
Abstract
Background
Among adult malignant tumors, renal cell carcinoma accounts for 2–3% and has the worst prognosis among common urologic tumors. Recently, an eosinophilic, solid and cystic (ESC) renal cell carcinoma (RCC) histological subtype has been described and proposed as an entity separate than the 16 subtypes described in the 2016 WHO classification. The aim of the present study is to share our experience of three such cases of this newly described entity.
Methods
We retrospectively reviewed our cases of renal cell carcinoma and describe the presentation, diagnosis and management with follow-up details of ESC RCC.
Results
Our three patients presented at an advanced stage with flank pain or mass, and one patient had metastasis. All patients underwent radical nephrectomy, diagnosis proved by histopathological examination with immunohistochemistry (IHC), with abundant eosinophilic cytoplasm in all three cases. On IHC, CK 20 was positive in two cases and one patient with CK 20, CK 7 negative and PAX 8 positive.
Conclusions
ESC RCC is a newly described entity, with increasing incidence probably due to better diagnosis. Previously, 66 such cases are described, with female predominance, lower stage and indolent behavior. Only four cases with metastasis are described.
Similar content being viewed by others


1 Background
Renal cell carcinoma (RCC), with the incidence of 400,000 new cases worldwide in 2018, is the ninth most commonly occurring cancer in men and fourteenth most commonly occurring cancer in women, accounting for 2–3% of adult malignant neoplasms [1]. It has the worst prognosis among common urologic malignancies. In the 2016 World Health Organization RCC classification, 16 distinct histological subtypes are described, with clear cell being the most common followed by papillary and chromophobe subtypes [2]. Eosinophilic solid and cystic renal cell carcinoma (ESC RCC) has been recently described, with specific clinical, pathological, immunohistochemical and molecular features [3,4,5]. Previously ESC RCC was grouped under “unclassified renal neoplasm with oncocytic or eosinophilic morphology” or “unclassified renal cell carcinoma” [5]. Majority of the ESC RCC is sporadic and non-syndromic, and fewer cases (< 10%) are reported in patients with tuberous sclerosis complex [5]. Majority of the ESC RCC is small, solitary, low stage tumors, more in females and having indolent behavior [5]. A current prevalence rate of 0.07 to 0.2% among RCC diagnoses is estimated [6]. Metastasis was identified in four cases (< 10%) cases and it was commonly to bone, liver, lungs and bone marrow [5]. Histologically, ESC RCC shows solid and cystic architecture, and cells have eosinophilic cytoplasm and have a focal expression of cytokeratin 20, are cytokeratin 7 negative and have inactivation of TSC genes in the cells [7]. To date, to the best of our knowledge, only 66 cases of ESC RCC have been described in the literature [6, 8, 9]. This report of three cases aims to add to the literature supporting the existence of this relatively new entity. We present and describe a series of three cases of ESC RCC with varied presentations than the ones classically described in the literature till now.
2 Methods
In the last decade from 2011 to 2021, a retrospective analysis of three cases diagnosed with ESC RCC at our center was done. The cases were studied with respect to patient demographics, clinical presentation, perioperative investigations, imaging (contrast-enhanced CT scan of abdomen, pelvis and chest), treatment received, histopathological diagnosis and follow-up.
3 Results
The factors that were studied were patient demographics such as patient age, gender, clinical presentation, imaging findings, tumor stage, treatment received in the form of surgery, chemotherapy, histopathological findings with a focus on immunohistochemistry (IHC) findings and follow-up details. The details are given in Table 1.
Of the three patients who were included in the study, only one was female. The mean age at presentation was 26.7 years (range 7–45 years). All cases had large tumors larger than 4 cm, underwent radical nephrectomy, grossly had hemorrhagic and cystic areas, and on microscopy had abundant eosinophilic cytoplasm.
The first patient was a 28-year-old female with complains of right flank pain, with 11 cm well-defined multiloculated cystic lesion from upper pole of left kidney, stage T2b, and had underwent left laparoscopic radical nephrectomy; the specimen was CK 20 positive with SDHB positive on IHC; and the follow-up contrast-enhanced CT (CECT) scan at 5 months was normal.
The second patient was 45-year-old male with complains of left flank pain, with 13.9 cm lesion in left kidney with IVC thrombus extending to infrahepatic level, enlarged retroperitoneal lymphnodes and liver and lung lesions suggestive of metastases, tumor stage being T3bN1M1, and had underwent left open radical nephrectomy with renal vein thrombectomy with CK 20 positive and SDHB positive on IHC and on pazopanib and follow-up CECT scan showing retroperitoneal lymphadenopathy, liver and bone lesions. He expired at 12 months after surgery.
The third patient was a 7-year-old male patient with complaints of left abdominal lump with CECT scan suggestive of multiple discrete heterogeneous lesions in both kidneys largest in midpole of left kidney of 4.7 cm and in lower pole of right kidney of 3.8 cm, with tumor stage being T2bN0; patient underwent left open radical nephrectomy with IHC suggestive of CK20 negative, CK7 negative and PAX8 positive; on follow-up, the patient was advised partial nephrectomy in contralateral kidney, but parents were not willing for the same and started alternative medicine.
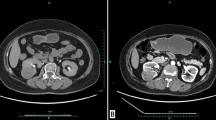
The radiological images and histopathological and immunohistochemistry images of each patient are shown in Figs. 1, 2 and 3.

A, B A large, well-defined, exophytic, heterogeneous, multiloculated, cystic lesion arising from upper pole of left kidney—9.4 * 8 * 11 cm size, showing thick heterogeneously enhancing internal septations and discrete foci of peripheral wall calcifications causing splaying of renal calyces with increased paralesional vascularity. C, D 15 * 9 * 7 cm specimen, 547 gm, left upper pole and mid pole tumor 9.5 * 8 * 8 cm, unifocal with cystic and hemorrhagic areas within E eosinophilic cells with solid and areas. F Section shoes tumor composed of solid and custic areas. G CK 20 positive. H CK 7 negative. I SDH B positive

A 9.6 * 8.9 * 13.9 cm heterogeneously enhancing lesion completely involving left renal parenchyma, perinephric fat stranding and nodularity, focal areas of extracapsular extension, involvement of renal pelvis and ureter, tumoral thrombus extending into infrahepatic IVC, no contrast excretion, enlarged L.N in preaortic and paraaortic, largest 2.1 * 1.4 cm, liver and lung lesions. B, C 21 * 12 * 10 cm gross kidney specimen, weighing 1103 gm. Diffusely infiltrative grayish pink tumors with areas of necrosis and focal calcification. D, E Low power and high power microscopic images showing predominantly papillary type 2 tumor composed of papillary fronds covered with eoisnophilic tumor cells. F CK 20 positive. G CK 7 negative. H CA 9 negative

A, B, C Multiple discrete variably sized heterogeneous hypodense cystic lesions in both kidneys, showing heterogeneous enhancement of solid component within lesion and septa, largest lesion in lower pole of right kidney measures 3.6 * 3.8 cm, largest lesion in mid pole of left kidney measures 4.7 * 3.6 cm. D, E 13.5 * 11 * 7.5 cm left nephrectomy specimen, on cut section, tumor mass shows partially solid and cystic masses filled with hemorrhagic fluid with focal area of hemorrhage and necrosis. F Tumor shows partially cystic areas filled with proteinaceous fluid and lined by tumor cells with eosinphilic cytoplasm. G Section from solid area shows tumor composed of nests and tubules composed of tumor cells with dense eosinophilic cytoplasm. H CK 20 negative. I CK 7 negative. J PAN-CK positive. K PAX 8 positive. L Vimentin positive
4 Discussion
Schreiner and colleagues, in 2010, first described ESC RCC [10]. It is not a new tumor subtype, it is just recognized recently. The present incidence is estimated at around 0.2% [5], but it is expected to increase as previously unclassified cases of RCC are being reviewed retrospectively. The incidence in our case series was 0.48%. The median age at diagnosis was 48.5 years with an age range of 14–85 years [9]. This is in contrast to the age of diagnosis of renal cell carcinoma which is between 60 and 70 years [11]. ESC RCC mainly affects females with 87% reported cases in females [9]. Initially it was described in patients with tuberous sclerosis (TSC); later, it was found that majority (80%) occur in patients who do not have TSC [9]. In our study, mean age of presentation was 26.7 years with only one out of three patients being male and none of the cases had a syndromic association. Only one study has focused on imaging findings of ESC RCC of two cases in detail [12]. ESC RCC tumors are typically solitary, unifocal tumors, small size and low stage [13]. 90% of tumors were stage T1 or T2 as organ-confined disease, as against 66.6% of all RCC [9]. Median size was 4.15 cm with a range of 0.5–15 cm [9]. Four cases of metastases are described [13] and hence the need for long-term surveillance. Of our patients, two patients had organ-confined disease, and one patient presented with metastasis who later died after surgery and mean size of tumor on presentation was 7.7 cm.
Grossly, as per the name of the tumor, there are multiple solid and cystic areas in the tumor specimen. Tumor is well delineated and circumscribed with a capsule. Cysts are macrocysts, multifocal, variably sized. Tumor surface is yellow/gray/tan [13]. Microscopically, cysts have epithelial lining in hobnail arrangement, cyst trabeculae are variable thickness, and scattered foamy histiocytes and lymphocytes are present. Cells are in acinar pattern, show eosinophilic, voluminous cytoplasm, coarse basophilic cytoplasmic stippling which are the aggregates of rough endoplasmic reticulum and have a low mitotic count. Nuclei are round to oval with prominent nucleoli focally, corresponding to WHO/ISUP grade 2 or 3 [13]. Cells may look like leishmania bodies due to the presence of scattered cells with densely eosinophilic globulles surrounded by delicate rims [5]. Our patients showed multiple solid and cystic areas in the gross specimen and microscopically showed cystic areas with proteinaceous fluid lined by tumor cells with eosinophilic cytoplasm and solid areas showed tumor composed of nests and tubules composed of tumor cells with dense eosinophilic cytoplasm.
Immunohistochemistry (IHC) shows CK 20 positive in 85–90% of ESC RCC, either focally or diffuse and this is usually paired with negative CK7 in 75% of cases. None of the cases show CK20 negative and CK 7 positive immunophenotype [13]. Other positive stains include PAX8, Vimentin, CK8/18, AE1/3, Cathepsin K and AMACR, and negative stains include CA9, HMB45, CD117 and melan A [13]. Negative CK20 staining does not rule out ESC RCC, specially if other positive markers are present.
Molecular analysis by next-generation sequencing (NGS) of ESC RCC showed recurrent and mutually exclusive somatic bi-allelic loss or mutation of TSC gene family, including TSC1 and TSC2 in 85% of the reported cases [13]. Molecular karyotyping of ESC has shown common and recurring genomic changes such as copy number (CN) gains at 16p13-16q23, 7p21-7q36, 13q14 and 19p12 and CN losses at Xp11.21 and 22q11, and loss of heterozygosity (LOH) was seen at 16p11.2-11.1, Xq11-13, Xq13-21, 11p11, 9q21-22 and 9q33 [13]. These genes were involved in regulation of mTOR signaling pathway and these genomic changes are distinct from commonly described RCC subtypes. Although these molecular changes were not specific for ESC RCC, taken together with the histopathological and immunohistopathologic features, they suggest a separate entity of RCC. The histopathological and immunohistochemistry features of ESC RCC are sufficient to distinguish ESC RCC from other renal tumors with eosinophilic cytoplasm such as oncocytoma, eosinophilic chromophobe RCC, epitheloid angiomyolipoma, MiTF RCC and SDH-deficient RCC [5].
In our cases, all three patients were having histopathological features of ESC RCC, with two patients having CK20 positivity and one patient with CK20 negative, but other markers were contributory to making the diagnosis of ESC RCC such as CK7 negative, PAN-CK positive, PAX 8 positive, EMA positive, Vimentin positive, RCC positive, C KIT negative, E cadherin negative, HMB 45 negative, Melan A negative and WT 1 negative.
Surgical therapy is curative in majority of the ESC RCC, and in metastatic disease, therapy with mTOR inhibitors may be more effective [14]. In our patients, surgery in the form of radical nephrectomy was done in all patients, with laparoscopic surgery done in one patient; one patient with metastasis was started on tyrosine kinase inhibitor with a plan to switch to mTOR inhibitor if there is disease progression who later died at 12 months post-surgery; one patient had tumor in contralateral kidney and was advised partial nephrectomy for the same, but patient the was not willing for the same and was undergoing an alternative therapy. Most of the cases are reported in the pathological literature, and even then, the available literature is less. So additional studies are required to determine the clinical significance and the biologic nature of this newly diagnosed subtype of ESC RCC which is more important from the urology point of view.
Limitations of the present study were the small patient population, retrospective study, variable presentation and short follow-up. Thus, the detection of significant differences of variables may not have been possible and definitive conclusions may not be possible.
5 Conclusion
ESC RCC is a newly described and distinct subtype of RCC with more cases being reported probably due to better diagnosis. It has a separate clinical presentation and it is also distinct histopathologically. It has a female predominance, lower stage, good prognosis tumor with indolent behavior and has a distinct morphological and immunohistochemistry and genetic profile. Our three patients in contrast had a varied presentation than the one classically described with male predominance, large tumor size, all having symptomatic presentation, one patient presenting with metastases and tumor thrombus to inferior venacava and one patient with bilateral renal tumors.
Availability of data and materials
The datasets generated in the current study are available from the corresponding author on reasonable request.
Abbreviations
- ESC:
-
Eosinophilic solid and cystic
- RCC:
-
Renal cell cancer
- CK:
-
Cytokeratin
- TSC:
-
Tuberous sclerosis
- IHC:
-
Immunohistochemistry
- NGS:
-
Next-generation sequencing
- LOH:
-
Loss of heterozygosity
References
Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A (2018) Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 68(6):394–424
Moch H, Cubilla AL, Humphrey PA, Reuter VE, Ulbright TM (2016) The 2016 WHO classification of tumours of the urinary system and male genital organs—part A: renal, penile, and testicular tumours. Eur Urol 70(1):93–105
Trpkov K, Hes O, Bonert M, Lopez JI, Bonsib SM, Nesi G et al (2016) Eosinophilic, solid, and cystic renal cell carcinoma. Am J Surg Pathol 40(1):60–71
Trpkov K, Abou-Ouf H, Hes O, Lopez JI, Nesi G, Comperat E et al (2017) Eosinophilic solid and cystic renal cell carcinoma (ESC RCC). Am J Surg Pathol 41(10):1299–1308
Trpkov K, Hes O (2019) New and emerging renal entities: a perspective post-WHO 2016 classification. Histopathology 74(1):31–59
Mohaghegh Poor SM, Mathur S, Kassier K, Rossouw J, Wightman R, Saranchuk J et al (2021) Two cases of sporadic eosinophilic solid and cystic renal cell carcinoma in manitoba population. Int J Surg Pathol 29:1066896921993229
Cho WC, Collins K, Mnayer L, Cartun RW, Earle JS (2019) Concurrent eosinophilic solid and cystic renal cell carcinoma and angiomyolipoma with epithelial cysts in the setting of tuberous sclerosis complex: a rare synchronous occurrence of 2 distinct entities. Int J Surg Pathol 27(7):804–811
Sharma R, Thirunavukkarasu B, Elhence P, Rodha MS, Sureka B (2021) Eosinophilic solid and cystic renal cell carcinoma: from unclassified to classified, a case report. Turk J Pathol 1(1):060–065
Yunker A, Holder L, Nething J (2020) Newly described eosinophilic, solid and cystic renal cell carcinoma: a case report and review of the literature. Arch Nephrol Urol 3(2):38–45
Schreiner A, Daneshmand S, Bayne A, Countryman G, Corless CL, Troxell ML (2010) Distinctive morphology of renal cell carcinomas in tuberous sclerosis. Int J Surg Pathol 18(5):409–418
Ljungberg B, Albiges L, Abu-Ghanem Y, Bensalah K, Dabestani S, Fernández-Pello S et al (2019) European association of urology guidelines on renal cell carcinoma: the 2019 update. Eur Urol 75(5):799–810
Fenelon SS, Santos JMMM, Faraj SF, Mattedi RL, Trpkov K, Nahas WC et al (2018) Eosinophilic solid and cystic renal cell carcinoma: imaging features of a novel neoplasm. Urology 114:e9–10
Siadat F, Trpkov K (2020) ESC, ALK, HOT and LOT: three letter acronyms of emerging renal entities knocking on the door of the WHO classification. Cancers 12(1):168
Palsgrove DN, Li Y, Lin M-T, Pallavajjalla A, Gocke C, De Marzo AM et al (2018) Eosinophilic solid and cystic (ESC) renal cell carcinomas harbor TSC mutations: molecular analysis supports an expanding clinicopathologic spectrum. Am J Surg Pathol 42(9):1166
Acknowledgements
Not applicable.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
NP, AS and PS did the material preparation, data collection and analysis. Histopathological information was provided by SS. The first draft of the manuscript was written by NP. AG, RS and MD commented on subsequent versions of the manuscript. All authors contributed to the study conception and design. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This retrospective study involving human participants was in accordance with the ethical standards of the institutional and national research committee and with the 1964 Helsinki Declaration and its later amendments. The Institutional Ethics Committee gave the approval for the study (MPSRNUEC). Number—EC/740/2021. Informed written consent was obtained from all individual participants included in the study, and for children less than 16 years old, informed written consent was obtained from the parents.
Consent for publication
Patients signed the informed written consent regarding publishing their data and photographs, and for children less than 16 years old, informed written consent was obtained from the parents.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Pathak, N.J., Singh, A.G., Jain, P.S. et al. Eosinophilic solid and cystic renal cell carcinoma: a single Indian tertiary center experience of three cases of a newly described entity. Afr J Urol 28, 48 (2022). https://doi.org/10.1186/s12301-022-00312-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12301-022-00312-8