
Abstract
Neuroplasticity is referred to the ability of the nervous system to change its structure or functions as a result of former stimuli. It is a plausible mechanism underlying a dynamic brain through adaptation processes of neural structure and activity patterns. Nevertheless, it is still unclear how the plastic neural systems achieve and maintain their equilibrium. Additionally, the alterations of balanced brain dynamics under different plasticity rules have not been explored either. Therefore, the present article primarily aims to review recent research studies regarding homosynaptic and heterosynaptic neuroplasticity characterized by the manipulation of excitatory and inhibitory synaptic inputs. Moreover, it attempts to understand different mechanisms related to the main forms of synaptic plasticity at the excitatory and inhibitory synapses during the brain development processes. Hence, this study comprised surveying those articles published since 1988 and available through PubMed, Google Scholar and science direct databases on a keyword-based search paradigm. All in all, the study results presented extensive and corroborative pieces of evidence for the main types of plasticity, including the long-term potentiation (LTP) and long-term depression (LTD) of the excitatory and inhibitory postsynaptic potentials (EPSPs and IPSPs).
Similar content being viewed by others

Introduction
Plasticity is referred to the ability of an individual organism or cell in adjusting its phenotype in response to its environmental alterations. In contrast to prior views, recent studies have highlighted the extraordinary plasticity of cells [1]. Plasticity is a common synaptic feature. Accordingly, disclosing the molecular and cellular mechanisms that lead to this phenomenon is a dynamic biology domain with promising therapeutic potentials. Neuroplasticity, otherwise known as brain plasticity or neural plasticity, is the capacity of the neural synapses and brain pathways to be modified by altered thoughts and emotions, as well as environmental, behavioral, and neural stimuli. These repeated modifications occur as the brain learns and retains new data during its development [2]. Synaptic pruning usually happens when the brain deletes unnecessary or useless neural connections; this process simultaneously reinforces the necessary synapses [3]. Generally, the reformations of the synaptic network are experience-dependent processes in which the nervous system fine-tunes itself for competence. Moreover, its restructuring could provoke physiological and anatomical changes. For instance, the brain activity associated with a particular function could be relocated in the brain [4]. Nevertheless, important progress has been achieved in recognizing the molecular mechanisms of the elementary plasticity processes. However, the necessity and adequacy of synaptic plasticity in rearranging dynamic cortical developments cannot be easily demonstrated.
In this review conducted based on selected articles data extraction, and will be firstly discussed, homosynaptic and heterosynaptic plasticity and then the synaptic plasticity reinforcement and depression processes. Also, the key synaptic plasticity mechanisms, including the effects of development, synapse type, brain regions, and dendrite biophysics, as well as the postsynaptic changes occurring at excitatory glutamatergic synapses on locus coeruleus neurons would be explored. Finally, differential expression of long-term plasticity will be reported. Methods for developing this review are outlined in Box 1.
Homosynaptic and heterosynaptic plasticity
Two plasticity types, homosynaptic and heterosynaptic ones, differ extensively in their necessity and respective presynaptic activity-dependency during the induction phase. Homosynaptic plasticity has shown whole-cell properties of neurons; however, heterosynaptic modulation remains restricted to individual synapses. Nevertheless, both processes interact at the level of single synapses [5, 6].
The Hebbian theory introduces three characteristics about synapses: homosynaptic plasticity, associativity, and input-specificity [7]. Accordingly, homosynaptic plasticity referred to as input-specific or associative plasticity, is induced at the directly activated synapses in a neuron during the brain’s developmental phase. To induce this type of plasticity at certain synapses, their presynaptic activation is required because they connect the postsynaptic neural firing to specific presynaptic neural activities [1].
In contrast to Heb theory, Kandel and Tauc [8] proposed a heterosynaptic rule for strengthening the synaptic connections. Experimental pieces of evidence have introduced the properties that were associated with heterosynaptic plasticity, including its induction at non-active synapses, weight-dependent direction and magnitude, and balanced potentiation and depression [9]. Therefore, it could occur at any cellular synapses following strong postsynaptic activities. Some differences between these two kinds of plasticity are shown in the following:
Effect of plasticity on learning and memory
The heterosynaptic plasticity have been involved in enhanced learning and relearning capacity, as well as the increased spreading of inputs with intrinsic connections of the neural network [10]. This form of plasticity may play a key role in maintaining the ability to learn various tasks and developmental processes [11]. In dead, heterosynaptic plasticity is necessary for the formation, refinement, and/or modification of intrinsic connectivity, as well as the development of response selectivity [11].
Evidence has revealed that some behavioral learning processes, like classical conditioning and sensitization, occur after a certain stimulus input [12]. Although the non-associative heterosynaptic modulation holds purely heterosynaptic properties, the associative type is activity-dependent due to the combined features of homosynaptic and heterosynaptic mechanisms [8]. Homosynaptic and heterosynaptic types of plasticity may both contribute to memory and learning processes, mainly by modifying the potency of neural connections. Since these two forms of plasticity have different types of computational properties, they affect learning differently. They have different properties and supply different functions, but they can both be provoked by classical protocols of inducing plasticity [13]. The input-specific properties of homosynaptic plasticity lead to changes in the synaptic strength, occurring only at specific postsynaptic neurons that are already stimulated and activated [7, 14]. By contrast, in heterosynaptic plasticity, specific neural stimulations lead to non-specific input alterations in the synaptic weight [5]. At times, their alterations are complementary forms of plasticity; hence, they are both required for normal neural actions in synaptic plasticity [15].
Duration of plasticity-induced alterations
Previously, Hebb [16] hypothesized homosynaptic rules for the long-term memory mechanisms; in this theory, those events that triggered synaptic reinforcement were proposed to have occurred at the same strengthened synapses [16]. According to the Hebbian hypothesis of homosynaptic plasticity, this process can always produce some distinct and short-term synaptic changes that cannot support long-term memory storage [5]. Therefore, the proposed mechanism might be used to explain learning and short-term memory; however, it may not recruit the required signaling pathways or transcriptional events for synaptic growth and long-term memory maintenance. Conversely, heterosynaptic facilitation could cause persistent changes when presented repeatedly by the transcriptional induction and new synaptic connections [5]. Also, the Hebbian homosynaptic and heterosynaptic modulatory mechanisms could recruit together in behavioral patterns [5]. Nevertheless, new synaptic plasticity categories could form due to their combination. Such joint mechanisms increase the duration of plastic changes in a non-additive way. Therefore, a greater level of synaptic specificity would be implicated that expands the nervous system’s ability to encode information [5]. Following the induction of homosynaptic changes, heterosynaptic plasticity was seen to be a common property of plastic synapses in the nervous systems. Heterosynaptic plasticity includes the neurons with operational stability that allow repetitive learning as well as the activation of dynamic features in sensory inputs [10]. In other words, the heterosynaptic changes may depend on postsynaptic firing and could associate with the homosynaptic plasticity induction; therefore, these changes demonstrate the intrinsic properties of synaptic plasticity.
Research studies indicate that Hebbian homosynaptic plasticity needs some modulatory transmitters to cause persistent changes. Also, homosynaptic action alone has not been sufficient to induce long-lasting plasticity. For instance, both homosynaptic and heterosynaptic processes are involved in classical conditioning. In conditional stimulus, the modulator neurons would release 5-HT with action on stimulated sensory neurons and undergo homosynaptic activity; then, the calcium influx into the sensory neurons increases the capability of 5-HT to activate adenylyl cyclase. Therefore, the temporal matching of heterosynaptic and homosynaptic activities causes an intense increase in cAMP concentrations and synaptic strength. Interestingly, these heterosynaptic and homosynaptic mechanisms have synergic effects. Therefore, the overall increase would be higher than the sum of both enhancements due to either heterosynaptic or homosynaptic processes alone. This event could be presented as a new plasticity class [5, 17, 18]: A combinatory mechanism that leads to a prolonged plasticity duration and ample synaptic specificity [5]. Additionally, the metaplastic effects of these combined mechanisms are associated with a new form of heterosynaptic synaptic depression, in which postsynaptic neural activity is simultaneous with weakened synaptic connections at the inactive synapses [19].
Distance-dependency of plasticity
Distance-dependency of heterosynaptic plasticity (from the stimulated synapses during the induction) leads to specific changes in its amplitude; the same/opposite-sign plasticity would, respectively, occur at shorter/longer distances [9, 20]. Moreover, this amplitude alteration pattern may cause lateral inhibitions at synapses. Due to this class of inhibitions, plasticity occurs at a local synaptic population whereas it may stimulate other synaptic populations against the neighboring ones. Moreover, the total synaptic weight would be preserved to a cell by balancing the homosynaptic potentiation or depression [21]. Heterosynaptic plasticity could also be induced by distance-independent mechanisms, without presynaptic stimulations, and by the increase in intracellular Ca2+ levels (evoked by photolytic release of Ca2+ reserves) [22, 23]. Nonetheless, heterosynaptic potentiation or depression does not have identical induction rules [24]. Apart from the distance-dependency of the activation sites during the plasticity induction, the homosynaptic plasticity sign is a contributory factor as well. The same-sign heterosynaptic plasticity is induced at shorter distances while the opposite one appears farther away from the focal activation point [24].
Plasticity latency
Overall, heterosynaptic and homosynaptic forms of plasticity (opposite terms) include different action durations. Homosynaptic plasticity has been reported to have needed 10 min for pairing, whereas heterosynaptic plasticity occurred 10–20 min after the pairing. The longer latency of heterosynaptic plasticity suggests that unpaired input changes serve as homeostatic modulators in synaptic exhaustion. However, homosynaptic plasticity is essential for the high performance of neural circuits [1]. While heterosynaptic plasticity is inhibited, homosynaptic plasticity could be preserved; therefore, heterosynaptic plasticity can exist in a non-stimulated pathway while a neighboring pathway is being stimulated [25].
Homosynaptic or heterosynaptic plasticity (Hebbian-type learning) characteristics and signal transduction are shown in Table 1.
Homeostatic effects of plasticity
Several forms of heterosynaptic plasticity are reported, among which the main form has a homeostatic role [25]. The ultra-structural aspects, such as the synapse size and surface area of the postsynaptic density (PSD) could represent homeostatic regulations. The coordinated changes of the PSD surface area in the hippocampal dendritic spines after LTP induction can be mentioned as an example. The increased PSD surface area at some synapses and formation of new synapses have been accompanied by corresponding changes in the PSD surface area at other synapses. Whether it was a compensatory decrease or complete elimination, the total amount of PSD surface area stays approximately constant. Similar rules could be seen at individual dendritic branches as well [26,27,28].
Region-specific plasticity in the brain
Different areas of the brain and nervous system could induce several forms of plasticity with a similar biological or experimental induction paradigm. Similar spike-timing-dependent plasticity (STDP) is a biological process that modulates the neural synaptic strength in the brain. This can lead to bidirectional corticostriatal (CS) and thalamostriatal (TS) STDP as anti-Hebbian CS-STDP and Hebbian TS-STDP [29] in physiological conditions without blocking the GABAergic transmission in the dorsolateral striatum [30,31,32,33].
In the somatosensory cortex, the deafferentation changes of capsaicin-induced C-fiber and the consequent peripheral inputs could cause cortical plasticity that would have been postsynaptic originally [34]. The electrophysiological analyses of nucleus tractus solitarii (NTS) neurons in the brainstem displayed hypertension-induced plasticity of GABAergic mechanisms [35]. In the raphe region of the brainstem, involved in cutaneous vasoconstriction due to hypothermia [36, 37], spatiotemporal developments and neural plasticity alterations occur in the serotonergic nuclei [38]. In another brainstem region, NTS neuroplasticity precedes the functional alterations in the autonomous adjustment of the arterial pressure [39].
Furthermore, the impact of thalamostriatal activity (through heterosynaptic plasticity) on shaping the corticostriatal plasticity maps in particular time scales could be significant. This heterosynaptic plasticity has a major role in shaping the corticostriatal plasticity map through the parafascicular thalamic nucleus (Pf) as well as the formation of flexible behaviors in procedural learning. Additionally, heterosynaptic plasticity at corticostriatal and thalamostriatal synapses has a significant impact on these plasticity maps. The slight precedence of cortical activation over the thalamic one or their simultaneous activation can either impose plasticity or disrupt corticostriatal plasticity. Also, thalamic inputs might strongly be modulated in corticostriatal plasticity maps through the heterosynaptic effects for specific timing patterns [29].
Certain signaling pathways, underlying the CS-STDP and TS-STDP, distinctively control the GABA levels. Moreover, the TS-STDP requires single molecular coincidence detectors (e.g., NMDA receptors or NMDARs), whereas CS-STDP needs both NMDARs and endocannabinoids (ECs) as distinct signaling pathways [33, 40, 41]. In this regard, there is evidence of inhibited GABAergic transmission in these excitatory synapses affecting the CS-STDP/TS-STDP polarity, and changing the bidirectional Hebbian TS-STDP to unidirectional anti-Hebbian STDP with LTD for the post–pre/pre–post pairings [29, 40]. At last, acetylcholine exerted a key role in the expression and polarity of both hippocampal and cortical NMDAR-mediated STDP; thus, the impacts of other neurotransmitters/modulators on similar mapping patterns need to be further explored [42, 43].
Homosynaptic and heterosynaptic plasticity mechanisms in different brain areas
Different brain areas have different mechanisms for homosynaptic or heterosynaptic plasticity; for example, homosynaptic and heterosynaptic forms of plasticity in the mouse auditory cortex and human temporal lobe of epileptic patients displayed different mechanisms. Moreover, in the intercalated neurons of the amygdala, synaptic potentiation in a pathway can result in the depression of non-stimulated pathways [9].
Conversely, the cortical and hippocampal neurons can express a different form of plasticity, known as homosynaptic inhibitory plasticity (LTPi or LTDi), which is observed in some brain areas and circuit development [44]. The plasticity of GABAergic synapses from an individual inhibitory neuron onto a postsynaptic excitatory one is a homosynaptic monosynaptic form of inhibitory plasticity [45, 46]. The induction and expression of this form of plasticity exhibit significant differences in the hippocampus and sensory neocortex [44].
Sensory information is primarily conveyed to layers 3, 4, and 6 [47] of the neocortex via thalamocortical axons. The response latency to sensory stimuli is distinguished in layer 4 neurons [48, 49]. The sensory information principally flows through layer 4 to layers 2/3, and then to layers 5–6 [50], or through layer 4 to layers 2/3/5, and then to layer 6 [48]. The layer classifications in the somatosensory cortex of rats and monkeys, as well as the visual cortex of cats, correspond with the size of their receptive fields as follows: layer 4 (the smallest one), supragranular layers, layer 3, and infragranular layers [48]. Occasionally, the sizes of layer 3 and infragranular layers are equal to the ones in the supragranular layers [51]. Neurons gather information from other neurons at the previous level with larger receptive fields and deviate them to the next level. In this way, larger and more integrated receptive fields are formed.
Homosynaptic LTPi and LTDi both depend on postsynaptic Ca2+ currents. Nevertheless, the Ca2+ influx sources and their mechanisms have not been thoroughly explored yet [46]. Unlike layer 5 of the primary visual cortex or hippocampus, this form of inhibitory plasticity does not seem to depend on the changes in potassium chloride cotransporter 2 (KCC2) activity or the activation of either GABAB receptors or NMDA ones [46].
The induction and expression of high-frequency LTPi in the visual cortex are dependent on intracellular Ca2+ storage, which is triggered by the activation of GABAB receptors. They are facilitated by the activation of serotoninergic (5-HT) or α-adrenergic receptors [52] and mediated through activating IP3 [53]. GABA release is mediated through a brain-derived neurotrophic factor and tropomyosin receptor kinase B (BDNF/TrkB) signaling cascade that is initiated by an intracellular Ca2+ release in the developing visual cortex [54] and hippocampus [55]; wherein, the high-frequency LTPi would be expressed presynaptically [44]. The maintenance of high-frequency LTPi in the visual cortex depends on persistent low-frequency stimulations (LFS). However, in the hippocampus, it is induced and maintained after the high-frequency stimulation (HFS) [56]. The ventral tegmental area (VTA) has a different mechanism. Its retrograde signaling pathways are mediated by nitric oxide (NO), guanylate cyclase (GC), and protein kinase G (PKG)-dependent pathways [57].
Also, the induction and expression mechanisms of heterosynaptic LTDi (long-term depression of IPSPs) illustrate significant differences in various circuits. For example, in layer 5 of the primary visual cortex, high-frequency LTDi depends on Ca2 + currents through NMDARs or L-type Ca2 + channels in postsynaptic excitatory neurons [58, 59]. The NMDAR-dependent LTDi produces a focal and restricted inhibitory depression, while the L-type Ca2+ channel-dependent LTDi depresses many inhibitory synapses that are related to the same postsynaptic neurons [58]. The ECs are also required for the high-frequency LTDi induction in layers 2/3 of the primary visual cortex and hippocampus [60].
The heterosynaptic LTPi of inhibitory postsynaptic potentials (IPSPs), has similar Ca2+-mediating signaling; nevertheless. However, it uses different sources of Ca2+ supply it has somewhat different underlying intracellular mechanisms for the induction and expression in the visual cortex [59], hippocampus [61], cerebellar nuclei [62], superior olivary complex [63], ventral tegmental area [64], brainstem [65], and other brain regions. For instance, the Ca2+ source is the voltage-gated calcium channels (VGCCs) in the neonatal hippocampus of rats [66], astrocytes in the young rat hippocampus [67], the postsynaptic intracellular reservoir for the visual cortical inhibitory synapses, and the postsynaptic NMDAR activation in the ventral tegmental area slices [68].
Moreover, low-frequency heterosynaptic LTDi has also been induced in several brain areas, including the VTA, amygdala, striatum, prefrontal cortex, and corticotectal cocultures [44]. Low-frequency LTDi is induced by the activation of glutamatergic axons, which can cause heterosynaptic depression in those GABAergic inputs that meet with the activated postsynaptic neurons and maintain their plasticity [69]. The EC release from the glutamatergic neurons to the postsynaptic ones affects the inhibitory synaptic strength [44]. The low-frequency LTDi requires the release and aggregation of Ca2+ in the presynaptic interneuron of the hippocampus. However, the presynaptic expression of low-frequency LTDi in the VTA occurs by GABA release in response to protein kinase A (PKA)-dependent modulations [60, 70].
Both homosynaptic and heterosynaptic inhibitory plasticity are involved in sensory processing, sound localization, neuropathic pain modulations, neural activity regulations after the brain injury, and pregnancy-induced neural excitability alterations [44]. Developing in-depth knowledge of different forms of plasticity is crucial to elucidate their role in brain functions in healthy subjects or the progression and treatment of diseases. Therefore, further investigations are necessary to identify their underlying mechanisms.
Long-term potentiation and depression
Both LTP and LTD are involved in circuit and memory improvement in the developing sensory neocortex [71]. Overall, long-term plasticity depends on different variables, including the baseline amplitude of synaptic strength, presynaptic and postsynaptic spiking frequencies, postsynaptic membrane potentials, and the dendritic location of synaptic inputs [24, 72,73,74]. Some factors that affect the induction of LTP or LTD are as the following:
-
1.
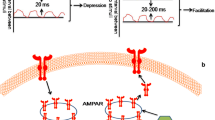
Different plasticity induction protocols have been used to define the direction and magnitude of homosynaptic plasticity and induce LTP and LTD, including afferent tetanization, pairing, and intracellular tetanization. The afferent tetanization is applied by simulating the presynaptic fibers using repeated electric pulses at a specified frequency or pattern with focal inputs that are decayed by distance. As such, low-frequency tetanization is given at 3 Hz and below, whereas high-frequency stimulation is received at 20 Hz and higher up to 50–200 Hz [24]. As such, in the afferent tetanization protocol, the change direction depends on the frequency. Therefore, the tetanic stimulations at higher frequencies (20 Hz and above) induce potentiation, but tetanization at lower frequencies (3 Hz and below) causes depression [24].
-
2.
LTP or LTD induction mainly depends on the timing of presynaptic activity in the pairing protocol which relates to the postsynaptic firing or current network activities [24]. The LTP or LTD magnitude, however, is determined by the frequency and number of postsynaptic potentials in each pairing burst, as well as the number of pairings in the pairing protocol [72,73,74]. Any increase in these parameters results in higher alterations in plasticity [24]. Conclusively, high-frequency afferent tetanization induces a characteristic response amplitude profile containing alterations (LTP at stimulated inputs surrounded by heterosynaptic LTD in the hippocampus and amygdala) [9, 20].
-
3.
Intracellular Ca2+ reserves play a major role in inducing heterosynaptic LTD and heterosynaptic LTP facilitation in the hippocampus [9]. Furthermore, inactive synapses have inverse sensitivity to local calcium signals [9]. Hence, higher levels of intracellular Ca2+ may lead to depression whereas the lower levels evoke potentiation at inactive synapses. This response profile corresponds with a hypothesis concerning the Ca2+-dependent LTP and LTD. Based on this evidence, the direction of synaptic alterations was related to the Ca2+ elevation amplitude and time course [75]. While fast and high-amplitude Ca2+ signals cause LTP induction, slow and low-amplitude signals could induce LTD. A brief and submicromolar increase in intracellular Ca2+ signals might lead to potentiation or depression changes [76].
-
4.
Unlike heterosynaptic LTP, the LTD is partly mediated by a decrease in release probability. Although plasticity is regulated by presynaptic changes, it is induced by postsynaptic spiking alone. This form of plasticity would necessarily require transsynaptic interactions by the postsynaptic release of a retrograde messenger that activates presynaptic receptors following a strong postsynaptic depolarization [77].
-
5.
Typically, LTP and LTD are produced by the postsynaptic activation of NMDA, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA), and metabotropic glutamate (mGlu) receptors [78]. Among the molecules that underlie synaptic plasticity, AMPA-type ionotropic glutamatergic receptors (AMPARs) play a key role in both LTP [79, 80] and LTD [81]. In neurons, AMPARs are highly flexible and undergo essential and activity-dependent trafficking [82]. The changes in synaptic AMPAR number are crucial during the experience-dependent synaptic modulations. For instance, LTP in the pyramidal CA3 and CA1 hippocampal neurons is associated with an increase of synaptic AMPARs in an activity-dependent manner [83]. Conversely, the reduced number of synaptic AMPARs occurs in LTD. However, it is not clear whether such alterations in the AMPAR number at one synapse would also affect neighboring synapses in a compensatory manner. The modification of synaptic AMPARs during the LTP and LTD induction can provoke compensatory heterosynaptic alterations that could rescale the synaptic strength of unstimulated synapses and modulate consequent activity-depended synaptic plasticity inductions. In LTP and LTD, the increase or decrease of synaptic AMPARs highly depends on the lateral diffusion of receptors [84].
-
6.
The interaction of retrograde messengers with specific receptors at the presynaptic membrane plays a role in inducing presynaptic LTP and LTD. In this way, the upregulation and downregulation of these receptors allow retrograde signaling to modify synaptic weight accordingly via certain proteins like protein kinase C (PKC) [85, 86]. Future expression of these postsynaptic receptors and the presynaptic receptors that correspond to them differ significantly concerning their development phases, synapse types, brain regions, and dendrite biophysics [78].
LTP and LTD of excitatory synapses
The depolarization pairing with the CA3 presynaptic inputs in the CA1 neurons enhances the EPSP amplitude. This phenomenon is called LTP. Concurrent presynaptic and postsynaptic neural activities cause potentiation of synaptic conduction. Therefore, the excitatory synapses should contain coincidence-detector neurons to display synchronized presynaptic and postsynaptic neural activities. NMDA receptors are ligand-gated calcium channels that act as such detectors of presynaptic and postsynaptic depolarization [87]. The ensuing transient increase in the intracellular Ca2+ concentrations activates Ca2+/calmodulin-dependent protein kinase II (CAMKII) and PKC. Subsequently, the enzyme-catalyzed phosphorylation of cAMP-response element-binding protein (CREB) generates CREB-dependent gene expression [88]. Presynaptic terminals mediate LTP, and retrograde messengers, such as NO and ECs convey messages to the presynaptic cells so that neurotransmitter release could be altered [89].
Three procedures lead to LTP induction: (a) pairing, intracellular postsynaptic depolarization paired with the LFS of afferent fibers, (b) theta-burst stimulation (TBS) of the afferent pathways (10 brief bursts, 5 bursts/s; each burst four pulses at 100 Hz), and (c) tetanic stimulation (100 Hz, 1 s) of afferent pathways. The physiological relevance of these protocols may differ significantly.
LTP that is induced by tetanus [90, 91] and pairing stimulation of the white-matter (WM) [92] can be produced in pyramidal neurons in layers 2/3, 5, and 6 [93]. Some factors increase the likelihood of LTP production: blocking GABAergic inhibition, removal of Mg2+, or taking slices from immature animals [90, 91]. Decrease of inhibition and increase of excitation both enhances the probability of LTP induction. According to Kirkwood and Bear [90], LTP could be induced in layers 2/3 by TBS of layer 4 with a success rate of over 80% [90]. Moreover, the visual cortex LTP mostly happens at synapses on layers 2/3, 4, and 5 [94, 95].
In a previous study by authors, corticogeniculate cells in layer 6 of the visual cortex received top-down synaptic inputs from cortical upper layers (Uls), and bottom-up synaptic inputs from the WM; also, the WM-induced and UL-induced plasticity could occur through NMDARs and mGluRs, respectively [93]. In LTD induction, repeated LFS reduces the synaptic efficacy [96]. However, in granular and agranular areas, homosynaptic LTD can be produced by layer 4 LFS (1 Hz for 15 min). LTD is induced by the LFS of the WM/layer 6 to layer 4 in immature animals in case of IPSP inhibition [97]. There is evidence that LTD in the visual cortex is mostly induced at synapses on layers 2/3, 4, and 5 [94, 95]. In our study regarding the CG cells of layer 6 (in the visual cortex), the cannabinoid type 1 receptors and calcineurin underlie the UL-induced and WM-induced heterosynaptic LTD, respectively. So, homosynaptic LTP and heterosynaptic LTD in corticogeniculate cells may modify the efficacy of synaptic transmission through different mechanisms [93].
LTP and LTD of inhibitory synapses
The central and peripheral nervous systems include a variety of inhibitory interneurons [98]. In a mature nervous system, and especially in the adult cortical networks, excitation is modulated by a complex set of inhibitory circuits [99, 100]. Gamma-aminobutyric acid (GABA) is the main neurotransmitter that has major inhibitory functions. Inhibition is critical for many neural functions, including spike generation, dendritic integration, synaptic plasticity, sleeping, learning, and prevention of pathological activities like epilepsy [101,102,103,104,105].
Twenty-five percent of neocortical neurons are GABAergic [106], and 20% of all synapses are GABAergic [107]. Inhibitory plasticity may play a critical role in cortical remapping [108]. Besides, GABA receptors would be downregulated following the visual or somatosensory cortex deafferentation [109] whereas they would be upregulated by chronic stimuli [110]. To avoid hyperactivity or hypoactivity in neurons and nervous networks during prolonged periods, inhibitory synapses should be calibrated or balanced by the relative strength of excitatory synapses. In the sensory cortex, inhibitory responses and excitatory–inhibitory balance are developed in early postnatal developments [111]. Since the experience-dependent regulation of excitatory synapses mandates corresponding alterations to inhibition, the dynamic excitatory–inhibitory balance should be maintained as well [112,113,114,115]. Previous studies have implied the plasticity of excitatory synapses on inhibitory neurons, resulting in the discovery of highly heterogeneous rules for plasticity induction in diverse types of interneurons [116, 117]. It is indicated that some excitatory synapses on inhibitory neurons have shown associative Hebbian-type plasticity. As such, the interneural activities could present either input-specific or input-non-specific types of plasticity at different excitatory synapses [117]. For instance, excitatory synapses on fast-spiking (FS) interneurons of the stratum pyramidal cells lacked input-specificity in the hippocampal CA1 [116]. Contrastingly, the excitatory synapses on interneurons of the stratum radiatum and stratum oriens expressed strict input-specific plasticity. In the former instance, there is LTP expression but no LTD changes. In the latter instance, there is Hebbian-type plasticity in stratum radiatum or anti-Hebbian plasticity in the stratum oriens [118].
The chief inhibitory neurons in the neocortex may associate with one of the two common classes of interneurons, the FS and non-FS (nFS) neurons. They show different functions and properties [119]. Also, some excitatory synapses on the inhibitory neurons could induce heterosynaptic plasticity, with weight-dependent properties, both in FS and nFS subtypes and also in all interneurons that are pooled together, such as the pyramidal neurons [117, 120].
Recently, weight-dependent heterosynaptic plasticity has been proposed as a novel understanding of plasticity at the excitatory synapses on inhibitory neurons. It is a widespread phenomenon that may not only participate in preventing runaway dynamics at excitatory synapses, but also exhibit potentiation or depression dispositions [25, 117]. Despite the weight-dependency of heterosynaptic plasticity in all interneurons, it displays different net effects in the FS and nFS cells [117]. Heterosynaptic changes in the FS neurons would contribute to the overall excitatory/inhibitory balance. Also, they balance the cortical network operation patterns [121,122,123] while facilitating the local rearrangement of neural activities and their synchronization [117].
In the nFS neurons, heterosynaptic plasticity may contribute to the operation maintenance for the inhibitory systems by preventing the elimination of the low-probability synapses. Pruning prevention by Hebbian-type plasticity preserves the functional inhibitory neurons that were activated by low-probability synapses. That is because these synapses tend to be facilitatory for these neurons and may operate as slowly activated ones through repeated firing in the network. In addition to GABA, traces of NO involvement are found in retrograde signaling during the heterosynaptic plasticity induction in the pyramidal and inhibitory neurons [116, 124].
Recordings of the linked pyramid-to-interneuron pairs confirmed the possibility of plasticity induction by purely postsynaptic protocols without any presynaptic spikes; thus, the induced plasticity type was heterosynaptic [117]. At GABAergic synapses on the pyramidal neurons, tetanus stimulation of layer 4 in the visual cortex of adult rats [56] could induce plasticity in presence of both NMDA and AMPA receptor blockers. Unlike the associative EPSP potentiation, the IPSP plasticity was membrane-potential-independent.
In our previous study, in the layers 2/3 of the mouse visual cortex, the tetanic activation of presynaptic FS-GABA neurons induced LTP for the unitary IPSCs (uIPSCs); whereas a similar activation of presynaptic nFS-GABA neurons could not produce LTP. This evidence indicated that long-term plasticity at the inhibitory synapses on FS-GABA neurons has pathway specificity. Also, it proposed that P/Q-type calcium channels may involve in the LTP induction at inhibitory synapses on FS-GABA interneurons [125]. In another study, tetanic stimulation of the sensory cortex induced LTP in motor cortical neurons [126].
Developmental plasticity
During postnatal developments in synaptic level, glutamatergic synapses become mature; also, various AMPAR and NMDAR activities occur. In the primary developmental stages, some brain synapses contain only NMDARs; therefore, the rapid addition of AMPARs into these synapses leads to their maturation. In our previous study, the postsynaptic alterations at excitatory glutamatergic synapses in the locus coeruleus of rats were analyzed to discover plasticity changes during the first postnatal weeks. The frequency and amplitude of NMDA sEPSCs and the frequency of AMPA sEPSCs in the locus coeruleus were increased during the second and third postnatal weeks compared to the first one [127]. Moreover, experience-dependent plasticity is a vital property of normal brain function that depends on regular LTP and is reduced in certain neurological and psychiatric disorders.
Experience-dependent plasticity can be mediated by the presynaptic NMDARs in the excitatory connections from the neurons of layer 4 and layers 2/3 in the visual cortex [128]. Also, the presynaptic NMDARs are involved in experience-dependent plasticity in excitatory connections from the neurons of layer 4 and layers 2/3 in the barrel cortex. However, in large excitatory spines of the CA1 hippocampal neurons, synapse-specific mediation of experience-dependent plasticity is facilitated by the activation of type-1 metabotropic glutamate receptors (mGluR1s) [128]. Synapse-type-specific variations of expressed proteins may be significant for synapse-type-specific plasticity (STSP); notably, proteins like CaMKII and mitogen-activated protein kinase (MAPK) are downstream signal pathways for neurotransmitter receptors [129]. However, postsynaptic signaling variations may control LTP. That is because the synapse-type-specific expression of the activity-related proteins like activity-regulated cytoskeleton-associated (ARC) proteins has affected homeostatic STSP [128].
According to previous pieces of evidence, enhancing NMDAR signaling could augment experience-dependent plasticity in the adult human brain [130].
Also, LTD in small immature dendritic spines only requires NMDAR activation, indicating the dynamic structure of dendritic spines in memory and cognition. However, in large mature dendritic spines, LTD needs activation of both NMDARs and mGluRs in addition to calcium release from internal reserves [131, 132]. Since large spines include more AMPARs [133], these findings correspond with metaplasticity, wherein previous activity could modify the following plasticity thresholds [134]. Sensory experience and neural activity adjust postsynaptic NMDAR subunit expression at many synapses of the brain [135].
Generally, age is a key factor in experience-dependent cortical plasticity. Significant alterations, as a result of stimulus-driven plasticity, occur primarily in critical stages of life [136]. These periods can later be revived according to a variety of elements, such as the damages of the peripheral sensory organs [136]. Such factors affect the plasticity changes in various life periods and do not function only in the critical periods of development. As such, these factors comprised myelin-associated proteins [137], inhibitory activities of parvalbumin-positive cells [115], and extracellular matrix components, including the perineuronal nets (PNNs) [136]. The number of parvalbumin (PV)- and somatostatin (SST)-positive interneurons deteriorates over time (as the subject ages). This indicates that different interneural subtypes would be affected differentially by aging. These results bear far-reaching consequences for developing rehabilitation plans targeted towards aging subjects [136].
Biophysics of dendrites and their structural plasticity
Dendrites (or, dendrons) are distinct neural sites where action potentials (APs) occur [138]. Therefore, many studies have focused on how biophysics of dendritic affect synaptic plasticity. As such, pyramidal neurons comprised apical and basal dendrites, extensively branching to secondary, tertiary, and fourth-degree dendrites. These branching patterns physically restrict biochemical signal transmissions that serve as defining factors for the signaling pattern. Dendrites of pyramidal neurons contain dendritic spines, or tiny protrusions emanating from the dendrite surface [139].
In neocortical layer 5 pyramidal neurons, the distal excitatory synapses present less LTP than the proximal synapses do [129]. That is because the back-propagation of APs, which establishes potentiation in these neurons, cannot reach distal arbors [73]. Nevertheless, each synaptic connection is formed by various synaptic contacts at distal and proximal locations [140], indicating the possible region-specificity of plasticity. Therefore, while distal synapses may show LTD, proximal synapses could be potentiated.
In the hippocampal CA1 area, axonally coupled spine pairs on the same dendrite have a similar size; the ones on different dendrites have unidentical sizes [141, 142]. The strict correlation between the spine volume and synaptic strength [143] could be considered the evidence for input-specificity of plasticity even at individual levels of synaptic contacts [144].
Neural dendrites generally receive information from many synapses (approx. 103–104) and process the received information in milliseconds [145, 146]. Dendritic spines are tiny actin-rich, micronized protrusions projected out of the dendritic shafts. Any modification in the spine size is accompanied by synaptic strengthening at the level of an individual spine. Besides, the actin dynamics in the dendritic spine have important roles in structural plasticity [147,148,149].
Overall, the biochemical signal transduction needs a longer time to modify synaptic strength, dendritic excitability, and electrical information. Synaptic strength and electrical properties of dendrites, known as neural plasticity, are regulated by changes in signaling state. These changes are regulated by the ion channels and transmitter receptors [145, 146]. Moreover, the dendritic structures and properties have significant effects on framing the spatiotemporal patterns of signal transduction in biochemical information processing. For instance, the time course and spatial spreading of synaptic inputs are determined by the passive cable properties of dendrites along with the distribution and functional state of voltage-gated channels [150].
Long and thin dendrite are cable-like structures. Each of them has a conducting cytoplasmic core and plasma that have membrane surface area with resistance and capacitance [151,152,153,154], displaying cable properties. These properties include current flow along the length of cable and across the membrane, as well as the drop of voltage across the membrane [73]. Particularly, the decrease of voltage is seen in the subthreshold regime of the long and thin dendrites that are related to a large axial resistance [155]. The asymmetric functional structure of dendritic trees affects the transient and steady-state voltage attenuation of subthreshold signals. Also, it results in an asymmetric activation of back- and forward propagating spikes [73]. Accordingly, the EPSP amplitude peak is attenuated by propagation at sites of origin to the soma (over 100-fold for most distal synapses in the neocortical pyramidal neurons of layer 5) [156]. This dendritic voltage decrement causes synapses at different dendritic locations to be influential (although not equally) on axonal spike outputs [157]. However, the synaptic charge attenuation in long and thin dendrites of pyramidal cells significantly reduces the amplitude of somatic EPSP (originating from distally located synapses on dendrites), compared to the proximally generated EPSPs with the same synaptic conductance. In the short spiny branchlets of Purkinje cells that are directly connected to the main thick dendrites, equal synaptic conductance was simulated on distal and proximal spiny branchlets with a similar somatic EPSP amplitude [158, 159]. The cable equations imply that electrotonic conduction of a somatic depolarization cannot fully preserve the somatic steady-state or transient depolarization as synaptic or action potential of all dendrites [160,161,162]. This problem is resolved by employing high-threshold VDCCs and their associated Na+ currents, instead of NMDARs or Ca+2 current [163, 164].
The dendrites could act as resistive and capacitive types of load on the axonal spike-initiation sites, causing difficult AP initiation. Therefore, dendritic morphology has a powerful impact on the neural input–output (I/O) function [73]. As such, the considerable increase of membrane area leads to large capacitive load with severe amplitude attenuation of fast-transient voltage; thus, it leads to the quick drop of AP, below the normal threshold of active propagation [165, 166]. Dendritic morphology determines which associations could occur between different synaptic inputs or input–output during the synaptic integration and plasticity [73]. Dendritic morphology also alters the coupling between somatic and dendritic spike-initiation sites in neurons [73, 167].
In addition to dendritic morphology, other dendritic properties, such as the kinetics, density, and spatial distribution of various voltage-gated conduction play major roles in spreading synaptic potentials, back-propagation of APs, initiation conditions, and forward propagation of dendritic spikes [168]. Also, regulation of channel properties and density, as well as the spatial gradients in these variables are functionally important neural characteristics. Besides, different kinds of voltage-gated conduction exist at different dendritic tree locations that may selectively modify the excitability of different neural types [168].
Some studies have proved the role of dendrites in input–output transformation and long-term synaptic plasticity. Therefore, local dendritic responses are presented as important factors that have a decisive impact on the nature and outcomes of synaptic plasticity [169]. Moreover, the biochemical synaptic transmissions reach postsynaptic density (PSD) in the dendritic spine and integrate with synaptic transmission [139]. Certain structural properties of the dendritic spines, such as the PSD surface area or spine head volume, have, respectively, influenced and correlated with the changes of synaptic efficacy [170]. Since the total surface area of PSD is approximately constant, any increase in the synaptic PSD surface area (i.e., new synapses) is balanced by a corresponding decrease in the number of other synapses, or by their complete elimination [27]. For instance, spine head volume is directly related to the PSD size; or long-term enlargement of spine size is associated with the LTP of synaptic transmission, so that spine head volume, PSD, and postsynaptic sensitivity to glutamate could increase [171, 172]. Any rapid increase in the sensitivity of the spine head volume or postsynaptic sensitivity to glutamate happens within few minutes despite the slow increase (in approximately 1 h) of PSD volume [172,173,174]. Accordingly, an opposite mechanism is also proposed for weak and prolonged stimulation, such as LTD and spine shrinkage [175,176,177].
Intracellular processes, as well as the extracellular signaling in dendritic spines, play significant roles in synaptic plasticity. These spines release BDNF through Ca2+-CaMKII-dependency. The released BDNF is bound to its TrkB receptors on the same spine and activates the receptors’ signaling to GTPase proteins Rac1 and Cdc42 to regulate the actin. Moreover, the extracellular signal-regulated kinase (ERK) and PKA pathways are involved in protein synthesis regulation in the activated dendritic spines [178,179,180,181]. Other than signaling protein activation (through rapid spine shape modification), also, activity-dependent protein synthesis in dendrites is contributory to the synaptic plasticity maintenance for more than several hours. Also, some GTPase proteins, such as Ras, RhoA, and Rac1 are important in facilitating the spine plasticity of adjacent spines. In other words, they lead to cooperative synaptic plasticity in adjacent spines [182, 183].
Since both passive and active dendritic features influence the local integration and forward propagation of evoked potentials, the effects of certain inputs on the spike output are determined by them. These passive and active properties also play a major role in synaptic plasticity activation as they establish both electrical and chemical signals received at each synapse and the interactions between the synapses. At last, the features of the dendritic trees could be modulated. Therefore, the electrical properties of dendrites could provide a vast range of mechanisms for plasticity modulation [73].
Conclusion
The homosynaptic and heterosynaptic plasticity are two key forms of plasticity that contribute to memory and learning processes with some differences in processes, mainly by modifying the potency of neural connections. They have different properties and supply different functions. Homosynaptic plasticity always produce some distinct and short-term synaptic changes that cannot support long-term memory storage, while heterosynaptic facilitation could cause persistent changes. The longer latency of heterosynaptic plasticity suggests that unpaired input changes serve as homeostatic modulators in synaptic exhaustion. However, homosynaptic plasticity is essential for the high performance of neural circuits. While heterosynaptic plasticity is inhibited, homosynaptic plasticity could be preserved; therefore, heterosynaptic plasticity can exist in a non-stimulated pathway while a neighboring pathway is being stimulated. In addition, different areas of the brain and nervous system are involved to form homo- and heterosynaptic plasticity. Heterosynaptic plasticity contributes to shaping the corticostriatal plasticity maps in particular time scales. This heterosynaptic plasticity has a major role in shaping the corticostriatal plasticity, and has a significant impact on the brain plasticity maps. Different brain areas have different mechanisms for homosynaptic or heterosynaptic plasticity that need to be more investigated and elucidated.
Availability of data and materials
Not applicable.
References
Chistiakova M, Bannon NM, Bazhenov M, Volgushev M (2014) Heterosynaptic plasticity: multiple mechanisms and multiple roles. Neuroscientist 20(5):483–498
Huttenlocher PR (2009) Neural plasticity. Harvard University Press, Cambridge
Kolb B, Gibb R (2011) Brain plasticity and behaviour in the developing brain. J Can Acad Child Adolesc Psychiatry 20(4):265
Nithianantharajah J, Hannan AJ (2006) Enriched environments, experience-dependent plasticity and disorders of the nervous system. Nat Rev Neurosci 7(9):697
Bailey CH, Giustetto M, Huang Y-Y, Hawkins RD, Kandel ER (2000) Is heterosynaptic modulation essential for stabilizing Hebbian plasticity and memory. Nat Rev Neurosci 1(1):11–20
Martin KC, Casadio A, Zhu H, Yaping E, Rose JC, Chen M et al (1997) Synapse-specific, long-term facilitation of aplysia sensory to motor synapses: a function for local protein synthesis in memory storage. Cell 91(7):927–938
Bliss TV, Collingridge GL (1993) A synaptic model of memory: long-term potentiation in the hippocampus. Nature 361(6407):31–39
Kandel ER, Tauc L (1965) Heterosynaptic facilitation in neurones of the abdominal ganglion of Aplysia depilans. J Physiol 181(1):1
Royer S, Paré D (2003) Conservation of total synaptic weight through balanced synaptic depression and potentiation. Nature 422(6931):518–522
Volgushev M, Chen J-Y, Ilin V, Goz R, Chistiakova M, Bazhenov M (2016) Partial breakdown of input specificity of STDP at individual synapses promotes new learning. J Neurosci 36(34):8842–8855
Zenke F, Agnes EJ, Gerstner W (2015) Diverse synaptic plasticity mechanisms orchestrated to form and retrieve memories in spiking neural networks. Nat Commun 6(1):1–13
Antonov I, Antonova I, Hawkins R (1999) Activity-dependent facilitation of monosynaptic sensory neuron-motor neuron PSPs contributes to classical conditioning of the Aplysia siphon-withdrawal reflex in a simplified preparation. Soc Neurosci Abstr 25:1129
Citri A, Malenka RC (2008) Synaptic plasticity: multiple forms, functions, and mechanisms. Neuropsychopharmacology 33(1):18-41
Malenka RC, Nicoll RA (1999) Long-term potentiation–a decade of progress? Science 285(5435):1870–1874
Jungenitz T, Beining M, Radic T, Deller T, Cuntz H, Jedlicka P et al (2018) Structural homo-and heterosynaptic plasticity in mature and adult newborn rat hippocampal granule cells. Proc Natl Acad Sci USA 115(20):E4670–E4679
Hebb DO (1949) An influential discussion on the neural control of perception and action, which includes a consideration of a homosynaptic (activity-dependent) rule for long-term memory. In: Hebb DO (ed) The organization of behavior: a neuropsychological theory. Wiley, New York
Hawkins RD, Kandel ER, Siegelbaum SA (1993) Learning to modulate transmitter release: themes and variations in synaptic plasticity. Annu Rev Neurosci. https://doi.org/10.1146/annurev.ne.16.030193.003205
Byrne JH (1987) Cellular analysis of associative learning. Physiol Rev 67(2):329–439
Young JZ, Nguyen PV (2005) Homosynaptic and heterosynaptic inhibition of synaptic tagging and capture of long-term potentiation by previous synaptic activity. J Neurosci 25(31):7221–7231
White G, Levy WB, Steward O (1990) Spatial overlap between populations of synapses determines the extent of their associative interaction during the induction of long-term potentiation and depression. J Neurophysiol 64(4):1186–1198
Schuman EM, Madison DV (1994) Locally distributed synaptic potentiation in the hippocampus. Science 263(5146):532–536
Yang S-N, Tang Y-G, Zucker RS (1999) Selective induction of LTP and LTD by postsynaptic [Ca2+] i elevation. J Neurophysiol 81(2):781–787
Neveu D, Zucker RS (1996) Long-lasting potentiation and depression without presynaptic activity. J Neurophysiol 75(5):2157–2160
Chistiakova M, Volgushev M (2009) Heterosynaptic plasticity in the neocortex. Exp Brain Res 199(3–4):377
Chistiakova M, Bannon NM, Chen J-Y, Bazhenov M, Volgushev M (2015) Homeostatic role of heterosynaptic plasticity: models and experiments. Front Comput Neurosci 9:89
Bourne JN, Harris KM (2011) Coordination of size and number of excitatory and inhibitory synapses results in a balanced structural plasticity along mature hippocampal CA1 dendrites during LTP. Hippocampus 21(4):354–373
Barnes SJ, Franzoni E, Jacobsen RI, Erdelyi F, Szabo G, Clopath C et al (2017) Deprivation-induced homeostatic spine scaling in vivo is localized to dendritic branches that have undergone recent spine loss. Neuron 96(4):871–82.e5
Triesch J, Vo AD, Hafner A-S (2018) Competition for synaptic building blocks shapes synaptic plasticity. Elife 7:e37836
Mendes A, Vignoud G, Perez S, Perrin E, Touboul J, Venance L (2020) Concurrent thalamostriatal and corticostriatal spike-timing-dependent plasticity and heterosynaptic interactions shape striatal plasticity map. Cereb Cortex 30(8):4381–4401
Paille V, Fino E, Du K, Morera-Herreras T, Perez S, Kotaleski JH et al (2013) GABAergic circuits control spike-timing-dependent plasticity. J Neurosci 33(22):9353–9363
Shen W, Flajolet M, Greengard P, Surmeier DJ (2008) Dichotomous dopaminergic control of striatal synaptic plasticity. Science 321(5890):848–851
Valtcheva S, Venance L (2016) Astrocytes gate Hebbian synaptic plasticity in the striatum. Nat Commun 7(1):1–17
Fino E, Paille V, Cui Y, Morera-Herreras T, Deniau JM, Venance L (2010) Distinct coincidence detectors govern the corticostriatal spike timing-dependent plasticity. J Physiol 588(16):3045–3062
Komaki A, Shahidi S, Sarihi A, Hasanein P, Lashgari R, Haghparast A et al (2013) Effects of neonatal C-fiber depletion on interaction between neocortical short-term and long-term plasticity. Basic Clin Neurosci 4(2):136
Zhang W, Mifflin S (2010) Plasticity of GABAergic mechanisms within the nucleus of the solitary tract in hypertension. Hypertension 55(2):201–206
Kourosh AM, Sarihi A, Behzadi G, Amiri I, Malacoti S, Vahabian M (2006) The effect of nucleus tractus solitarius nitric oxidergic neurons on blood pressure in diabetic rats. Iran Biomed J 10:1
Malakouti SM, Kourosh AM, Sarihi A, Hajizadeh S, Behzadi G, Shahidi S et al (2008) Reversible inactivation and excitation of nucleus raphe magnus can modulate tail blood flow of male wistar rats in response to hypothermia. Iran Biomed J 12:203
Ekström P (1994) Developmental changes in the brain-stem serotonergic nuclei of teleost fish and neural plasticity. Cell Mol Neurobiol 14(4):381–393
Fu GJ, Huber DA, Schreihofer AM (2013) Neuroplasticity in the nucleus tractus solitarius precedes development of functional changes in autonomic regulation of arterial pressure in obese Zucker rats. FASEB J. https://doi.org/10.1096/fasebj.27.1_supplement.699.16
Ellender T, Harwood J, Kosillo P, Capogna M, Bolam J (2013) Heterogeneous properties of central lateral and parafascicular thalamic synapses in the striatum. J Physiol 591(1):257–272
Wu Y-W, Kim J-I, Tawfik VL, Lalchandani RR, Scherrer G, Ding JB (2015) Input-and cell-type-specific endocannabinoid-dependent LTD in the striatum. Cell Rep 10(1):75–87
Foncelle A, Mendes A, Jędrzejewska-Szmek J, Valtcheva S, Berry H, Blackwell KT et al (2018) Modulation of spike-timing dependent plasticity: towards the inclusion of a third factor in computational models. Front Comput Neurosci 12:49
Brzosko Z, Mierau SB, Paulsen O (2019) Neuromodulation of spike-timing-dependent plasticity: past, present, and future. Neuron 103(4):563–581
Maffei A (2011) The many forms and functions of long term plasticity at GABAergic synapses. Neural Plast. https://doi.org/10.1155/2011/254724
Maffei A, Nataraj K, Nelson SB, Turrigiano GG (2006) Potentiation of cortical inhibition by visual deprivation. Nature 443(7107):81–84
Holmgren CD, Zilberter Y (2001) Coincident spiking activity induces long-term changes in inhibition of neocortical pyramidal cells. J Neurosci 21(20):8270–8277
Garraghty PE, Sur M (1990) Morphology of single intracellularly stained axons terminating in area 3b of macaque monkeys. J Comp Neurol 294(4):583–593
Armstrong-James M, Fox K, Das-Gupta A (1992) Flow of excitation within rat barrel cortex on striking a single vibrissa. J Neurophysiol 68(4):1345–1358
Welker E, Armstrong-James M, Loos H, Kraftsik R (1993) The mode of activation of a barrel column: response properties of single units in the somatosensory cortex of the mouse upon whisker deflection. Eur J Neurosci 5(6):691–712
Schwark H, Jones E (1989) The distribution of intrinsic cortical axons in area 3b of cat primary somatosensory cortex. Exp Brain Res 78(3):501–513
Sur M, Garraghty PE, Bruce CJ (1985) Somatosensory cortex in macaque monkeys: laminar differences in receptive field size in areas 3b and 1. Brain Res 342(2):391–395
Komatsu Y (1996) GABAB receptors, monoamine receptors, and postsynaptic inositol trisphosphate-induced Ca2+ release are involved in the induction of long-term potentiation at visual cortical inhibitory synapses. J Neurosci 16(20):6342–6352
Komatsu Y, Yoshimura Y (2011) Long-term modification at inhibitory synapses in developing visual cortex. In: Arianna M, Melanie AW (eds) Inhibitory synaptic plasticity. Springer, New York, pp 17–27
Inagaki T, Begum T, Reza F, Horibe S, Inaba M, Yoshimura Y et al (2008) Brain-derived neurotrophic factor-mediated retrograde signaling required for the induction of long-term potentiation at inhibitory synapses of visual cortical pyramidal neurons. Neurosci Res 61(2):192–200
Kuczewski N, Porcher C, Ferrand N, Fiorentino H, Pellegrino C, Kolarow R et al (2008) Backpropagating action potentials trigger dendritic release of BDNF during spontaneous network activity. J Neurosci 28(27):7013–7023
Komatsu Y, Yoshimura Y (2000) Activity-dependent maintenance of long-term potentiation at visual cortical inhibitory synapses. J Neurosci 20(20):7539–7546
Nugent FS, Niehaus JL, Kauer JA (2009) PKG and PKA signaling in LTP at GABAergic synapses. Neuropsychopharmacology 34(7):1829–1842
Kurotani T, Yoshimura Y, Komatsu Y (2003) Postsynaptic firing produces long-term depression at inhibitory synapses of rat visual cortex. Neurosci Lett 337(1):1–4
Komatsu Y, Iwakiri M (1993) Long-term modification of inhibitory synaptic transmission in developing visual cortex. NeuroReport 4(7):907–910
Chevaleyre V, Castillo PE (2003) Heterosynaptic LTD of hippocampal GABAergic synapses: a novel role of endocannabinoids in regulating excitability. Neuron 38(3):461–472
McLean H, Caillard O, Ben-Ari Y, Gaiarsa J (1996) Bidirectional plasticity expressed by GABAergic synapses in the neonatal rat hippocampus. J Physiol 496(2):471–477
Ouardouz M, Sastry BR (2000) Mechanisms underlying LTP of inhibitory synaptic transmission in the deep cerebellar nuclei. J Neurophysiol 84(3):1414–1421
Kotak VC, Sanes DH (2000) Long-lasting inhibitory synaptic depression is age-and calcium-dependent. J Neurosci 20(15):5820–5826
Madhavan A, Bonci A, Whistler JL (2010) Opioid-Induced GABA potentiation after chronic morphine attenuates the rewarding effects of opioids in the ventral tegmental area. J Neurosci 30(42):14029–14035
Glaum SR, Brooks PA (1996) Tetanus-induced sustained potentiation of monosynaptic inhibitory transmission in the rat medulla: evidence for a presynaptic locus. J Neurophysiol 76(1):30–38
Caillard O, Ben-Ari Y, Gaiarsa J-L (1999) Long-term potentiation of GABAergic synaptic transmission in neonatal rat hippocampus. J Physiol 518(Pt 1):109
Kang J, Jiang L, Goldman SA, Nedergaard M (1998) Astrocyte-mediated potentiation of inhibitory synaptic transmission. Nat Neurosci 1(8):683–692
Nugent FS, Penick EC, Kauer JA (2007) Opioids block long-term potentiation of inhibitory synapses. Nature 446(7139):1086–1090
Heifets BD, Castillo PE (2009) Endocannabinoid signaling and long-term synaptic plasticity. Annu Rev Physiol 71:283–306
Azad SC, Monory K, Marsicano G, Cravatt BF, Lutz B, Zieglgänsberger W et al (2004) Circuitry for associative plasticity in the amygdala involves endocannabinoid signaling. J Neurosci 24(44):9953–9961
Nabavi S, Fox R, Proulx CD, Lin JY, Tsien RY, Malinow R (2014) Engineering a memory with LTD and LTP. Nature 511(7509):348
Markram H, Lübke J, Frotscher M, Sakmann B (1997) Regulation of synaptic efficacy by coincidence of postsynaptic APs and EPSPs. Science 275(5297):213–215
Sjostrom PJ, Rancz EA, Roth A, Hausser M (2008) Dendritic excitability and synaptic plasticity. Physiol Rev 88(2):769–840
Birtoli B, Ulrich D (2004) Firing mode-dependent synaptic plasticity in rat neocortical pyramidal neurons. J Neurosci 24(21):4935–4940
Evans R, Blackwell K (2015) Calcium: amplitude, duration, or location? Biol Bull 228(1):75–83
Mulkey RM, Malenka RC (1992) Mechanisms underlying induction of homosynaptic long-term depression in area CA1 of the hippocampus. Neuron 9(5):967–975
Zilberter Y, Kaiser KM, Sakmann B (1999) Dendritic GABA release depresses excitatory transmission between layer 2/3 pyramidal and bitufted neurons in rat neocortex. Neuron 24(4):979–988
Larsen RS, Rao D, Manis PB, Philpot BD (2010) STDP in the developing sensory neocortex. Front Synaptic Neurosci 2:9
Calabresi P, Pisani A, Mercuri N, Bernardi G (1992) Long-term potentiation in the striatum is unmasked by removing the voltage-dependent magnesium block of NMDA receptor channels. Eur J Neurosci 4(10):929–935
Burnashev N, Schoepfer R, Monyer H, Ruppersberg JP, Gunther W, Seeburg PH et al (1992) Control by asparagine residues of calcium permeability and magnesium blockade in the NMDA receptor. Science 257(5075):1415–1419
Czarnecki A, Birtoli B, Ulrich D (2007) Cellular mechanisms of burst firing-mediated long-term depression in rat neocortical pyramidal cells. J Physiol 578(2):471–479
Henley JM, Wilkinson KA (2013) AMPA receptor trafficking and the mechanisms underlying synaptic plasticity and cognitive aging. Dialogues Clin Neurosci 15(1):11
Shimshek DR, Bus T, Schupp B, Jensen V, Marx V, Layer LE et al (2017) Different forms of AMPA receptor mediated LTP and their correlation to the spatial working memory formation. Front Mol Neurosci 10:214
Antunes G, Simoes-de-Souza F (2018) AMPA receptor trafficking and its role in heterosynaptic plasticity. Sci Rep 8(1):1–14
Pelkey KA, Lavezzari G, Racca C, Roche KW, McBain CJ (2005) mGluR7 is a metaplastic switch controlling bidirectional plasticity of feedforward inhibition. Neuron 46(1):89–102
Crosby KM, Inoue W, Pittman QJ, Bains JS (2011) Endocannabinoids gate state-dependent plasticity of synaptic inhibition in feeding circuits. Neuron 71(3):529–541
Cooke S, Bliss T (2006) Plasticity in the human central nervous system. Brain 129(7):1659–1673
Ahmed T, Frey S, Frey J (2004) Regulation of the phosphodiesterase PDE4B3-isotype during long-term potentiation in the area dentata in vivo. Neuroscience 124(4):857–867
Wilson RI, Nicoll RA (2002) Endocannabinoid signaling in the brain. Science 296(5568):678–682
Kirkwood A, Bear MF (1994) Hebbian synapses in visual cortex. J Neurosci 14(3):1634–1645
Tsumoto T (1993) Long-term depression in cerebral cortex: a possible substrate of “forgetting” that should not be forgotten. Neurosci Res 16(4):263–270
Frégnac Y, Burke JP, Smith D, Friedlander MJ (1994) Temporal covariance of pre- and postsynaptic activity regulates functional connectivity in the visual cortex. J Neurophysiol 71(4):1403–1421
Arami MK, Sohya K, Sarihi A, Jiang B, Yanagawa Y, Tsumoto T (2013) Reciprocal homosynaptic and heterosynaptic long-term plasticity of corticogeniculate projection neurons in layer VI of the mouse visual cortex. J Neurosci 33(18):7787–7798
Cooke SF, Bear MF (2010) Visual experience induces long-term potentiation in the primary visual cortex. J Neurosci 30(48):16304–16313
Jiang B, Trevino M, Kirkwood A (2007) Sequential development of long-term potentiation and depression in different layers of the mouse visual cortex. J Neurosci 27(36):9648–9652
Dudek SM, Bear MF (1993) Bidirectional long-term modification of synaptic effectiveness in the adult and immature hippocampus. J Neurosci 13(7):2910–2918
Dudek SM, Friedlander MJ (1996) Developmental down-regulation of LTD in cortical layer IV and its independence of modulation by inhibition. Neuron 16(6):1097–1106
Thayer JF (2006) On the importance of inhibition: central and peripheral manifestations of nonlinear inhibitory processes in neural systems. Dose Response. https://doi.org/10.2203/dose-response.004.01.002.Thayer
Constantinidis C, Williams GV, Goldman-Rakic PS (2002) A role for inhibition in shaping the temporal flow of information in prefrontal cortex. Nat Neurosci 5(2):175–180
Nakayama H, Miyazaki T, Kitamura K, Hashimoto K, Yanagawa Y, Obata K et al (2012) GABAergic inhibition regulates developmental synapse elimination in the cerebellum. Neuron 74(2):384–396
Cossart R, Dinocourt C, Hirsch J, Merchan-Perez A, De Felipe J, Ben-Ari Y et al (2001) Dendritic but not somatic GABAergic inhibition is decreased in experimental epilepsy. Nat Neurosci 4(1):52–62
Hattori R, Kuchibhotla KV, Froemke RC, Komiyama T (2017) Functions and dysfunctions of neocortical inhibitory neuron subtypes. Nat Neurosci 20(9):1199
Isaacson JS, Scanziani M (2011) How inhibition shapes cortical activity. Neuron 72(2):231–243
Oliveira MS, Pacheco LF, Mello CF, Cavalheiro EA, Garrido-Sanabria ER (2011) Epileptiform activity in the limbic system. Front Biosci 3:565–593
Scharfman HE, Brooks-Kayal AR (2014) Is plasticity of GABAergic mechanisms relevant to epileptogenesis? In: Scharfman HE, Buckmaster PS (eds) Issues in clinical epileptology: a view from the bench. Springer, Dordrecht, pp 133–150
Ren J, Aika Y, Heizmann C, Kosaka T (1992) Quantitative analysis of neurons and glial cells in the rat somatosensory cortex, with special reference to GABAergic neurons and parvalbumin-containing neurons. Exp Brain Res 92(1):1–14
Beaulieu C, Kisvarday Z, Somogyi P, Cynader M, Cowey A (1992) Quantitative distribution of GABA-immunopositive and-immunonegative neurons and synapses in the monkey striate cortex (area 17). Cereb Cortex 2(4):295–309
Jones E (1993) GABAergic neurons and their role in cortical plasticity in primates. Cereb Cortex 3(5):361–372
Garraghty PE, Lachica EA, Kaas JH (1991) Injury-induced reorganization of somatosensory cortex is accompanied by reductions in GABA staining. Somatosens Mot Res 8(4):347–354
Welker E, Soriano E, Dörfl J, Van der Loos H (1989) Plasticity in the barrel cortex of the adult mouse: transient increase of GAD-immunoreactivity following sensory stimulation. Exp Brain Res 78(3):659–664
Field RE, D’amour JA, Tremblay R, Miehl C, Rudy B, Gjorgjieva J et al (2020) Heterosynaptic plasticity determines the set point for cortical excitatory-inhibitory balance. Neuron. https://doi.org/10.1016/j.neuron.2020.03.002
Dorrn AL, Yuan K, Barker AJ, Schreiner CE, Froemke RC (2010) Developmental sensory experience balances cortical excitation and inhibition. Nature 465(7300):932–936
Froemke RC, Debanne D, Bi G-Q (2010) Temporal modulation of spike-timing-dependent plasticity. Front Synaptic Neurosci 2:19
House DR, Elstrott J, Koh E, Chung J, Feldman DE (2011) Parallel regulation of feedforward inhibition and excitation during whisker map plasticity. Neuron 72(5):819–831
Kuhlman SJ, Olivas ND, Tring E, Ikrar T, Xu X, Trachtenberg JT (2013) A disinhibitory microcircuit initiates critical-period plasticity in the visual cortex. Nature 501(7468):543–546
Jt L, Li C-Y, Zhao JP, Poo M-M, Zhang X-H (2007) Spike-timing-dependent plasticity of neocortical excitatory synapses on inhibitory interneurons depends on target cell type. J Neurosci 27(36):9711–9720
Chistiakova M, Ilin V, Roshchin M, Bannon N, Malyshev A, Kisvárday Z et al (2019) Distinct heterosynaptic plasticity in fast spiking and non-fast-spiking inhibitory neurons in rat visual cortex. J Neurosci 39(35):6865–6878
Lamsa K, Heeroma JH, Kullmann DM (2005) Hebbian LTP in feed-forward inhibitory interneurons and the temporal fidelity of input discrimination. Nat Neurosci 8(7):916–924
Druckmann S, Hill S, Schürmann F, Markram H, Segev I (2013) A hierarchical structure of cortical interneuron electrical diversity revealed by automated statistical analysis. Cereb Cortex 23(12):2994–3006
Chen J-Y, Lonjers P, Lee C, Chistiakova M, Volgushev M, Bazhenov M (2013) Heterosynaptic plasticity prevents runaway synaptic dynamics. J Neurosci 33(40):15915–15929
Cardin JA, Carlén M, Meletis K, Knoblich U, Zhang F, Deisseroth K et al (2009) Driving fast-spiking cells induces gamma rhythm and controls sensory responses. Nature 459(7247):663–667
Sohal VS, Zhang F, Yizhar O, Deisseroth K (2009) Parvalbumin neurons and gamma rhythms enhance cortical circuit performance. Nature 459(7247):698–702
Allen K, Monyer H (2015) Interneuron control of hippocampal oscillations. Curr Opin Neurobiol 31:81–87
Markram H, Wang Y, Tsodyks M (1998) Differential signaling via the same axon of neocortical pyramidal neurons. Proc Natl Acad Sci 95(9):5323–5328
Sarihi A, Mirnajafi-Zadeh J, Jiang B, Sohya K, Safari M-S, Arami MK et al (2012) Cell type-specific, presynaptic LTP of inhibitory synapses on fast-spiking GABAergic neurons in the mouse visual cortex. J Neurosci 32(38):13189–13199
Iriki A, Pavlides C, Keller A, Asanuma H (1989) Long-term potentiation in the motor cortex. Science 245(4924):1385–1387
Arami MK, Semnanian S, Javan M, Hajizadeh S, Sarihi A (2011) Postnatal developmental alterations in the locus coeruleus neuronal fast excitatory postsynaptic currents mediated by ionotropic glutamate receptors of rat. Physiol Pharmacol 14(4):337–348
Larsen RS, Sjöström PJ (2015) Synapse-type-specific plasticity in local circuits. Curr Opin Neurobiol 35:127–135
Colgan LA, Yasuda R (2014) Plasticity of dendritic spines: subcompartmentalization of signaling. Annu Rev Physiol 76:365–385
Forsyth JK, Bachman P, Mathalon DH, Roach BJ, Asarnow RF (2015) Augmenting NMDA receptor signaling boosts experience-dependent neuroplasticity in the adult human brain. Proc Natl Acad Sci USA 112(50):15331–15336
Holbro N, Grunditz Å, Oertner TG (2009) Differential distribution of endoplasmic reticulum controls metabotropic signaling and plasticity at hippocampal synapses. Proc Natl Acad Sci USA 106(35):15055–15060
Oh WC, Hill TC, Zito K (2013) Synapse-specific and size-dependent mechanisms of spine structural plasticity accompanying synaptic weakening. Proc Natl Acad Sci USA 110(4):E305–E312
Kasai H, Fukuda M, Watanabe S, Hayashi-Takagi A, Noguchi J (2010) Structural dynamics of dendritic spines in memory and cognition. Trends Neurosci 33(3):121–129
Hulme SR, Jones OD, Abraham WC (2013) Emerging roles of metaplasticity in behaviour and disease. Trends Neurosci 36(6):353–362
Paoletti P, Bellone C, Zhou Q (2013) NMDA receptor subunit diversity: impact on receptor properties, synaptic plasticity and disease. Nat Rev Neurosci 14(6):383–400
Voss P, Thomas ME, Cisneros-Franco JM, de Villers-Sidani É (2017) Dynamic brains and the changing rules of neuroplasticity: implications for learning and recovery. Front Psychol 8:1657
McGee AW, Yang Y, Fischer QS, Daw NW, Strittmatter SM (2005) Experience-driven plasticity of visual cortex limited by myelin and Nogo receptor. Science 309(5744):2222–2226
Vetter P, Roth A, Häusser M (2001) Propagation of action potentials in dendrites depends on dendritic morphology. J Neurophysiol 85(2):926–937
Yasuda R (2017) Biophysics of biochemical signaling in dendritic spines: implications in synaptic plasticity. Biophys J 113(10):2152–2159
Markram H, Lübke J, Frotscher M, Roth A, Sakmann B (1997) Physiology and anatomy of synaptic connections between thick tufted pyramidal neurones in the developing rat neocortex. J Physiol 500(2):409–440
Bartol TM, Bromer C, Kinney JP, Chirillo MA, Bourne JN, Harris KM et al (2015) Hippocampal spine head sizes are highly precise. Biorxiv. https://doi.org/10.1101/016329
Sorra KE, Harris KM (1993) Occurrence and three-dimensional structure of multiple synapses between individual radiatum axons and their target pyramidal cells in hippocampal area CA1. J Neurosci 13(9):3736–3748
Murthy VN, Schikorski T, Stevens CF, Zhu Y (2001) Inactivity produces increases in neurotransmitter release and synapse size. Neuron 32(4):673–682
Koester HJ, Johnston D (2005) Target cell-dependent normalization of transmitter release at neocortical synapses. Science 308(5723):863–866
Azeloglu EU, Iyengar R (2015) Signaling networks: information flow, computation, and decision making. Cold Spring Harb Perspect Biol 7(4):a005934
Bromberg KD, Ma’ayan A, Neves SR, Iyengar R (2008) Design logic of a cannabinoid receptor signaling network that triggers neurite outgrowth. Science 320(5878):903–909
Hotulainen P, Hoogenraad CC (2010) Actin in dendritic spines: connecting dynamics to function. J Cell Biol 189(4):619–629
Korobova F, Svitkina T (2010) Molecular architecture of synaptic actin cytoskeleton in hippocampal neurons reveals a mechanism of dendritic spine morphogenesis. Mol Biol Cell 21(1):165–176
Okamoto K-I, Nagai T, Miyawaki A, Hayashi Y (2004) Rapid and persistent modulation of actin dynamics regulates postsynaptic reorganization underlying bidirectional plasticity. Nat Neurosci 7(10):1104–1112
Jaffe DB, Carnevale NT (1999) Passive normalization of synaptic integration influenced by dendritic architecture. J Neurophysiol 82(6):3268–3285
Jack JJB, Noble D, Tsien RW (1975) Electric current flow in excitable cells. Clarendon Press, Oxford
Koch C (2004) Biophysics of computation: information processing in single neurons. Oxford University Press, Oxford
Rall W (1962) Theory of physiological properties of dendrites. Ann NY Acad Sci 96(4):1071–1092
Schwindt PC, Crill WE (1997) Local and propagated dendritic action potentials evoked by glutamate iontophoresis on rat neocortical pyramidal neurons. J Neurophysiol 77(5):2466–2483
Nevian T, Larkum ME, Polsky A, Schiller J (2007) Properties of basal dendrites of layer 5 pyramidal neurons: a direct patch-clamp recording study. Nat Neurosci 10(2):206–214
Stuart G, Spruston N (1998) Determinants of voltage attenuation in neocortical pyramidal neuron dendrites. J Neurosci 18(10):3501–3510
Häusser M (2001) Synaptic function: dendritic democracy. Curr Biol 11(1):R10–R12
De Schutter E, Bower JM (1994) Simulated responses of cerebellar Purkinje cells are independent of the dendritic location of granule cell synaptic inputs. Proc Natl Acad Sci USA 91(11):4736–4740
Roth A, Häusser M (2001) Compartmental models of rat cerebellar Purkinje cells based on simultaneous somatic and dendritic patch-clamp recordings. J Physiol 535(2):445–472
Stuart G, Spruston N, Sakmann B, Häusser M (1997) Action potential initiation and backpropagation in neurons of the mammalian CNS. Trends Neurosci 20(3):125–131
Korogod S, Kopysova I, Bras H, Gogan P, Tyc-Dumont S (1996) Differential back-invasion of a single complex dendrite of an abducens motoneuron by N-methyl-D-aspartate-induced oscillations: a simulation study. Neuroscience 75(4):1153–1163
Hoffman DA, Magee JC, Colbert CM, Johnston D (1997) K+ channel regulation of signal propagation in dendrites of hippocampal pyramidal neurons. Nature 387(6636):869–875
Jaffe D, Johnston D (1990) Induction of long-term potentiation at hippocampal mossy-fiber synapses follows a Hebbian rule. J Neurophysiol 64(3):948–960
Urban NN, Barrionuevo G (1996) Induction of hebbian and non-hebbian mossy fiber long-term potentiation by distinct patterns of high-frequency stimulation. J Neurosci 16(13):4293–4299
Gentet LJ, Williams SR (2007) Dopamine gates action potential backpropagation in midbrain dopaminergic neurons. J Neurosci 27(8):1892–1901
Häusser M, Stuart G, Racca C, Sakmann B (1995) Axonal initiation and active dendritic propagation of action potentials in substantia nigra neurons. Neuron 15(3):637–647
Schaefer AT, Larkum ME, Sakmann B, Roth A (2003) Coincidence detection in pyramidal neurons is tuned by their dendritic branching pattern. J Neurophysiol 89(6):3143–3154
Migliore M, Shepherd GM (2002) Emerging rules for the distributions of active dendritic conductances. Nat Rev Neurosci 3(5):362–370
Ariav G, Polsky A, Schiller J (2003) Submillisecond precision of the input-output transformation function mediated by fast sodium dendritic spikes in basal dendrites of CA1 pyramidal neurons. J Neurosci 23(21):7750–7758
Harris KM, Stevens JK (1989) Dendritic spines of CA 1 pyramidal cells in the rat hippocampus: serial electron microscopy with reference to their biophysical characteristics. J Neurosci 9(8):2982–2997
Chang J-Y, Parra-Bueno P, Laviv T, Szatmari EM, Lee S-JR, Yasuda R (2017) CaMKII autophosphorylation is necessary for optimal integration of Ca2+ signals during LTP induction, but not maintenance. Neuron 94(4):800–8.e4
Matsuzaki M, Honkura N, Ellis-Davies GC, Kasai H (2004) Structural basis of long-term potentiation in single dendritic spines. Nature 429(6993):761–766
Lee S-JR, Escobedo-Lozoya Y, Szatmari EM, Yasuda R (2009) Activation of CaMKII in single dendritic spines during long-term potentiation. Nature 458(7236):299–304
Bosch M, Castro J, Saneyoshi T, Matsuno H, Sur M, Hayashi Y (2014) Structural and molecular remodeling of dendritic spine substructures during long-term potentiation. Neuron 82(2):444–459
Hayama T, Noguchi J, Watanabe S, Takahashi N, Hayashi-Takagi A, Ellis-Davies GC et al (2013) GABA promotes the competitive selection of dendritic spines by controlling local Ca 2+ signaling. Nat Neurosci 16(10):1409
Zhou Q, Homma KJ, Poo MM (2004) Shrinkage of dendritic spines associated with long-term depression of hippocampal synapses. Neuron 44(5):749–757
Wiegert JS, Oertner TG (2013) Long-term depression triggers the selective elimination of weakly integrated synapses. Proc Natl Acad Sci USA 110(47):E4510–E4519
Edelmann E, Cepeda-Prado E, Franck M, Lichtenecker P, Brigadski T, Leßmann V (2015) Theta burst firing recruits BDNF release and signaling in postsynaptic CA1 neurons in spike-timing-dependent LTP. Neuron 86(4):1041–1054
Harward SC, Hedrick NG, Hall CE, Parra-Bueno P, Milner TA, Pan E et al (2016) Autocrine BDNF–TrkB signalling within a single dendritic spine. Nature 538(7623):99–103
Tanaka J-I, Horiike Y, Matsuzaki M, Miyazaki T, Ellis-Davies GC, Kasai H (2008) Protein synthesis and neurotrophin-dependent structural plasticity of single dendritic spines. Science 319(5870):1683–1687
Panja D, Bramham CR (2014) BDNF mechanisms in late LTP formation: a synthesis and breakdown. Neuropharmacology 76:664–676
Harvey CD, Yasuda R, Zhong H, Svoboda K (2008) The spread of Ras activity triggered by activation of a single dendritic spine. Science 321(5885):136–140
Hedrick NG, Harward SC, Hall CE, Murakoshi H, McNamara JO, Yasuda R (2016) Rho GTPase complementation underlies BDNF-dependent homo-and heterosynaptic plasticity. Nature 538(7623):104–108
Maejima T, Hashimoto K, Yoshida T, Aiba A, Kano M (2001) Presynaptic inhibition caused by retrograde signal from metabotropic glutamate to cannabinoid receptors. Neuron 31(3):463–475
Zhang LI, Tao HW, Holt CE, Harris WA, Poo M-M (1998) A critical window for cooperation and competition among developing retinotectal synapses. Nature 395(6697):37–44
Caillard O, Ben-Ari Y, Gaiarsa JL (1999) Long-term potentiation of GABAergic synaptic transmission in neonatal rat hippocampus. J Physiol 518(1):109–119
Gubellini P, Ben-Ari Y, Gaïarsa JL (2001) Activity-and age-dependent GABAergic synaptic plasticity in the developing rat hippocampus. Eur J Neurosci 14(12):1937–1946
Gaiarsa J-L, Caillard O, Ben-Ari Y (2002) Long-term plasticity at GABAergic and glycinergic synapses: mechanisms and functional significance. Trends Neurosci 25(11):564–570
Sivakumaran S, Mohajerani MH, Cherubini E (2009) At immature mossy-fiber–CA3 synapses, correlated presynaptic and postsynaptic activity persistently enhances GABA release and network excitability via BDNF and cAMP-dependent PKA. J Neurosci 29(8):2637–2647
Yoshimura Y, Inaba M, Yamada K, Kurotani T, Begum T, Reza F et al (2008) Involvement of T-type Ca2+ channels in the potentiation of synaptic and visual responses during the critical period in rat visual cortex. Eur J Neurosci 28(4):730–743
Nugent FS, Kauer JA (2008) LTP of GABAergic synapses in the ventral tegmental area and beyond. J Physiol 586(6):1487–1493
Wardle RA, Poo M-m (2003) Brain-derived neurotrophic factor modulation of GABAergic synapses by postsynaptic regulation of chloride transport. J Neurosci 23(25):8722–8732
Woodin MA, Ganguly K, Poo M-m (2003) Coincident pre-and postsynaptic activity modifies GABAergic synapses by postsynaptic changes in Cl− transporter activity. Neuron 39(5):807–820
Anwyl R (2006) Induction and expression mechanisms of postsynaptic NMDA receptor-independent homosynaptic long-term depression. Prog Neurobiol 78(1):17–37
Fiumelli H, Woodin MA (2007) Role of activity-dependent regulation of neuronal chloride homeostasis in development. Curr Opin Neurobiol 17(1):81–86
Ormond J, Woodin MA (2009) Disinhibition mediates a form of hippocampal long-term potentiation in area CA1. PLoS ONE 4(9):e7224
Kirkwood A, Bear MF (1994) Homosynaptic long-term depression in the visual cortex. J Neurosci 14(5):3404–3412
Kameyama K, Lee H-K, Bear MF, Huganir RL (1998) Involvement of a postsynaptic protein kinase A substrate in the expression of homosynaptic long-term depression. Neuron 21(5):1163–1175
Massey PV, Bhabra G, Cho K, Brown M, Bashir Z (2001) Activation of muscarinic receptors induces protein synthesis-dependent long-lasting depression in the perirhinal cortex. Eur J Neurosci 14(1):145–152
Heynen AJ, Yoon B-J, Liu C-H, Chung HJ, Huganir RL, Bear MF (2003) Molecular mechanism for loss of visual cortical responsiveness following brief monocular deprivation. Nat Neurosci 6(8):854–862
Malenka RC, Bear MF (2004) LTP and LTD: an embarrassment of riches. Neuron 44(1):5–21
Bocklisch C, Pascoli V, Wong JC, House DR, Yvon C, De Roo M et al (2013) Cocaine disinhibits dopamine neurons by potentiation of GABA transmission in the ventral tegmental area. Science 341(6153):1521–1525
Jones S, Kornblum JL, Kauer JA (2000) Amphetamine blocks long-term synaptic depression in the ventral tegmental area. J Neurosci 20(15):5575–5580
Gutlerner JL, Penick EC, Snyder EM, Kauer JA (2002) Novel protein kinase A-dependent long-term depression of excitatory synapses. Neuron 36(5):921–931
Bellone C, Lüscher C (2005) mGluRs induce a long-term depression in the ventral tegmental area that involves a switch of the subunit composition of AMPA receptors. Eur J Neurosci 21(5):1280–1288
Pignatelli M, Bonci A (2015) Role of dopamine neurons in reward and aversion: a synaptic plasticity perspective. Neuron 86(5):1145–1157
Wood J, Garthwaite J (1994) Models of the diffusional spread of nitric oxide: implications for neural nitric oxide signalling and its pharmacological properties. Neuropharmacology 33(11):1235–1244
Castillo PE, Chiu CQ, Carroll RC (2011) Long-term plasticity at inhibitory synapses. Curr Opin Neurobiol 21(2):328–338
Chiu CQ, Puente N, Grandes P, Castillo PE (2010) Dopaminergic modulation of endocannabinoid-mediated plasticity at GABAergic synapses in the prefrontal cortex. J Neurosci 30(21):7236–7248
Pan B, Hillard CJ, Liu Q-s (2008) Endocannabinoid signaling mediates cocaine-induced inhibitory synaptic plasticity in midbrain dopamine neurons. J Neurosci 28(6):1385–1397
Acknowledgements
We would like to thank INSF profusely for their support in conducting this research.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
MKA, NH, and AK were responsible for the study concept and design. MKA and NH drafted the manuscript. All authors critically reviewed the content and approved the final version for publication. All authors have read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
All authors fully agree with the submission and publication of this manuscript in this journal.
Competing interests
The authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Kourosh-Arami, M., Hosseini, N. & Komaki, A. Brain is modulated by neuronal plasticity during postnatal development. J Physiol Sci 71, 34 (2021). https://doi.org/10.1186/s12576-021-00819-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12576-021-00819-9