
Abstract
FOXP1 syndrome is a neurodevelopmental disorder caused by mutations or deletions that disrupt the forkhead box protein 1 (FOXP1) gene, which encodes a transcription factor important for the early development of many organ systems, including the brain. Numerous clinical studies have elucidated the role of FOXP1 in neurodevelopment and have characterized a phenotype. FOXP1 syndrome is associated with intellectual disability, language deficits, autism spectrum disorder, hypotonia, and congenital anomalies, including mild dysmorphic features, and brain, cardiac, and urogenital abnormalities. Here, we present a review of human studies summarizing the clinical features of individuals with FOXP1 syndrome and enlist a multidisciplinary group of clinicians (pediatrics, genetics, psychiatry, neurology, cardiology, endocrinology, nephrology, and psychology) to provide recommendations for the assessment of FOXP1 syndrome.
Similar content being viewed by others

Introduction
FOXP1 (forkhead box P1) (OMIM#605515) is a member of the forkhead box family of transcriptional factors that play important roles in the regulation of gene transcription from early development through adulthood [1]. The FOXP1 gene, located on chromosome 3p14.1, codes for a transcriptional repressor protein. The FOXP1 protein is widely expressed in human tissues and is involved in regulating the development of the brain, heart, lung, esophagus, immune system, and spinal motor neurons [1,2,3,4,5,6,7]. It has also been suggested that the FOXP1 protein plays a critical role in striatal regulation and vocal communication, and when disrupted, may contribute to expressive language deficits [8].
In 2009, Pariani and colleagues reported a child with intellectual disability (ID) and a multigenic deletion including the FOXP1 gene, and hypothesized that FOXP1 contributed to ID [9]. In 2010, Hamdan and colleagues described two cases with ID and features of autism spectrum disorder (ASD) in whom a deletion involving FOXP1 and a sequence variant in FOXP1 were found [10]. Since then, many more cases have been reported and FOXP1 syndrome is now a recognizable entity [11]. FOXP1 syndrome (FOXP1S) is associated with “intellectual disability with language impairment and with or without autistic features” (OMIM #613670) and is caused by FOXP1 gene deletions and mutations (nonsense, missense, and in-frame deletions) [11].
There have been at least 30 case reports, case series, and cohort studies published in the literature that taken together describe more than 100 individuals with FOXP1S. The majority of information has been collected retrospectively from medical genetic evaluations. The objective of this review is to describe the clinical phenotype of individuals with FOXP1S and establish recommendations to assess the medical, developmental, and behavioral features.
Methods
We conducted a comprehensive review of the literature published between March 2009, when Pariani and colleagues hypothesized the association of FOXP1 and ID, and July 2020. We searched for articles on FOXP1S on PubMed and Scopus. Keywords used in PubMed included FOXP1, 3p14.1 [All Fields] and FOXP1 or 3p14.1 and Autism [All Fields], while keywords used in Scopus included FOXP1, 3p14.1 [TITLE-ABS-KEY] and FOXP1 or 3p14.1 and Autism [TITLE-ABS-KEY].
Results
There were at least ten studies that screened individuals with autism spectrum disorder (ASD) and intellectual disability (ID) for FOXP1 deletions and mutations. In most of these studies, the clinical features of the patients were not described in detail other than the presence of ASD, learning disability, and/or neurodevelopmental disorder broadly. Other studies screened for FOXP1 variants in patients with other medical problems such as cardiac and renal malformations [12, 13]. For the purpose of this review, we focused on clinical case reports and case series where insights into the phenotype could be feasibly ascertained.
We identified a total of 62 independent individuals described in 21 case reports and case series, [9,10,11, 14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31]. Overlapping cases were excluded [32, 33]. Furthermore, one case of a child who had a known FOXP1 variant and a PTCH1 pathogenic mutation was also excluded [34]. The data were extracted and sent to multiple clinical specialists for interpretation and to provide multidisciplinary recommendations.
The 62 independent individuals described were predominantly male (male 41: female 21) and ranged in age from 4 months to 31 years old (mean = 11.4 years, SD = 7.1). For most features, 62 cases had neurobehavioral information (Table 1), 59 cases had dysmorphology descriptions (Table 2), and 60 cases had medical histories (Table 3). Unless otherwise specified in the original studies, we used these denominators and assume that if a feature was not recorded, it was absent. We also use these denominators to avoid inflation of prevalence which can affect recommendations for assessment and monitoring.
Clinical genetics
In this review, 18 cases had a deletion of the FOXP1 gene identified by chromosomal microarray analysis (CMA) and 44 had a sequence variant identified by next generation sequencing (Table 1). CMA is used as a first-tier test for individuals with ID, ASD, or developmental delay [35] and identifies individuals with FOXP1S due to 3p14 deletions which include the FOXP1 gene. The deletions at 3p14 occurred de novo in the vast majority of patients, although in one case, an affected parent with a complex inversion with a breakpoint disrupting FOXP1 transmitted the rearrangement to an affected child [36]. In another case, a healthy parent carrying a balanced intra-arm (intrachromosomal) insertion transmitted an unbalance rearrangement to an affected child [19].
Fifty-nine cases described in the literature included dysmorphology evaluations. The most common dysmorphic features reported among any cases where exams were performed were prominent forehead (48/59; 81%), short nose with a broad tip or base (41/59; 69%), down slanting palpebral fissures (24/59; 41%), ptosis (22/59; 37%), thick vermillion (18/59; 31%), ocular hypertelorism (17/59; 29%), and frontal hair upsweep (16/59; 27%). Other less common dysmorphic features were single palmar crease (14/56; 25%), clinodactyly (13/56; 23%), pointed chin (12/57; 21%), high-arched palate (10/59; 17%), malformed ears (10/59; 17%), macrocephaly (9/59; 15%), and broad nasal bridge (8/59; 14%) (Table 2).
Recommendations
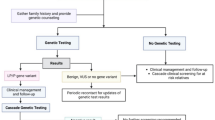
Since CMA does not detect balanced structural chromosome rearrangements, chromosome analysis (karyotype) and/or metaphase fluorescence in situ hybridization (FISH) should be performed in the biological parents to rule out an insertion or inversion which may help to determine recurrence risk. A balanced insertion or inversion involving chromosome 3p14 in a parent significantly increases the risk of recurrence in families, and siblings of the proband should also be tested when relevant. Parental testing is also performed to confirm de novo status. The recurrence risk for future pregnancies is low for apparent de novo aberrations, but it is marginally greater than for the general population (1%) because parents may have germline mosaicism (found specifically within the gamete cells) [35]. Genetic testing for individuals with ID, ASD, or developmental delay, in addition to CMA, involves next-generation sequencing [35], which should be used to test for FOXP1 sequence variants. Many clinical laboratories offer clinical sequencing (whole exome sequencing and whole genome sequencing) and autism focused sequencing panels, which include the FOXP1 gene.
Clinical genetics evaluations and dysmorphology exams should be performed to assess growth, head circumference, craniofacial features, pubertal development, and screen for organ malformations (such as congenital heart defects and urogenital abnormalities) and to determine appropriate referrals to other subspecialties. Most patients with FOXP1S have multiple dysmorphic features, although none are specific (Fig. 1).

Facial characteristic of two individuals with FOXP1 sequence variants, ages 9 and 15, including prominent forehead, wide nasal bridge with a broad tip, down slanting palpebral fissures, mild ptosis, thick vermillion, and wide spacing between teeth
Psychology and psychiatry
Mild to moderate ID or global developmental delay was present in 90% (55/61) of cases evaluated and the Full-Scale Intelligence Quotient (FSIQ) ranged from 20 to 93 (mean = 50.04, SD = 9.5). Gross and fine motor delays were present in virtually all cases (59/61; 97%). The mean age of walking unaided for the first time was 24.4 months (range, 16 to 48 months). Speech and language delays were reported in all the cases (60/60; 100%); thus, language impairment may represent a core feature of FOXP1S. Early in life, infants with FOXP1S have hypotonia (18/62; 29%) and feeding issues (13/62; 21%). Early sucking and feeding difficulties may be related to hypotonia. Orofacial hypotonia can also interfere with speech production and cause dysarthria and articulation difficulties later in life. The mean age of first word spoken was 33 months (range, 17 to 42 months). Two thirds of individuals described in the literature had articulation problems (32/48; 67%) (Table 1). Some reports note that expressive language is more impaired than receptive language [12, 37]; however, these findings were largely based on caregiver report rather than standardized assessments. One study prospectively applied norm-referenced assessments (Expressive Vocabulary Test, 2nd Edition; Peabody Picture Vocabulary Tests, 4th Edition for seven and eight children respectively) and the results indicated stronger expressive language skills in comparison to receptive language skills [24].
About 57% (32/56) had documented psychiatric comorbidities, including attention deficit/hyperactivity disorder (ADHD), aggression, obsessive-compulsive traits, mood disorders, and anxiety. ASD symptoms were reported in 50% (28/56) (e.g., social skills deficits, sensory symptoms, and repetitive behaviors/interests). Five of 10 cases assessed using gold-standard diagnostic testing, including the Diagnostic and Statistical Manual for Mental Disorders, Fifth Edition (DSM-5), the Autism Diagnostic Observation Schedule 2 (ADOS-2), and the Autism Diagnostic Interview-Revised (ADI-R) also met criteria for ASD.
Recommendations
All patients with FOXP1S should be referred for comprehensive neuropsychological evaluations by licensed clinical psychologists with expertise in the assessment of patients with neurodevelopmental disorders. Evaluations should include measures of cognitive functioning, adaptive behavior, ASD symptomatology, expressive and receptive language, visual-motor integration, academic achievement, executive functioning, and global behavioral functioning (e.g., internalizing and externalizing symptoms). Testing batteries should be tailored to an individual’s age and level of functioning. Evaluations should include a combination of clinician-administered assessments and caregiver report questionnaires. Full re-evaluations are recommended at a minimum of every 3 years to assess progress and update treatment recommendations. Therapists and teachers should measure progress regularly and individuals failing to make expected gains should be evaluated more frequently.
Speech and language therapy, occupational therapy, and behavior therapy are often critical components to treatment plans. We recommend comprehensive evaluations by therapists with experience treating individuals with neurodevelopmental disorders to assess need and frequency of services required. Augmentative and alternative communication evaluations should be provided to all individuals who are nonverbal or minimally verbal. Applied behavior analysis (ABA) is recommended to improve communication, social skills, attention, and learning. ABA should also target activities of daily living and challenging behavior. Families should work with behavior analysts to develop individualized treatment plans. School-age children with FOXP1S often require an individualized education program to ensure an appropriate educational setting, adequate services and appropriate accommodations. In the case of psychiatric comorbidities such as ADHD, anxiety, or irritability/aggression, patients should be referred to a child and adolescent psychiatrist, developmental pediatrician, pediatric neurologist, or geneticist with experience in medication management for individuals with neurodevelopmental disorders.
Neurology
Hypertonia/muscle spasms (20/58; 34%) and contractures (16/56; 29%) were present in about a third of cases. Contractures of upper extremities can significantly impair activities of daily living and require assessment and treatment spinal defects were also reported (2/60; 3%) (Table 3).
Recommendations
A neurology and/or developmental pediatrics referral to evaluate muscle tone, joint mobility, fine and gross motor skills, gait, cranial nerves, and deep tendon reflexes, as well as speech, language, and behavior. These consultations should be followed, as indicated, by appropriate referrals to pediatric physiatry and orthopedics. The possibility of various interventions should be explored including physical and occupational therapy, feeding therapy, orthotics, and bracing. It is important to distinguish between speech delays caused by oromotor hypotonia and language abnormality related to cognitive delays—as both can be present in FOXP1S. Therapies should be initiated as early as possible, and we recommend these interventions begin prior to the onset of missed milestones in cases detected early by genetic methods. Sufficient frequency of interventions is critical to ensure individuals reach their full potential. All children with FOXP1S are candidates for early intervention programs.
Seizure assessment
Seizures were reported in some cases (7/59; 12%). Details about the seizures were provided in three cases and included staring episodes [15], febrile [23], and tonic-clonic seizures [28]; other reports did not include a description [11, 20]. Among the seven patients with seizures, only four had abnormal findings on electroencephalogram (EEG).
Recommendations
If there are clinical concerns about seizures, EEG is recommended. Overnight video-EEG is preferred using the standard 10–20 system with 64 inputs and online automated spike and seizure detection programs. Sedation for the EEG is not generally recommended but can be considered if an EEG cannot be otherwise performed.
Anticonvulsants used for the treatment of seizures in FOXP1S are the same as those used for the treatment of seizure disorders in general. There is insufficient information about the severity of seizures in FOXP1S or about whether certain anticonvulsant medications are more effective than others. EEG can be beneficial in defining the type of seizures and determining treatment. We recommend adopting a low threshold for repeating an EEG with new onset seizures, change in seizure pattern, behavioral changes, or if new neurological signs emerge.
Brain imaging
Brain abnormalities were evident in about a half of total cases (29/58) although imaging was only explicitly reported in 45 cases. Abnormalities included dilated lateral ventricles, white matter abnormalities, arachnoid cysts, large cisterna magna, corpus callosum defects, moderate frontal atrophy, cerebellar defects, and Chiari I malformation (Table 3).
Recommendations
New assessments of individuals affected with FOXP1S should include brain imaging to rule out structural brain defects. Brain imaging may need to be repeated if indicated, such as for new onset or focal neurological symptoms, new seizures, or excessive head growth. Sedation is very frequently needed in children in order to acquire meaningful imaging studies, and the risks of the procedure must be balanced with the potential benefit.
Endocrinology
Short stature (8/60; 13%) and obesity (3/60; 5%) were described in some patients with FOXP1S. Hypothyroidism (2/60; 3%) and type 2 diabetes (1/60; 2%) were rarely described (Table 3).
Recommendations
Endocrine abnormalities should be assessed following the same guidelines for the general population. Emphasis on proper nutrition is essential, especially in the setting of patients with restricted diets or feeding issues. Pubertal development must be clinically evaluated as per the general population screening. Pituitary hormone deficiencies may also be considered because of other brain anomalies such as corpus callosum defects and ventriculomegaly in patients with FOXP1S [38]
Cardiology
Our review of the literature showed that almost a third of cases (17/56; 30%) had heart abnormalities, including patent ductus arteriosus, patent foramen ovale, pulmonary stenosis, and/or atrial septal defect (Table 3). Chang and colleagues described a patient with atrioventricular septal defect and hypoplastic left ventricle who had a deletion of FOXP1; subsequently in 82 patients with atrioventricular septal defect or hypoplastic left heart syndrome, two patients with FOXP1 mutations were identified (one had hypoplastic left ventricle with mitral valve and aortic valve atresia, and another patient had atrioventricular septal defect, pulmonary atresia and single ventricle in the setting of heterotaxy syndrome) [12].
Recommendations
Cardiac defects in patients with FOXP1S may or may not require medical and surgical intervention. A standard cardiac evaluation, including echocardiography and electrocardiography in all patients.
Nephrology
In our review, genitourinary malformations were reported in some cases, including horseshoe kidney (3/46; 7%), cryptorchidism (9/41 males; 22%), and micropenis (3/41 males; 7%).
This is consistent with findings from Bekheirnia and colleagues which described eight unrelated individuals with de novo mutations in FOXP1; four had genitourinary abnormalities, including unilateral renal agenesis, hydronephrosis, hypospadias, and a duplicated renal collecting system [13].
Recommendations
Genitourinary tract anomalies may not require universal sonography, but it is indicated for any symptomatic patient (e.g., urinary tract infection; voiding dysfunction) or for those with an externally noted abnormality such as hypospadias.
Ophthalmology and hearing
Visual refractory issues such as hypermetropia, amblyopia, myopia (29/58; 50%), and strabismus (11/60; 18%) were reported in a significant number of cases. Other severe eye abnormalities (3/60; 5%) included retinitis pigmentosa, cerebral visual impairment, aniridia, microphthalmia, and coloboma. Hearing loss was also reported (8/48; 17%). Recurrent ear infections were reported in some cases (7/60; 12%).
Recommendations
Hearing and vision evaluations for all individuals with FOXP1S and corrective measures should be implemented promptly.
Other medical problems
Other medical problems reported were recurrent upper respiratory infections (7/60; 12%), constipation (5/60; 8%), sleep difficulties (3/60; 5%), hip dysplasia (2/60; 3%), recurrent skin infections (2/60; 3%), and iron deficiency anemia (2/60; 3%). Isolated reports of medical problems included gut atresia (1/60), absent gallbladder (1/60), vocal cord paralysis (1/60), craniosynostosis (1/60), recurrent fever episodes (1/60), asthma (1/60), and neuroendocrine hyperplasia of infancy (1/60) (Table 3).
Recommendations
Based on our clinical experience with FOXP1S, some of the described medical problems may be more frequent than noted in the literature, particularly constipation, sleep difficulties, and frequent infections. As such, ongoing monitoring in primary care and appropriate referrals to gastroenterology and sleep specialists, for example, are necessary. Dental problems are also important to assess and are associated with high-arched palate; routine dental care is also recommended.
Discussion
This literature review revealed a clinically heterogeneous phenotype of FOXP1S. Clinical features consistently reported include global developmental delay/intellectual disability, speech delay and articulation problems, ASD, and mild dysmorphic features. Cardiac, brain, and renal malformations, as well as other medical problems, were reported in some individuals affected by FOXP1S. Severe cardiac defects were uncommon, although studies of individuals with FOXP1S with cardiac malformations are currently underway. Affected individuals are more likely to have hearing deficits, strabismus, visual refractive errors, and recurrent infections. Frequent upper respiratory infections during childhood [24] may be related to the role of FOXP1 in transcriptional regulation of B cell development and T cell suppression [3, 39]. Studies are also currently underway to examine the presence, frequency, and type of infections in children with FOXP1S.
Results from this review are limited by the small number of cases, lack of detailed clinical information, and the variability of assessments used. It is possible that some of the medical features are incidental findings, particularly those observed at a lower rate; further studies are necessary to clarify the phenotype and associated medical problems in FOXP1S.
Robust genotype-phenotype associations have not been reported in FOXP1S. Among cases with the same recurring variant (c.1573C>T, p. Arg525*) different phenotypes have been described. In addition, individuals with FOXP1 deletions, truncating variants, and missense variants did not have significant differences in the severity of developmental delay [11]. However, individuals with large 3p deletions may be at risk of a more severe clinical presentation as compared to those in whom only the FOXP1 gene is disrupted [11].
As genetic testing becomes more accessible, the population of individuals with FOXP1S will continue to increase. CMA is a first-tier testing method for individuals with ID and ASD, and sequencing of FOXP1 is now included in many clinically available neurodevelopmental testing panels. In addition, improved access to whole exome and whole genome sequencing will identify additional cases.
Conclusions
To date, information about the clinical presentation of individuals with FOXP1S relies mainly on individual case reports and small case series but reveals a heterogeneous phenotype with global developmental delay, intellectual disability, speech impairment, and associated medical features. Advancing knowledge about the clinical features of FOXP1S and providing medical recommendations for assessment is critical for this population. Table 4 includes a summary of the recommended practice parameters for medical assessment. Comprehensive assessment and longitudinal follow up of individuals with FOXP1S are necessary to better understand the clinical phenotype and natural history of FOXP1S and to clarify the efficacy of treatments for affected individuals.
Availability of data and materials
This is a literature review. All data generated and analyzed during this study are included in this published article.
Abbreviations
- ADOS-2:
-
Autism Diagnostic Observation Schedule 2
- ADI-R:
-
Autism Diagnostic Interview-Revised
- ABA:
-
Applied behavior analysis
- ASD:
-
Autism spectrum disorder
- ADHD:
-
Attention deficit/hyperactivity disorder
- BMI:
-
Body mass index
- CMA:
-
Chromosomal microarray analysis
- DSM-5:
-
Diagnostic and Statistical Manual of Mental Disorders, 5th edition
- EEG:
-
Electroencephalography
- FOXP1S:
-
FOXP1 syndrome
- ID:
-
Intellectual disability
- MRI:
-
Magnetic resonance imaging
References
Shu W, Yang H, Zhang L, Lu MM, Morrisey EE. Characterization of a new subfamily of winged-helix/forkhead (Fox) genes that are expressed in the lung and act as transcriptional repressors. J Biol Chem. 2001;276(29):27488–97.
Dasen JS, De Camilli A, Wang B, Tucker PW, Jessell TM. Hox repertoires for motor neuron diversity and connectivity gated by a single accessory factor, FoxP1. Cell. 2008;134(2):304–16.
Hu H, Wang B, Borde M, Nardone J, Maika S, Allred L, et al. Foxp1 is an essential transcriptional regulator of B cell development. Nat Immunol. 2006;7(8):819–26.
Jepsen K, Gleiberman AS, Shi C, Simon DI, Rosenfeld MG. Cooperative regulation in development by SMRT and FOXP1. Genes Dev. 2008;22(6):740–5.
Palmesino E, Rousso DL, Kao TJ, Klar A, Laufer E, Uemura O, et al. Foxp1 and lhx1 coordinate motor neuron migration with axon trajectory choice by gating Reelin signalling. PLoS Biol. 2010;8(8):e1000446.
Shi C, Zhang X, Chen Z, Sulaiman K, Feinberg MW, Ballantyne CM, et al. Integrin engagement regulates monocyte differentiation through the forkhead transcription factor Foxp1. J Clin Invest. 2004;114(3):408–18.
Wang B, Weidenfeld J, Lu MM, Maika S, Kuziel WA, Morrisey EE, et al. Foxp1 regulates cardiac outflow tract, endocardial cushion morphogenesis and myocyte proliferation and maturation. Development. 2004;131(18):4477–87.
Araujo DJ, Anderson AG, Berto S, Runnels W, Harper M, Ammanuel S, et al. FoxP1 orchestration of ASD-relevant signaling pathways in the striatum. Genes Dev. 2015;29(20):2081–96.
Pariani MJ, Spencer A, Graham JM Jr, Rimoin DL. A 785kb deletion of 3p14.1p13, including the FOXP1 gene, associated with speech delay, contractures, hypertonia and blepharophimosis. Eur J Med Genet. 2009;52(2-3):123–7.
Hamdan FF, Daoud H, Rochefort D, Piton A, Gauthier J, Langlois M, et al. De novo mutations in FOXP1 in cases with intellectual disability, autism, and language impairment. Am J Hum Genet. 2010;87(5):671–8.
Meerschaut I, Rochefort D, Revencu N, Petre J, Corsello C, Rouleau GA, et al. FOXP1-related intellectual disability syndrome: a recognisable entity. J Med Genet. 2017;54(9):613–23.
Chang SW, Mislankar M, Misra C, Huang N, Dajusta DG, Harrison SM, et al. Genetic abnormalities in FOXP1 are associated with congenital heart defects. Hum Mutat. 2013;34(9):1226–30.
Bekheirnia MR, Bekheirnia N, Bainbridge MN, Gu S, Coban Akdemir ZH, Gambin T, et al. Whole-exome sequencing in the molecular diagnosis of individuals with congenital anomalies of the kidney and urinary tract and identification of a new causative gene. Genet Med. 2017;19(4):412–20.
Horn D, Kapeller J, Rivera-Brugues N, Moog U, Lorenz-Depiereux B, Eck S, et al. Identification of FOXP1 deletions in three unrelated patients with mental retardation and significant speech and language deficits. Hum Mutat. 2010;31(11):E1851–60.
Carr CW, Moreno-De-Luca D, Parker C, Zimmerman HH, Ledbetter N, Martin CL, et al. Chiari I malformation, delayed gross motor skills, severe speech delay, and epileptiform discharges in a child with FOXP1 haploinsufficiency. Eur J Hum Genet. 2010;18(11):1216–20.
Ţuţulan-Cunită AC, Papuc SM, Arghir A, Rötzer KM, Deshpande C, Lungeanu A, et al. 3p interstitial deletion: novel case report and review. J Child Neurol. 2012;27(8):1062–6.
Palumbo O, D'Agruma L, Minenna AF, Palumbo P, Stallone R, Palladino T, et al. 3p14.1 de novo microdeletion involving the FOXP1 gene in an adult patient with autism, severe speech delay and deficit of motor coordination. Gene. 2013;516(1):107–13.
Le Fevre AK, Taylor S, Malek NH, Horn D, Carr CW, Abdul-Rahman OA, et al. FOXP1 mutations cause intellectual disability and a recognizable phenotype. Am J Med Genet A. 2013;161A(12):3166–75.
Lloveras E, Vendrell T, Fernández A, Castells N, Cueto A, del Campo M, et al. Intrachromosomal 3p insertion as a cause of reciprocal pure interstitial deletion and duplication in two siblings: further delineation of the emerging proximal 3p deletion syndrome. Cytogenet Genome Res. 2014;144(4):290–3.
Dimitrov BI, Ogilvie C, Wieczorek D, Wakeling E, Sikkema-Raddatz B, van Ravenswaaij-Arts CM, et al. 3p14 deletion is a rare contiguous gene syndrome: report of 2 new patients and an overview of 14 patients. Am J Med Genet A. 2015;167(6):1223–30.
Song H, Makino Y, Noguchi E, Arinami T. A case report of de novo missense FOXP1 mutation in a non-Caucasian patient with global developmental delay and severe speech impairment. Clin Case Rep. 2015;3(2):110–3.
Sollis E, Graham SA, Vino A, Froehlich H, Vreeburg M, Dimitropoulou D, et al. Identification and functional characterization of de novo FOXP1 variants provides novel insights into the etiology of neurodevelopmental disorder. Hum Mol Genet. 2016;25(3):546–57.
Sollis E, Deriziotis P, Saitsu H, Miyake N, Matsumoto N, Hoffer MJV, et al. Equivalent missense variant in the FOXP2 and FOXP1 transcription factors causes distinct neurodevelopmental disorders. Hum Mutat. 2017;38(11):1542–54.
Siper PM, De Rubeis S, Trelles MDP, Durkin A, Di Marino D, Muratet F, et al. Prospective investigation of FOXP1 syndrome. Mol Autism. 2017;8:57.
Myers A, du Souich C, Yang CL, Borovik L, Mwenifumbo J, Rupps R, et al. FOXP1 haploinsufficiency: phenotypes beyond behavior and intellectual disability? Am J Med Genet A. 2017;173(12):3172–81.
Johnson TB, Mechels K, Anderson RH, Cain JT, Sturdevant DA, Braddock S, et al. Characterization of a recurrent missense mutation in the forkhead DNA-binding domain of FOXP1. Sci Rep. 2018;8(1):16161.
Yamamoto-Shimojima K, Okamoto N, Matsumura W, Okazaki T, Yamamoto T. Three Japanese patients with 3p13 microdeletions involving FOXP1. Brain and Development. 2019;41(3):257–62.
Vuillaume ML, Cogné B, Jeanne M, Boland A, Ung DC, Quinquis D, et al. Whole genome sequencing identifies a de novo 2.1 Mb balanced paracentric inversion disrupting FOXP1 and leading to severe intellectual disability. Clin Chim Acta. 2018;485:218–23.
Mutlu-Albayrak H, Karaer K. Vocal cord immobility as a cause of aphonia in a child with 3p13p12 deletion syndrome encompassing FOXP1 gene. Int J Pediatr Otorhinolaryngol. 2019;117:179–81.
Urreizti R, Damanti S, Esteve C, Franco-Valls H, Castilla-Vallmanya L, Tonda R, et al. A de novo FOXP1 truncating mutation in a patient originally diagnosed as C syndrome. Sci Rep. 2018;8(1):694.
Blanco Sánchez T, Duat Rodríguez A, Cantarín Extremera V, Lapunzina P, Palomares Bralo M, Nevado Blanco J. Clinical phenotype of a patient with FOXP1 deletion. An Pediatr (Barc). 2015;82(4):280–1.
O'Roak BJ, Deriziotis P, Lee C, Vives L, Schwartz JJ, Girirajan S, et al. Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat Genet. 2011;43(6):585–9.
Lozano R, Vino A, Lozano C, Fisher SE, Deriziotis P. A de novo FOXP1 variant in a patient with autism, intellectual disability and severe speech and language impairment. Eur J Hum Genet. 2015;23(12):1702–7.
Zombor M, Kalmár T, Maróti Z, Zimmermann A, Máté A, Bereczki C, et al. Co-occurrence of mutations in FOXP1 and PTCH1 in a girl with extreme megalencephaly, callosal dysgenesis and profound intellectual disability. J Hum Genet. 2018;63(11):1189–93.
Schaefer GB, Mendelsohn NJ. Clinical genetics evaluation in identifying the etiology of autism spectrum disorders: 2013 guideline revisions. Genet Med. 2013;15(5):399–407.
Schluth-Bolard C, Diguet F, Chatron N, Rollat-Farnier PA, Bardel C, Afenjar A, et al. Whole genome paired-end sequencing elucidates functional and phenotypic consequences of balanced chromosomal rearrangement in patients with developmental disorders. J Med Genet. 2019;56(8):526–35.
Stenson PD, Mort M, Ball EV, Shaw K, Phillips A, Cooper DN. The Human Gene Mutation Database: building a comprehensive mutation repository for clinical and molecular genetics, diagnostic testing and personalized genomic medicine. Hum Genet. 2014;133(1):1–9.
D.Carmichael J. Anterior pituitary failure. In: The Pituitary. 4th ed; 2017. p. 245–7.
Ren J, Han L, Tang J, Liu Y, Deng X, Liu Q, et al. Foxp1 is critical for the maintenance of regulatory T-cell homeostasis and suppressive function. PLoS Biol. 2019;17(5):e3000270.
Acknowledgements
The authors would like to thank Dr. John M Graham for this thoughtful comments and edits to this paper.
Funding
This work was supported by the Beatrice and Samuel A. Seaver Foundation (AK, JB, PS, RL), the Friedman Brain Institute (AK, RL), the NIH (GM082773 to RL), Harold Amos Faculty Development Award/ Robert Wood Johnson Foundation (RL), and the FOXP1 RareConnect group.
Author information
Authors and Affiliations
Contributions
RL and AK: design, data collection, analysis, writing, and edits. CG and LT: data collection, analysis, and edits. PS, SS, JS, ED, YF, and JB: analysis and edits. The authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
The manuscript includes two pictures of patients; we used our institutional consent forms which are available upon request.
Competing interests
AK receives research support from AMO Pharma and consults to Ovid Therapeutics, Acadia, Ritrova, and Alkermes. The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Lozano, R., Gbekie, C., Siper, P.M. et al. FOXP1 syndrome: a review of the literature and practice parameters for medical assessment and monitoring. J Neurodevelop Disord 13, 18 (2021). https://doi.org/10.1186/s11689-021-09358-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s11689-021-09358-1