at least 30 min to obtain stable hemodynamics. During the waiting period, CSP was servo-controlled to mimic instantaneous AP. Although pressure waveforms did not exactly match between the CSP and AP signals due to the limited performance of the servo controller, the baroreflex negative feedback loop was effectively closed.
To estimate the open-loop static characteristics of the carotid sinus baroreflex, CSP was first decreased to 60 mmHg for 5 min, and then increased stepwise to 180 mmHg in increments of 20 mmHg every minute. The stepwise CSP input was repeated, and the sequences were referred to as S1 to S6 (Fig. 1, left panels). One minute after the completion of S2, low-dose (2 μg/kg) clonidine (clonidine hydrochloride, Tokyo Chemical Industry; 2 μg/mL dissolved in physiological saline) was administered from the left femoral vein. One minute after the completion of S4, high-dose (5 μg/kg) clonidine (5 μg/mL dissolved in physiological saline) was added. The effect of high-dose clonidine may need to be interpreted as a cumulative one.

Time series of carotid sinus pressure (CSP), sympathetic nerve activity (SNA), arterial pressure (AP), and heart rate (HR) obtained in one rat. In the left panels, stepwise CSP changes were repeated and referred to as S1 to S6. One minute after the completion of S2, low-dose (2 μg/kg) clonidine was intravenously administered. One minute after the completion of S4, high-dose (5 μg/kg) clonidine was intravenously administered. The baroreflex response in S2 was treated as the control, whereas the responses in S4 and S6 were used to evaluate the effects of low-dose and high-dose clonidine, respectively. In the right panels, hexamethonium bromide (C6) was intravenously administered. CSP was servo-controlled to mimic instantaneous AP during the C6 protocol. The CSP and AP signals are virtually the same at this time resolution. SNA was depicted as a 10-Hz resampled signal (gray) and a 2-s moving averaged signal (black). AP and HR were depicted as 200-Hz resampled signals (gray) and 2-s moving averaged signals (black)
At the end of the protocol, CSP was again servo-controlled to mimic instantaneous AP. Under the baroreflex closed-loop condition, hexamethonium bromide, a ganglionic blocker, was intravenously administered (60 mg/kg), and the noise level of SNA was assessed. Approximately 3 min after the injection of hexamethonium bromide, mean AP was obtained as a 10-s averaged value.
Data analysis
Data were recorded on a laboratory computer system using a 16-bit analog-to-digital converter. We treated the baroreflex response in S2 as a control and evaluated the effects of low-dose and high-dose clonidine from the responses in S4 and S6, respectively. In each of S2, S4, and S6, the SNA, AP, and HR data were averaged for the last 10 s at each CSP level. As the absolute amplitude of SNA varied among animals depending on the recording conditions, SNA was normalized in each animal based on the value at the CSP of 60 mmHg in S2 (100%) and the value after ganglionic blockade (0%). The relationships between CSP and AP (the total reflex arc), between CSP and HR, and between CSP and SNA (the neural arc) were analyzed using a four-parameter logistic function as follows [11, 12]:
where y denotes the output variable, and P1, P2, P3, and P4 represent the response range, slope coefficient, midpoint pressure on the CSP axis, and the lower asymptote of the sigmoid curve, respectively.
The relationship between SNA and AP (the peripheral arc) was quantified using linear regression analysis [12]:
where b0 and b1 represent the intercept and slope of the regression line, respectively.
The operating point of the carotid sinus baroreflex was determined from the intersection point between the fitted neural and peripheral arcs on a baroreflex equilibrium diagram [12,13,14]. The operating-point AP values were estimated under conditions of control, after low-dose clonidine, and after high-dose clonidine. To exclude the peripheral effect of clonidine, the intersection points after clonidine were also estimated by reflecting changes in the neural arc alone.
Statistical analysis
Data are presented as box and whisker plots except otherwise specified. The fitted parameter values of the baroreflex static characteristics were compared among conditions of control, after low-dose clonidine, and after high-dose clonidine using one-way repeated-measures analysis of variance (ANOVA) followed by a Tukey test. The Geisser-Greenhouse's correction was applied (Prism 8, GraphPad Software, San Diego, CA, USA). The operating-point AP values estimated under conditions of control, after low-dose clonidine (with or without the peripheral effect), and after high-dose clonidine (with or without the peripheral effect) were compared using one-way repeated-measures ANOVA followed by a Tukey test. The AP value obtained after the injection of hexamethonium bromide was compared with the intercepts of the peripheral arcs determined under the control condition and after high-dose clonidine using one-way repeated-measures ANOVA followed by a Tukey test. The differences were considered statistically significant at P < 0.05.
Results
The left panels in Fig. 1 represent the time series obtained from one rat. Under baseline conditions (S1 and S2), keeping CSP at 60 mmHg increased SNA, AP, and HR. Thereafter, the increase in CSP decreased SNA, AP, and HR. Administration of low-dose clonidine one minute after the completion of S2 (indicated by the down arrow at 2 μg/kg) decreased SNA and HR but transiently increased AP. The transient AP increase subsided by the end of S3. Although the maximum AP was lower in S4 than in S2, the minimum AP was higher in S4 than in S2. The addition of high-dose clonidine one minute after the completion of S4 (indicated by the down arrow at 5 μg/kg) further decreased SNA and HR with a transient increase in AP. The SNA was nearly silenced after high-dose clonidine. The elevated AP gradually declined toward the end of S5. The maximum AP was lower in S6 than in S2, whereas the minimum AP was higher in S6 than in S2. The right panels in Fig. 1 indicate that the remaining small burst activity of SNA (depicted in gray) disappeared after the administration of hexamethonium bromide. The top panel was virtually the same as the third panel at this time resolution because CSP was servo-controlled to mimic instantaneous AP.
Figure 2 illustrates the open-loop static characteristics of the total reflex arc (Fig. 2A), HR control (Fig. 2B), neural arc (Fig. 2C), and peripheral arc (Fig. 2D). The corresponding parameter values were displayed using box and whisker plots with the mean values superimposed in cross symbols (Fig. 2E–2H). The total reflex arc approximated inverse sigmoidal curves (Fig. 2A). The response range, P1, decreased as the dose of clonidine increased (Fig. 2E). No significant effect of clonidine was observed in the slope coefficient, P2, or the midpoint pressure, P3. The lower asymptote, P4, was significantly higher after clonidine administration than before. The baroreflex-mediated HR response also approximated inverse sigmoidal curves (Fig. 2B). The response range and the lower asymptote of HR decreased as the dose of clonidine increased (Fig. 2F). The neural arc approximated inverse sigmoidal curves (Fig. 2C). The response range, midpoint pressure, and lower asymptote showed a reduction dependent on the dose of clonidine (Fig. 2G). The peripheral arc approximated straight lines (Fig. 2D). The intercept, b0, was significantly higher after clonidine administration than before, whereas the slope, b1, did not differ significantly among the three conditions (Fig. 2H).

Open-loop static characteristics of the total reflex arc (A), HR control (B), neural arc (C), and peripheral arc (D) together with their parameter values (E–H). CSP carotid sinus pressure, AP arterial pressure, HR heart rate, SNA sympathetic nerve activity, P1 response range, P2 slope coefficient; P3 midpoint input pressure, P4 lower asymptote of the fitted sigmoid curve. Labels C, L, H denote the data under control conditions, after low-dose (2 μg/kg) clonidine, and after high-dose (5 μg/kg) clonidine, respectively. In panels A–D, data are expressed as mean ± SE values (n = 8). The smooth curve or the line was drawn using mean values of fitted parameters. In panels E–H, data are expressed as box and whisker plots (n = 8). The box indicates the 25th–75th percentile range. The horizontal line in the box indicates the median. The values above the 75th percentile plus 1.5 times the interquartile range (IQR) or those below the 25th percentile minus 1.5 times IQR are defined as outliers (filled circles). The whiskers indicate the minimum and maximum data points, excluding the outliers. In each plot, the mean of data points was superimposed in a cross symbol. *P < 0.05, **P < 0.01, and ***P < 0.001 by a Tukey test following one-way repeated-measures analysis of variance
The baroreflex equilibrium diagram was obtained by plotting the neural and peripheral arcs on a pressure–SNA plane. The operating-point AP, which was determined from the intersection point between the neural and peripheral arcs, decreased with the increasing dose of clonidine (Fig. 3A, the left arrowheads). The operating-point AP was also determined with the peripheral arc fixed at its control position, thereby excluding the peripheral effect of clonidine (Fig. 3B, the left arrowheads; PX, the exclusion of the peripheral effect). The operating-point AP after low-dose clonidine did not differ significantly with or without the peripheral effect (Fig. 3C). By contrast, the operating-point AP after high-dose clonidine was significantly higher with the peripheral effect than without.

Baroreflex equilibrium diagrams constructed from fitted neural and peripheral arcs with (A) or without (B) the peripheral effect of clonidine. PX, the exclusion of the peripheral effect. Labels C, L, H denote the data under control conditions, after low-dose (2 μg/kg) clonidine, and after high-dose (5 μg/kg) clonidine, respectively. The operating-point AP was defined as the AP at the intersection point between the neural and peripheral arcs (the left arrowheads). In panel C, the operating-point AP values are expressed as the box and whisker plots (n = 8). The box indicates the 25th–75th percentile range. The horizontal line in the box indicates the median. The whiskers indicate the minimum and maximum data points. The mean of data points was superimposed in a cross symbol. *P < 0.05, **P < 0.01, and ***P < 0.001 by a Tukey test following one-way repeated-measures analysis of variance
The mean AP observed after hexamethonium was 79.3 (75.7–81.2) mmHg [median (25th–75th percentile range)]. This value remained significantly higher than the intercept of the peripheral arc estimated under the control condition [53.2 (46.8–63.2) mmHg, P < 0.001], whereas it was not significantly different from the intercept estimated after high-dose clonidine [74.7 (70.0–82.4) mmHg].
Discussion
We have shown that the intravenous administration of clonidine dose-dependently suppressed SNA (Fig. 2C and 2G). Clonidine significantly increased the intercept of the peripheral arc without a statistically significant change in the slope (Fig. 2D and 2H). Although the lower asymptote of the neural arc was significantly lower after clonidine administration than before, the lower asymptote of the total reflex arc was significantly higher after clonidine administration than before (Fig. 2A and 2E).
Effect of clonidine on baroreflex-mediated sympathetic AP regulation
Agonists for α2-adrenergic receptors, such as clonidine and guanfacine, are known to inhibit SNA. Studies on mice that lack specific subtypes of α2-adrenergic receptors indicate that the α2A subtype is responsible for central sympathetic suppression [5], whereas the α2C subtype contributes to peripheral vasoconstriction [15]. However, in our previous study, guanfacine, a selective α2A-adrenergic agonist, demonstrated a significant peripheral effect, which nearly cancels the centrally mediated reduction of the operating-point AP [7]. By contrast, the peripheral effect of clonidine did not affect the operating-point AP after the low-dose administration and only partly offset the reduction of the operating-point AP after the high-dose administration (Fig. 3C). While guanfacine and clonidine are recognized as α2-adrenergic agonists, α1-adrenergic receptors may be involved in the peripheral effect. For instance, guanfacine and clonidine exerted a vasoconstrictive effect on a rabbit aortic strip, and this vasoconstriction was prevented by prazosin, an α1-adrenergic antagonist [16]. In that study, the maximum contractile effect of guanfacine was close to that of norepinephrine, whereas the maximum contractile effect of clonidine was less than a half of that of norepinephrine. The pressor effect of clonidine was also observed in pithed rabbits in vivo [17], suggesting that the pressor effect is independent of ongoing SNA. In the same study, prazosin partly inhibited the pressor response to clonidine. In addition, yohimbine, an α2-adrenergic antagonist, blunted the pressor response to clonidine.
Moxonidine and rilmenidine are second-generation central antihypertensive agents that preferentially act on I1-imidazoline receptors to reduce unwanted side effects of clonidine, such as dry mouth and sedation, resulting from α2-adrenergic stimulation [1, 2, 4]. Nevertheless, it has been demonstrated that an expression of α2A-adrenergic receptors is a prerequisite for the cardiovascular effect of moxonidine and rilmenidine [18]. It is understood that I1-imidazoline receptors are located upstream from α2-adrenergic receptors in the rostral ventrolateral medulla. Activation of I1-imidazoline receptors increases the release of norepinephrine, which suppresses SNA by activating α2-adrenergic receptors [2, 19]. By contrast, sedation is mediated by α2-adrenergic stimulation in the locus coeruleus [20], and imidazoline receptors are only indirectly involved in the stimulation of the locus coeruleus neurons [21].
In our previous study, moxonidine exhibited a strong peripheral effect that attenuated the reduction of the operating-point AP by more than a half magnitude. While it is not classified as an α1-adrenergic agonist, moxonidine increases the tension of the rat tail artery in a manner dependent on α1-adrenergic receptors [22]. The maximum tension development was near 90% of that induced by phenylephrine, and the tension development of moxonidine was inhibited by urapidil, an α1-adrenergic antagonist. Clonidine also increased the tension of the rat tail artery with a maximum tension development of approximately 60% of that induced by phenylephrine [22]. After the high-dose administration, the vasoconstrictive effect clonidine attenuated the centrally mediated reduction of the operating-point AP (Fig. 3C).
Although classification based on receptor affinities helps understand the action of a given drug, it can lead to oversimplification in the interpretation of drug effects. Once a given drug is classified as a central antihypertensive, investigators are prone to attribute an observed AP change to its central effect. The present study demonstrated the importance of interpreting the AP change in terms of the relative potency of the central and peripheral effects in vivo.
Effect of clonidine on baroreflex-mediated sympathetic HR control
In the present study, the HR response was mediated by the sympathetic system because of vagotomy. The lower asymptote of HR decreased as the dose of clonidine increased (Fig. 2F), which agrees with changes in the lower asymptote of the SNA response (Fig. 2G). Although the sympathetic HR response depends mainly on β1-adrenergic receptors, the sympathetic HR control can be affected by presynaptic inhibition via α2-adrenergic receptors. In our previous study using rabbits, clonidine infusion attenuated the release of norepinephrine during cardiac sympathetic nerve stimulation and attenuated the dynamic HR response [23]. Furthermore, clonidine exhibited an inhibitory effect on hyperpolarization-activated cyclic nucleotide-gated (HCN) channels and reduced HR in α2ABC-knockout mice [24]. Since the inhibition of HCN channels by ivabradine does not narrow the range of the baroreflex-mediated HR response [25], the HCN channel inhibition may not account for the reduced response range of HR after clonidine administration. On the other hand, HCN channel inhibition by clonidine may partially contribute to the reduction of the lower asymptote of HR after clonidine administration.
Implication of the study
Sympathetic overactivity can aggravate cardiovascular diseases, such as chronic heart failure (CHF). Inhibition of peripheral sympathetic effects by β-blockers [26], angiotensin converting enzyme inhibitors [27], and angiotensin II receptor blockers [28] is widely used for the treatment of CHF. Although central sympathetic suppression may be an alternative approach for treating sympathetic overactivity, a clinical trial of moxonidine resulted in worse outcomes in CHF patients [29]. The reason for the unsuccessful treatment may be multifactorial, including the high dose of moxonidine without a slow titration period. In addition, we speculate that a peripheral effect of moxonidine might have antagonized the expected beneficial effect of afterload reduction on the failing heart [6]. In support of the interpretation, moxonidine exerted a beneficial effect in a rat model of hypertensive heart failure when administered centrally to avoid its peripheral effect [30]. Judging from the reduction of the operating-point AP (Fig. 3C), the peripheral effect of clonidine may be weaker than that of moxonidine. Although oral administration of clonidine improved survival in a rat model of CHF after myocardial infarction [31], clonidine is not ideal for the treatment of CHF because of side effects associated with α2-adrenergic stimulation. Further studies are required to identify central antihypertensive agents with minimal side effects.
Limitations
First, the present results cannot account for any chronic effects of clonidine under conscious conditions because of the acute nature of the protocol. Second, there may be a species difference and a regional difference with regard to the peripheral effect relative to the central sympatholytic effect. Third, the AP response to SNA includes both the vascular and cardiac effects. Although the vascular properties play a predominant role in the baroreflex-mediated AP regulation under vagotomy [32], further studies are required to separate the vascular and cardiac effects of clonidine. Finally, we recorded splanchnic SNA as a proxy of systemic SNA. There is a possibility that sympathetic nerves other than the splanchnic sympathetic nerve responded differently to clonidine and contributed to the rise in the intercept of the peripheral arc drawn using splanchnic SNA as an abscissa. However, the total sympathetic blockade by hexamethonium at the end of the experiment did not significantly reduce AP compared with the intercept of the peripheral arc observed after high-dose clonidine. Hence, the rise in the intercept of the peripheral arc observed after high-dose clonidine likely represents a direct peripheral effect of clonidine rather than a differential regional modulation of sympathetic control.
Conclusions
Clonidine is a first-generation central antihypertensive, and its use has declined because of side effects, such as dry mouth and sedation. However, when the baroreflex-mediated sympathetic AP regulation was in focus, clonidine represented a weak peripheral effect relative to its central sympatholytic effect. These characteristics of clonidine may serve as a reference to the understanding of other central antihypertensive agents.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- ANOVA:
-
Analysis of variance
- AP:
-
Arterial pressure
- CHF:
-
Chronic heart failure
- CSP:
-
Carotid sinus pressure
- HR:
-
Heart rate
- SNA:
-
Sympathetic nerve activity
References
Khan ZP, Ferguson CN, Jones RM (1999) Alpha-2 and imidazoline receptor agonists. Their pharmacology and therapeutic role. Anaesthesia 54:146–165. https://doi.org/10.1046/j.1365-2044.1999.00659.x
Nikolic K, Agbaba D (2012) Imidazoline antihypertensive drugs: selective I1-imidazoline receptors activation. Cardiovasc Ther 30:209–216. https://doi.org/10.1111/j.1755-5922.2011.00269.x
Tomassoni AJ (2016) Alpha-2 adrenergic and imidazoline receptor agonists: clonidine, dexmedetomidine, and related antihypertensives, decongestants, and sedatives. In: Brent J, Burkhart K, Dargan P, Hatten B, Megarbane B, Palmer R (eds) Critical care toxicology. Springer, Cham. https://doi.org/10.1007/978-3-319-20790-2_27-1
Edwards LP, Brown-Bryan TA, McLean L, Ernsberger P (2012) Pharmacological properties of the central antihypertensive agent, moxonidine. Cardiovasc Ther 30:199–208. https://doi.org/10.1111/j.1755-5922.2011.00268.x
MacMillan LB, Hein L, Smith MS, Piascik MT, Limbird LE (1996) Central hypotensive effects of the α2a-adrenergic receptor subtype. Science 273:801–803. https://doi.org/10.1126/science.273.5276.801
Kawada T, Shimizu S, Yamamoto H, Miyamoto T, Shishido T, Sugimachi M (2017) Peripheral versus central effect of intravenous moxonidine on rat carotid sinus baroreflex-mediated sympathetic arterial pressure regulation. Life Sci 190:103–109. https://doi.org/10.1016/j.lfs.2017.09.038
Kawada T, Shimizu S, Turner MJ, Fukumitsu M, Yamamoto H, Sugimachi M (2016) Systematic understanding of acute effects of intravenous guanfacine on rat carotid sinus baroreflex-mediated sympathetic arterial pressure regulation. Life Sci 149:72–78. https://doi.org/10.1016/j.lfs.2016.02.051
Kawada T, Hayama Y, Nishikawa T, Yamamoto H, Tanaka K, Sugimachi M (2019) Even weak vasoconstriction from rilmenidine can be unmasked in vivo by opening the baroreflex feedback loop. Life Sci 219:144–151. https://doi.org/10.1016/j.lfs.2019.01.009
Shoukas AA, Callahan CA, Lash JM, Haase EB (1991) New technique to completely isolate carotid sinus baroreceptor regions in rats. Am J Physiol 260:H300–H303. https://doi.org/10.1152/ajpheart.1991.260.1.H300
Sato T, Kawada T, Miyano H, Shishido T, Inagaki M, Yoshimura R, Tatewaki T, Sugimachi M, Alexander J Jr, Sunagawa K (1999) New simple methods for isolating baroreceptor regions of carotid sinus and aortic depressor nerves in rats. Am J Physiol 276:H326–H332. https://doi.org/10.1152/ajpheart.1999.276.1.H326
Kent BB, Drane JW, Blumenstein B, Manning JW (1972) A mathematical model to assess changes in the baroreceptor reflex. Cardiology 57:295–310. https://doi.org/10.1159/000169528
Kawada T, Sugimachi M (2016) Open-loop static and dynamic characteristics of the arterial baroreflex system in rabbits and rats. J Physiol Sci 66:15–41. https://doi.org/10.1007/s12576-015-0412-5
Mohrman DE, Heller LJ (2010) Cardiovascular physiology, 7th edn. McGraw-Hill, New York, pp 246–250
Sato T, Kawada T, Inagaki M, Shishido T, Takaki H, Sugimachi M, Sunagawa K (1999) New analytic framework for understanding sympathetic baroreflex control of arterial pressure. Am J Physiol 276:H2251–H2261. https://doi.org/10.1152/ajpheart.1999.276.6.H2251
Link RE, Desai K, Hein L, Stevens ME, Chruscinski A, Bernstein D, Barsh GS, Kobilka BK (1996) Cardiovascular regulation in mice lacking α2-adrenergic receptor subtypes b and c. Science 273:803–805. https://doi.org/10.1126/science.273.5276.803
Takeuchi K, Kogure M, Hashimoto T (1987) Comparison of agonistic and antagonistic actions of guanabenz and guanfacin on α1 and α2-adrenoceptors in isolated smooth muscles. J J Pharmacol 43:267–275. https://doi.org/10.1254/jjp.43.267
Pompermayer K, Salgado MCSO, Feldman J, Bousquet P (1999) Cardiovascular effects of clonidine-like drugs in pithed rabbits. Hypertension 34:1012–1015. https://doi.org/10.1161/01.HYP.34.4.1012
Zhu QM, Lesnick JD, Jasper JR, MacLennan SJ, Dillon MP, Eglen RM, Blue DR Jr (1999) Cardiovascular effects of rilmenidine, moxonidine and clonidine in conscious wild-type and D79N α2A-adrenoceptor transgenic mice. Br J Pharmacol 126:1522–1530. https://doi.org/10.1038/sj.bjp.0702429
Head GA, Burke SL (2000) I1 imidazoline receptors in cardiovascular regulation: the place of rilmenidine. Am J Hypertens 13:89S-98S. https://doi.org/10.1016/s0895-7061(00)00224-7
De Sarro GB, Ascioti C, Froio F, Libri V, Nisticò G (1987) Evidence that locus coeruleus is the site where clonidine and drugs acting at α1- and α2-adrenoceptors affect sleep and arousal mechanisms. Br J Pharmacol 90:675–685. https://doi.org/10.1111/j.1476-5381.1987.tb11220.x
Ruiz-Ortega JA, Ugedo L (1997) The stimulatory effect of clonidine on locus coeruleus neurons of rats with inactivated α2-adrenoceptors: involvement of imidazoline receptors located in the nucleus paragigantocellularis. Naunyn-Schmiedeberg’s Arch Pharmacol 355:288–294. https://doi.org/10.1007/pl00004945
Kennedy WB, Crane L, Gonzalez RR, George OK, Edwards LP (2006) Centrally acting imidazolines stimulate vascular alpha 1A-adrenergic receptors in rat-tail artery. Cell Mol Neurobiol 26:645–657. https://doi.org/10.1007/s10571-006-9109-x
Miyamoto T, Kawada T, Yanagiya Y, Akiyama T, Kamiya A, Mizuno M, Takaki H, Sunagawa K, Sugimachi M (2008) Contrasting effects of presynaptic α2-adrenergic autoinhibition and pharmacologic augmentation of presynaptic inhibition on sympathetic heart rate control. Am J Physiol Heart Circ Physiol 295:H1855–H1866. https://doi.org/10.1152/ajpheart.522.2008
Knaus A, Zong X, Beetz N, Jahns R, Lohse MJ, Biel M, Hein L (2007) Direct inhibition of cardiac hyperpolarization-activated cyclic nucleotide-gated pacemaker channels by clonidine. Circulation 115:872–880. https://doi.org/10.1161/CIRCULATIONAHA.106.667675
Yamamoto H, Kawada T, Shimizu S, Uemura K, Inagaki M, Kakehi K, Iwanaga Y, Fukuda K, Miyamoto T, Miyazaki S, Sugimachi M (2018) Ivabradine does not acutely affect open-loop baroreflex static characteristics and spares sympathetic heart rate control in rats. Int J Cardiol 257:255–261. https://doi.org/10.1016/j.ijcard.2017.11.115
Foody JM, Farrell MH, Krumholz HM (2002) β-Blocker therapy in heart failure: scientific review. JAMA 287:883–889. https://doi.org/10.1001/jama.287.7.883
Flather MD, Yusuf S, Køber L, Pfeffer M, Hall A, Murray G, Torp-Pedersen C, Ball S, Pogue J, Moyé L, Braunwald E (2000) Long-term ACE-inhibitor therapy in patients with heart failure or left-ventricular dysfunction: a systematic overview of data from individual patients. ACE-inhibitor Myocardial Infarction Collaborative Group. Lancet 355:1575–1581. https://doi.org/10.1016/s0140-6736(00)02212-1
Pitt B, Poole-Wilson PA, Segal R, Martinez FA, Dickstein K, Camm AJ, Konstam MA, Riegger G, Klinger GH, Neaton J, Sharma D, Thiyagarajan B (2000) Effect of losartan compared with captopril on mortality in patients with symptomatic heart failure: randomized trial—the Losartan Heart Failure Survival Study ELITE II. Lancet 355:1582–1587. https://doi.org/10.1016/s0140-6736(00)02213-3
Cohn JN, Pfeffer MA, Rouleau J, Sharpe N, Swedberg K, Straub M, Wiltese C, Wright TJ (2003) MOXCON investigators. Adverse mortality effect of central sympathetic inhibition with sustained-release moxonidine in patients with heart failure (MOXCON). Eur J Heart Fail 5:659–667. https://doi.org/10.1016/s1388-9842(03)00163-6
Honda N, Hirooka Y, Ito K, Matsukawa R, Shinohara K, Kishi T, Yasukawa K, Utsumi H, Sunagawa K (2013) Moxonidine-induced central sympathoinhibition improves prognosis in rats with hypertensive heart failure. J Hypertens 31:2300–2308. https://doi.org/10.1097/HJH.0b013e328364a2a1
Zhang Y, Cheng Z (2000) Sympathetic inhibition with clonidine prolongs survival in experimental chronic heart failure. Int J Cardiol 73:157–162. https://doi.org/10.1016/s0167-5273(00)00213-8
Sakamoto T, Kakino T, Sakamoto K, Tobushi T, Tanaka A, Saku K, Hosokawa K, Onitsuka K, Murayama Y, Tsutsumi T, Ide T, Sunagawa K (2015) Changes in vascular properties, not ventricular properties, predominantly contribute to baroreflex regulation of arterial pressure. Am J Physiol Heart Circ Physiol 308:H49–H58. https://doi.org/10.1152/ajpheart.00552.2014
Acknowledgements
None.
Funding
This study was partly supported by the Grant-in-Aid for Scientific Research [JSPS KAKENHI grants 18K10695 and 20K20622] and Salt Science Research Foundation [No. 2029 and 2125]. Part of this work was carried out by the research grant from NTT research, Inc. The authors confirm that these parties did not influence the study design, contents of the article, or selection of this journal.
Author information
Authors and Affiliations
Contributions
TK and MS conceived and designed the study. TK performed the experiments and analyzed data. TK, TN, YH, ML, CZ, KU, KS, TM, and MS interpreted the results of the experiments. TK prepared the figures and drafted the manuscript. TK edited and revised the manuscript. All the authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Animal care was provided in strict accordance with the Guiding Principles for the Care and Use of Animals in the Field of Physiological Sciences, which has been approved by the Physiological Society of Japan. The experimental protocols were reviewed and approved by the Animal Subject Committee of the National Cerebral and Cardiovascular Center (No. 20008, 21009).
Consent for publication
Not applicable.
Competing interests
The authors declare no conflict of interest regarding this study.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Kawada, T., Nishikawa, T., Hayama, Y. et al. Quantitative assessment of the central versus peripheral effect of intravenous clonidine using baroreflex equilibrium diagrams. J Physiol Sci 71, 39 (2021). https://doi.org/10.1186/s12576-021-00824-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12576-021-00824-y