
Abstract
Background
Migraine is a disorder associated with neuropeptide release, pain and inflammation. Tau protein has recently been linked to inflammatory diseases and can be influenced by neuropeptides such as CGRP, a key neurotransmitter in migraine. Here, we report serum concentrations of total-tau protein in migraine patients and healthy controls.
Methods
In this cross-sectional study, interictal blood samples from n = 92 patients with episodic migraine (EM), n = 93 patients with chronic migraine (CM), and n = 42 healthy matched controls (HC) were studied. We assessed serum total-tau protein (t-tau) and for comparison neurofilament light chain protein (NfL), glial fibrillary acidic protein (GFAP), and ubiquitin carboxy-terminal hydrolase L (UCH-L1) concentrations using the Neurology 4-plex kit, on a single molecule array HD-X Analyzer (Quanterix Corp Lexington, MA). Matched serum/cerebrospinal fluid (CSF) samples were used for post-hoc evaluations of a central nervous system (CNS) source of relevant findings. We applied non-parametric tests to compare groups and assess correlations.
Results
Serum t-tau concentrations were elevated in EM [0.320 (0.204 to 0.466) pg/mL] and CM [0.304 (0.158 to 0.406) pg/mL] patients compared to HC [0.200 (0.114 to 0.288) pg/mL] (p = 0.002 vs. EM; p = 0.025 vs. CM). EM with aura [0.291 (0.184 to 0.486 pg/mL); p = 0.013] and EM without aura [0.332 (0.234 to 0.449) pg/mL; p = 0.008] patients had higher t-tau levels than HC but did not differ between each other. Subgroup analysis of CM with/without preventive treatment revealed elevated t-tau levels compared to HC only in the non-prevention group [0.322 (0.181 to 0.463) pg/mL; p = 0.009]. T-tau was elevated in serum (p = 0.028) but not in cerebrospinal fluid (p = 0.760). In contrast to t-tau, all proteins associated with cell damage (NfL, GFAP, and UCH-L1), did not differ between groups.
Discussion
Migraine is associated with t-tau elevation in serum but not in the CSF. Our clinical study identifies t-tau as a new target for migraine research.
Similar content being viewed by others

Introduction
Migraine is a highly prevalent primary headache disorder with a multitude of debilitating symptoms [1]. The pathophysiology of migraine is complex and multifaceted but it is known that one of the key anatomical structures, which is involved in pain generation, is the trigeminal nervous system [2]. Trigeminally mediated inflammation has been identified as a component in the pathophysiology of migraine [3,4,5]. Anti-inflammatory drugs such as cyclooxygenase (COX) inhibitors abort migraine attacks successfully and serotonin 5-HT1B/D/F receptor agonists (triptans) block sterile neuro-inflammation in experimental animal models [6,7,8,9,10]. Nevertheless, a deeper understanding of the underlying molecular and pathophysiological mechanisms leading to migraine attacks is crucial for the development of more effective therapeutic strategies. This paper aims to explore the potential role of tau protein, a microtubule-associated protein crucial for neuronal stability and function i.e. axonal transport in migraine pathophysiology.
Neuropeptides play a crucial role in pain and inflammation. The dysregulation of certain neuropeptides such as neuropeptide Y in combination with abnormal tau proteins has been reported [11], but the direct link between tau and these neuropeptide abnormalities remains to be elucidated. Studies show that misfolded or phosphorylated tau proteins have the capacity to induce neuroinflammation and initiate the breakdown of the bloodbrain barrier [12,13,14,15,16]. For example, substance P reduced tau phosphorylation in a rat animal model of concussion, which could be reverted by a NK1 receptor antagonist [17]. Microtubule alterations could therefore potentially contribute to neurological disorders linked with inflammation including migraine or cluster headache. In line, a recent study showed that tau protein is increased in inflammatory neuropathies such as Guillain–Barre syndrome [18, 19].
Another central neuropeptide in the pathogenesis of migraine is Calcitonin Gene-Related Peptide (CGRP) [20]. It is a potent vasodilator and pain signal transmitter. In migraine, microtubule instability could alter axonal function and affect the release of CGRP from peripheral trigeminal neurons [21, 22]. The possibility of a role of tau-related microtubule stability in CGRPmediated disorders represents an interesting target for investigation. In fact, an experimental animal study showed that CGRP receptor antagonists have effects on neuroinflammation in tau-mediated disorders [23]. Additionally, CGRP was found to inhibit tau hyperphosphorylation and thereby reduced total tau levels in a focal cerebral ischemia/reperfusion model [24]. Along another line of evidence, CGRP is elevated during Cortical Spreading Depression (CSD) a wave of neuronal and glial depolarization that is thought to be the underlying pathophysiological correlate of migraine aura [25, 26]. The role of tau in neuroinflammation has been described in tauopathies, for example, misfolded or phosphorylated tau might have different capacities to induce neuroinflammation [12,13,14].
Based on the role of tau in maintaining neuronal structure and potentially influencing neuronal function, it is possible that tau dysregulation could affect CSD and consequently influence CGRP release, thus contributing to migraine pathophysiology.
Pituitary adenylate cyclase-activating polypeptide (PACAP) is also implicated in migraine pathophysiology as initially proposed by Schytz et al. [27], and recently confirmed in a phase II clinical trial (NCT05133323). In this study for the prevention of migraine the antibody Lu AG09222, which blocks PACAP, effectively reduced monthly migraine days superior to placebo [27, 28]. The PAC-1 receptor, a binding site of PACAP, is expressed in the human trigeminal ganglion as well as in the brain [29]. Interestingly, the activation of the same receptor (PAC-1) prevents the accumulation of aggregate-prone tau in transgenic tauopathy mice brain pointing to a role of the PAC-1 receptor in both diseases [30].
Most importantly, tau protein dysregulation is a well-established factor in several neurodegenerative disorders such as Alzheimer´s disease, but several other markers for neurodegeneration such as neurofilament light chain (NfL) [31], glial fibrillary acidic protein (GFAP) [32], and ubiquitin carboxy-terminal hydrolase L1 (UCH-L1) exist [33].
The exploration of the link between tau and migraine could help better understand the pathophysiology of this disease. This investigation aims to explore tau protein serum levels in the whole spectrum of patients with migraine in a cross-sectional case-control study. To assure that potential tau elevation in migraine is not a consequence of neuronal cell damage, a phenomenon that is typically associated with tau increase, we also assessed typical markers of neurodegeneration in this study. Additional serum and CSF samples were used for the evaluation of a central or peripheral origin of elevated biomarkers.
Materials and methods
Design
This is an observational cross-sectional study with a case–control design, which was approved by the ethical committee of Charité Universitätsmedizin Berlin (EA4/149/18). Written informed consent was obtained from all participants before study inclusion.
The study consisted of three study groups: individuals with episodic migraine (EM), with chronic migraine (CM), and healthy controls (HC). The migraine groups were recruited from the headache outpatient clinic of Charité – Universitätsmedizin Berlin between June 2019 and February 2021. All patients have been diagnosed according to the ICHD-3 guidelines [34]. We recruited healthy control participants using the intranet platform of Charité Universitätsmedizin Berlin and notice boards.
Inclusion & exclusion criteria
Patients were eligible if they had an existing ICHD-3 diagnosis for EM or CM, with or without aura with an age between 18 and 65 years. Patients were excluded if they suffered from any other neurological, psychiatric, or other chronic or inflammatory diseases or other primary headache disorder with the exception of infrequent episodic tension-type headache.
While we included CM patients with and without prophylaxis, in the EM group migraine prophylactic medications were not allowed, regardless of indication. In detail, the following medications led to study exclusion in the EM group: Metoprolol, Propranolol, Bisoprolol, Amitriptyline, Topiramate, Candesartan, Flunarizine, a CGRP monoclonal antibody, or a CGRP-receptor monoclonal antibody [35].
Healthy controls were eligible if they were between 18 and 65 years of age and if they did not suffer from any headache disorders or any other neurological disease. A positive family history of migraine, dementia, any chronic disease, a history of stroke or head trauma, or a history of or present moderate or severe depression as determined by the Beck Depression Inventory led to exclusion. The occurrence of any headache in the past three months also led to exclusion.
Assessment
For blood sampling, participants (including healthy controls) were in a non-fasting state and had to be free of any headache at the time of the blood withdrawal. The last headache attack of participants with migraine had to be completely resolved at least 12 h prior to blood withdrawal. Blood samples were taken from each participant using a 5 mL serum tube containing CAT Serum Sep Clot Activator (VACUETTE: 456010) from the cubital vein. Samples were centrifuged after blood withdrawal for 10 min at 800g at room temperature. The serum was stored within two hours at -80°C until sample analysis. We analyzed t-tau, NfL, GFAP, and UCH-L1 concentrations in serum using the Neurology 4-plex assay (HD‐1/HD‐X Item 102153) (Quanterix Corp, Lexington, MA) on a single molecule array (Simoa®) HD-X Analyzer (Quanterix Corp) at the Departments of Biomedicine and Clinical Research, University Hospital and University of Basel, Basel, Switzerland. Values under the limit of detection (LOD) of t-tau, NfL, GFAP, and UCH-L1 were set to 0.007, 0.025, 0.042, and 0.120 pg/ml, respectively. Inter-assay mean coefficient of variation (CV) was established with one serum and two assay kit controls. The mean CVs were 7.4%, 2.8%, 3.2% and 10.7% for t-tau, NfL, GFAP and UCH-L1 respectively.
Outcomes
The primary endpoint of the study was the difference in the concentrations of t-tau, NfL, GFAP, and UCH-L1 between the HC group and both migraine (EM/CM) groups. Secondary endpoints were the differences of t-tau, NfL, GFAP, and UCH-L1, across HC and EM with (EMA) and without aura (EMO), as well as between HC and CM with (CM +)/ without (CM-) prophylactic treatment. Further secondary endpoints were the correlations between the biomarkers and the demographic variables age and body mass index (BMI) and variables related to headaches: days since resolution of the last headache attack, days since last migraine attack, monthly headache days and monthly migraine days.
Variables
Data for monthly headache days (MHD), monthly migraine days (MMD), days since the last headache/migraine day, and monthly days with acute medication (AMD) were collected from the headache diaries. These variables were determined on the 28-day period before blood collection.
A migraine headache was defined as a headache with or without aura lasting for at least 30 min meeting at least one of the following criteria [34]: (A) ≥ 2 of the following pain features: unilateral, throbbing, moderate to severe, exacerbated by exercise/physical activity; (B) ≥ 1 of the following associated symptoms: nausea and/or vomiting, and photophobia and phonophobia; (C) Intake of migraine-specific acute medication, which was effective. We also collected the demographic characteristics of age, sex, ethnicity, body mass index (BMI; kg/m2), and alcohol and tobacco consumption.
CSF/serum analysis
In a post-hoc analysis, we assessed matched CSF/serum samples. These were acquired during routine in-hospital diagnostic work-up of headache disorders (patients eventually diagnosed with migraine) and after exclusion of CNS pathology in psychosomatic disorders (controls) at the Greifswald University Medicine, Germany. The control group patients did not suffer from any headache. Patients provided written informed consent for their samples to be used for research purposes. The use of the biospecimens for headache research was approved by the Ethical Committee, Greifswald University Medicine, Germany (BB 161/18). All samples were stored at -80°C within 24 h after sample collection. Patients were diagnosed with migraine according to ICHD-3 criteria and had no other diagnosis of any systemic chronic or neurological diseases. The samples from migraine patients were taken during the diagnostic work up (n = 23) and from controls (n = 16). Samples were analyzed for t-tau, NfL, GFAP, and UCH-L1 concentrations in serum and CSF using the Neurology 4-plex assay (SR-X Item 102,153) (Quanterix Corp, Lexington, MA) on a single molecule array (Simoa®) SR-X Analyzer (Quanterix Corp) at the Greifswald University Medicine.
Sample-size and statistical analysis
Due to the novelty of this approach, we did not calculate the sample size a priori for the comparison of serum t-tau and therefore our current sample size was determined by convenience. The biomarker values are reported as median (Md) interquartile range (IQR), other continuous variables are reported as mean ± standard deviation (SD), and counts and percentages are used for categorical variables. Statistical analyses were carried out using SPSS 28.0.1.0 (IBM Corp., Armonk, NY, USA). To test the normal distribution of our data, we used the Kolmogorov–Smirnov Test. Since our data remained non-normal after log-transformation, we chose a nonparametric approach. To compare groups, we used the Independent-Samples Kruskal–Wallis Test with a Dunn’s Post-Hoc test with Bonferroni correction for multiple comparisons. For post-hoc analyses of matched CSF/serum samples, we used the Mann–Whitney test (unpaired for the hypothesis of confirmation of findings). To correct for the covariates age, sex, and BMI, we used a Quade non-parametric ANCOVA.
Spearman's rho test was used for the testing of correlations. To analyze whether these biomarkers are able to distinguish between HC and migraine patients, receiver operating characteristics (ROC) statistics and area under the curve (AUC) values were calculated. A p-value below 0.05 was considered statistically significant.
Data availability
The data that support the findings of this study are available from the corresponding author, upon reasonable request.
Results
Characteristics of the study population
For this study, n = 288 participants were screened. Forty-four participants did not meet the eligibility criteria and 17 participants did not sign informed consent. Data from the remaining 227 participants (n = 42 healthy controls, n = 92 patients with EM, and n = 93 patients with CM) were included for analyses. Age, gender, and BMI did not differ between groups (for all p > 0.05). Migraine patients were more likely to consume less alcohol compared to healthy controls (p < 0.001). Tobacco consumption did not differ between groups (p = 0.119), Table 1. The characteristics of our subgroups can be found in Supplementary Table 1.
On average (SD), EM patients had 6.2 (3.0) monthly migraine days, while CM patients had 12.2 (6.7) monthly migraine days. Further headache characteristics are shown in Table 1.
The matched CSF/serum samples (n = 39) were mostly from females (83%), with a mean age of 37.4 ± 13.1 years.
Episodic and chronic migraine
Serum t-tau concentrations were different across HC, EM, and CM patients, H(2) = 11.643, p = 0.003, Fig. 1. Serum t-tau concentrations were higher in blood samples of EM patients (Md = 0.320 [0.204 to 0.466] pg/mL) and CM patients (Md = 0.304 [0.158 to 0.406] pg/mL) compared to HC (Md = 0.200 [0.114 to 0.288] pg/mL). Pairwise comparisons showed a significant difference between HC and EM (p = 0.002, r = 0.289) and HC and CM (p = 0.025, r = 0.230). T-tau levels from patients with EM and CM did not differ from each other (p = 1.000, r = 0.071), Table 2.

Blood serum concentrations of total-tau, NfL, GFAP, and UCH-L1 in pg/mL for patients with EM and CM and healthy controls. Legend: t-tau = total tau; NfL = neurofilament-light; GFAP = glial fibrillary acidic protein; UCH-L1 = ubiquitin C-terminal hydrolase L1; HCs = healthy controls; EM = episodic migraine; CM = chronic migraine. a p-value for between-group difference as estimated with the Independent-Samples Kruskal–Wallis Test for continuous variables, corrected with the Bonferroni correction for multiple comparisons. P-values < 0.05 are depicted in bold
No statistically significant differences of NfL, GFAP, and UCH-L1 were observed across HC, EM, and CM patients (p = 0.507, p = 0.850, and p = 0.195, respectively). The results are shown in Fig. 1. Serum concentrations of the biomarkers are illustrated in Table 2.
Episodic migraine with and without aura
Stratification in EM for aura revealed differences in serum t-tau concentrations across HC, EMO (n = 49), and EMA (n = 43), H(2) = 11.180, (p = 0.004). Serum t-tau concentrations were higher in migraine patients with and without aura (Md = 0.332 [0.234 to 0.449] pg/mL for EMO and Md = 0.291 [0.184 to 0.486] pg/mL for EMA) in comparison to HC (Md = 0.200 [0.114 to 0.288] pg/mL). Pairwise comparisons showed significant differences between HC and EMO (p = 0.008, r = 0.315) and HC and EMA (p = 0.013, r = 0.305. These differences remained significant after the correction for age, sex, and BMI (p = 0.001 and p = 0.003, respectively) but not between patients with EMO and EMA (p = 1.000, r = 0.004). Our analysis did not find any differences for NfL, GFAP, and UCH-L1 between groups, Table 3a.
T-tau was correlated with BMI only in the subgroup of patients with episodic migraine without aura (ρ = -0.307, p = 0.032, n = 49). There was no association between t-tau and any of the following parameters in the EM group: age, number of monthly headache days, and monthly migraine days.
Chronic migraine with (CM +) and without (CM-) prophylaxis
The stratification of CM patients according to current prophylactic treatment (no prophylaxis: CM-; n = 48 vs prophylaxis: CM + ; n = 45), revealed differences in t-tau concentrations across HC, CM-, and CM + , H(2) = 8.838, p = 0.012. T-tau was elevated in CM- patients (Md = 0.322 [0.181 to 0.463]) compared with HC (Md = 0.200 [0.114 to 0.288]) (p = 0.009, r = 0.307). This difference remained significant after the adjustment for age, sex, and BMI (p = 0.004). There was no difference of serum t-tau concentrations between CM + (Md = 0.293 [0.126 to 0.375]) patients and HC (p = 0.289, r = 0.184). Patients with CM- and CM + were not different from each other (p = 0.576, r = 0.140). Our analysis did not show any differences for NfL, GFAP, and UCH-L1 between groups, Table 3b.
Correlations of t-tau with headache characteristics
For an unbiased analysis, we included solely migraine patients without prophylactic treatment in the assessment of the association between serum markers and headache characteristics (n = 140). Complete headache dairies were available from n = 122 (87%) patients. We did not find correlations between t-tau and the frequency of headache days or migraine days, and time between sample collection and last migraine day. Also, the number of days with acute medication was not correlated with t-tau.
ROC curve analysis of t-tau
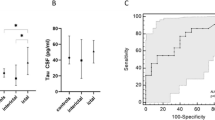
In order to assess whether t-tau levels can help to distinguish between migraine patients and healthy controls we performed ROC analysis. The data show that t-tau protein is able to distinguish between patients with migraine and healthy controls in our study at a cut-off value of 0.294 pg/ml (p < 0.001) with a sensitivity of 54% and a specificity of 81%, Fig. 2.

Receiver Operating Characteristics (ROC) curve analysis of serum total-tau concentrations for the prediction of migraine. Legend: AUC = area under the curve, CI = confidence interval
Post-hoc analyses of matched CSF/serum samples
In order to determine the source of t-tau changes, CSF and serum samples obtained during routine in-hospital diagnostic work-up of headache in patients eventually diagnosed with migraine (n = 23) and in patients presenting for exclusion of CNS pathology in psychosomatic disorders (controls; n = 16) were investigated using the same methodology. CSF samples were obtained by lumbar puncture and serum samples were drawn concomitantly. There was no difference in the albumin ratio (Qalb) between both groups (migraine 5.1 ± 2.0 × 10–3 vs. control 5.8 ± 2.7 × 10–3; p = 0.330). The mean age of the migraine group was 34.9 (22.4 to 42.7) years and included n = 20 (87%) females. For the control group the mean age was 27.0 (24.0 to 41.0) and included n = 12 (63%) females. Serum t-tau concentrations were elevated in migraine patients (Md = 0.69 pg/mL [0.39 to 1.06]) compared to samples taken in controls (Md = 0.43 pg/mL [0.33 to 0.54]); p = 0.028, r = 0.339), Fig. 3A. In contrast, CSF t-tau concentrations did not differ between migraine (Md = 51.17 [36.22 to 64.82]) and control samples (Md = 42.96 pg/ml [35.13 to 70.57], p = 0.760, r = 0.047), Fig. 3B.

Serum (A) and CSF (B) t-tau concentration in migraine patients and controls. Legend p-values for between-group difference as estimated with the Independent-Samples Mann–Whitney U test for continuous variables. p-values < 0.05 are depicted in bold
Discussion
This cross-sectional observational study assessed t-tau concentrations in serum samples of 227 participants with migraine and age- and sex-matched healthy controls. T-tau was elevated in patients with EM and CM compared to controls. Stratification for aura did not show any differences between patients with migraine with and without aura. CM patients without preventive medications had higher t-tau serum concentrations than HC, which could not be observed in CM patients with preventive medication. T-tau elevation was not observed in the CSF of migraine patients in comparison to controls. NfL, GFAP, and UCH-L1 were not different from controls and interictal migraine patients. These findings may point to a pathophysiological role of tau in migraine pathophysiology.
T-tau levels in EM patients were not different between migraine without and with aura. The latter is the clinical correlate of cortical neuronal depolarization [36]. The lack of a difference in both groups indicates that cortical activity is not relevant for tau release and implies that rather neuronal activity associated with pain, e.g. the trigeminal nerve or the ganglion is associated with tau release. However, our study is not suited to determine the origin of tau release in patients with migraine with certainty. Animal studies with a focus on the interplay of tau, PACAP and the PAC-1 receptor are needed to shed light on this question. Of note, no other markers associated with neuronal or glial damage were elevated in serum.
Tau is a microtubule-associated protein, which is involved in axonal transport as it stabilizes neuronal microtubules. Moreover, tau has a role in the formation, maintenance, and repetition of myelin by activating Fyn-kinase rafts. Only a few studies link tau protein, neuropeptides, and inflammation. Most recently, the small molecule CGRP receptor antagonist BIBN-4096, a drug that successfully aborts migraine attacks, reduced neuroinflammation in an experimental model of Alzheimer´s disease [23]. Tau pathology is a key component of this disease [37]. Tau was also elevated in inflammatory peripheral neuronal diseases such as Guillain-Barré syndrome as recently described [23, 38]. Interestingly, tau concentrations were significantly higher in inflammatory neuropathies than in non-inflammatory neuropathies, which indicates a pathophysiological link between tau protein and inflammation. In inflammatory neuropathies, tau elevation was seen along with a rise of neurofilament light chain (NfL) levels, which confirms structural damage. Inflammation in migraine is thought to be independent of structural damage. As inflammation and neuropeptide release has been described in migraine in the meninges and trigeminal ganglion (for review see Edvinsson, 2019), these structures could also be relevant for t-tau elevation in this disorder [36]. Markers associated with cell damage such as NfL, GFAP or UCHL-1 were normal in serum samples in this study. This observation is in line with our aforementioned hypothesis of neuro-inflammation and t-tau release and a rather functional role of tau in migraine. In line, in tau knock-out mice the neuronal response to noxious stimuli was reduced while the response to tonic painful stimuli was increase along enhanced evoked c-fos expression in the dorsal horn [39]. The latter observation supports a functional role of tau in pain conditions.
The estimated area under the curve (AUC) indicates that serum t-tau concentration could distinguish between patients with migraine and healthy controls in two-thirds of cases (p < 0.001). However, the low AUC (0.662) suggests an inadequate ability to discriminate between individuals with and without migraine. Therefore, the utility of serum t-tau levels as a diagnostic biomarker needs to be explored in future studies. More specifically, it remains to be determined whether t-tau levels can differentiate between different primary headaches.
At this stage, we cannot determine the clinical significance of tau in migraine. It is also worth noting that t-tau levels in migraine are significantly lower than in e.g. inflammatory neuropathies, brain trauma or neurodegenerative disorders. However, CGRP and PACAP levels in peripheral blood in migraine patients are also in the picogram range and nevertheless play a crucial role in migraine pathophysiology [40, 41]. A possible role of tau in migraine pathophysiology and the relationship between CGRP, PACAP, or the PAC-1 receptor with tau needs to be determined in future studies.
Tau is not different between EM and CM in this study indicating that migraine frequency is not a determining factor for tau levels. We show a moderate negative correlation of tau levels over time as determined from the last migraine day before blood withdrawal. The lack of a difference might be explained by relatively low tau levels in this study and the lack of accumulation of tau with increasing number of attacks. Whether pain intensity or the duration of migraine episodes play a role for elevated tau levels in blood remains to be determined.
The different t-tau concentrations observed in CM patients with prophylaxis compared to CM patients without prophylactic medication use is unlikely to be caused by lower number of MHD (mean 12.8 vs 17.0, respectively) since the number of MHD of EM patients is much lower (mean 7.2). It is possible that prophylactic medication reduces tau levels by other mechanisms than reducing headache frequency. A longitudinal study is necessary to provide evidence for any causality.
Alcohol consumption differed between migraine patients and controls in this study. Lower alcohol consumption in migraine patients has been observed before [42]. Accumulating evidence indicates that alcohol consumption leads to elevated t-tau concentrations [43]. In our cohort, no effect of alcohol consumption on serum t-tau concentrations was observed, nor did the correction for alcohol use affect the results (data not shown). This might be explained by the fact that none of our participants are considered heavy drinkers.
A strength of our study is the large sample size, which provides accurate and robust findings. Our strict in- and exclusion criteria helped to select patients and controls in a consistent, reliable, uniform, and objective way to avoid confounding or biasing factors. Another strength is the method of sample analyses. The single-molecule array (Simoa®) technology is an ultrasensitive method using antibodies, which is superior to the enzyme-linked immunosorbent assay (ELISA) technology. Therefore, our measurements are more accurate and precise than those from ELISA.
This study also has limitations. First, tau elevation in serum is not specific for any disease. The different types of headache diaries (paper/electronic) led to missing information on headache intensity and the precise duration of migraine attacks in hours. We did not check if patients had an attack immediately after blood collection and therefore, we cannot exclude the possibility that the patient was in the prodromal migraine phase. Based on the fact that premonitory symptoms are typical central CNS phenomena and that we found a significant tau increase in the periphery during attacks, we believe that an influence of a premonitory migraine stage on these findings is improbable.
In summary, this is the first report of t-tau elevation in a primary headache disorder. Patients with migraine have elevated t-tau levels in blood serum in the interictal state, but not in CSF compared to age- and sex-matched controls. Our findings indicate that tau is derived from a peripheral source in migraine. Future studies need to establish the precise role of tau in migraine.
Availability of data and materials
We will make data available upon request to UR.
References
Collaborators; GDaIIaP (2017) Global, regional, and national incidence, prevalence, and years lived with disability for 328 diseases and injuries for 195 countries, 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet 390(10100):1211–1259
Migraine AM (2020) N Engl J Med 383(19):1866–1876
Edvinsson L, Haanes KA, Warfvinge K (2019) Does inflammation have a role in migraine? Nat Rev Neurol 15(8):483–490
Biscetti L, Cresta E, Cupini LM, Calabresi P, Sarchielli P (2023) The putative role of neuroinflammation in the complex pathophysiology of migraine: from bench to bedside. Neurobiol Dis 180:106072
Ashina M, Hansen JM, Do TP, Melo-Carrillo A, Burstein R, Moskowitz MA (2019) Migraine and the trigeminovascular system-40 years and counting. Lancet Neurol 18(8):795–804
Pardutz A, Schoenen J (2010) NSAIDs in the acute treatment of migraine: a review of clinical and experimental data. Pharmaceuticals (Basel) 3(6):1966–1987
Cameron C, Kelly S, Hsieh SC, Murphy M, Chen L, Kotb A et al (2015) Triptans in the acute treatment of migraine: a systematic review and network meta-analysis. Headache 55(Suppl 4):221–235
Spekker E, Tanaka M, Szabó Á, Vécsei L (2021) Neurogenic inflammation: the participant in migraine and recent advancements in translational research. Biomedicines 10(1):76
Johnson KW, Schaus JM, Durkin MM, Audia JE, Kaldor SW, Flaugh ME et al (1997) 5-HT1F receptor agonists inhibit neurogenic dural inflammation in guinea pigs. NeuroReport 8(9–10):2237–2240
Hadjikhani N, Albrecht DS, Mainero C, Ichijo E, Ward N, Granziera C et al (2020) Extra-axial inflammatory signal in parameninges in migraine with visual aura. Ann Neurol 87(6):939–949
Beal MF, Mazurek MF, Chattha GK, Svendsen CN, Bird ED, Martin JB (1986) Neuropeptide Y immunoreactivity is reduced in cerebral cortex in Alzheimer’s disease. Ann Neurol 20(3):282–288
Laurent C, Buée L, Blum D (2018) Tau and neuroinflammation: what impact for alzheimer’s disease and tauopathies? Biomed J 41(1):21–33
Shi Y, Yamada K, Liddelow SA, Smith ST, Zhao L, Luo W et al (2017) ApoE4 markedly exacerbates Tau-mediated neurodegeneration in a mouse model of Tauopathy. Nature 549(7673):523–527
Didonna A, Cantó E, Shams H, Isobe N, Zhao C, Caillier SJ et al (2019) Sex-specific Tau methylation patterns and synaptic transcriptional alterations are associated with neural vulnerability during chronic neuroinflammation. J Autoimmun 101:56–69
Gurney KJ, Estrada EY, Rosenberg GA (2006) Blood-brain barrier disruption by stromelysin-1 facilitates neutrophil infiltration in neuroinflammation. Neurobiol Dis 23(1):87–96
Yamanaka G, Suzuki S, Morishita N, Takeshita M, Kanou K, Takamatsu T et al (2021) Role of neuroinflammation and blood-brain barrier permutability on migraine. Int J Mol Sci 22(16):8929
Corrigan F, Cernak I, McAteer K, Hellewell SC, Rosenfeld JV, Turner RJ et al (2021) NK1 antagonists attenuate tau phosphorylation after blast and repeated concussive injury. Sci Rep 11(1):8861
Jin K, Takeda A, Shiga Y, Sato S, Ohnuma A, Nomura H et al (2006) CSF tau protein: a new prognostic marker for Guillain-Barré syndrome. Neurology 67(8):1470–1472
Wang XK, Zhang HL, Meng FH, Chang M, Wang YZ, Jin T et al (2013) Elevated levels of S100B, tau and pNFH in cerebrospinal fluid are correlated with subtypes of Guillain-Barré syndrome. Neurol Sci 34(5):655–661
Edvinsson L (2019) Role of CGRP in Migraine. Handb Exp Pharmacol 255:121–130
Bertels Z, Mangutov E, Conway C, Siegersma K, Asif S, Shah P et al (2022) Migraine and peripheral pain models show differential alterations in neuronal complexity. Headache 62(7):780–791
Bertels Z, Singh H, Dripps I, Siegersma K, Tipton AF, Witkowski WD et al (2021) Neuronal complexity is attenuated in preclinical models of migraine and restored by HDAC6 inhibition. Elife 10:e63076
Na H, Gan Q, McParland L, Yang JB, Yao H, Tian H et al (2020) Characterization of the effects of calcitonin gene-related peptide receptor antagonist for Alzheimer’s disease. Neuropharmacology 168:108017
Zhang ZH, Fang XB, Xi GM, Li WC, Ling HY, Qu P (2010) Calcitonin gene-related peptide enhances CREB phosphorylation and attenuates tau protein phosphorylation in rat brain during focal cerebral ischemia/reperfusion. Biomed Pharmacother 64(6):430–436
Zhang XC, Kainz V, Burstein R, Levy D (2011) Tumor necrosis factor-α induces sensitization of meningeal nociceptors mediated via local COX and p38 MAP kinase actions. Pain 152(1):140–149
Tozzi A, de Iure A, Di Filippo M, Costa C, Caproni S, Pisani A et al (2012) Critical role of calcitonin gene-related peptide receptors in cortical spreading depression. Proc Natl Acad Sci U S A 109(46):18985–18990
Schytz HW, Birk S, Wienecke T, Kruuse C, Olesen J, Ashina M (2009) PACAP38 induces migraine-like attacks in patients with migraine without aura. Brain 132(Pt 1):16–25
H. Lundbeck A/S (2023) Lundbeck announces positive phase II Proof of Concept results with Lu AG09222 in migraine prevention (Corporate Release No 740). Retrieved from: https://mb.cision.com/Main/18215/3754245/1995698.pdf
Knutsson M, Edvinsson L (2002) Distribution of mRNA for VIP and PACAP receptors in human cerebral arteries and cranial ganglia. NeuroReport 13(4):507–509
Schaler AW, Runyan AM, Clelland CL, Sydney EJ, Fowler SL, Figueroa HY et al (2021) PAC1 receptor-mediated clearance of tau in postsynaptic compartments attenuates tau pathology in mouse brain. Sci Transl Med 13(595):eaba7394
Gaetani L, Blennow K, Calabresi P, Di Filippo M, Parnetti L, Zetterberg H (2019) Neurofilament light chain as a biomarker in neurological disorders. J Neurol Neurosurg Psychiatry 90(8):870–881
Lamers KJ, Vos P, Verbeek MM, Rosmalen F, van Geel WJ, van Engelen BG (2003) Protein S-100B, neuron-specific enolase (NSE), myelin basic protein (MBP) and glial fibrillary acidic protein (GFAP) in cerebrospinal fluid (CSF) and blood of neurological patients. Brain Res Bull 61(3):261–264
Setsuie R, Wada K (2007) The functions of UCH-L1 and its relation to neurodegenerative diseases. Neurochem Int 51(2–4):105–111
(2018) Headache Classification Committee of the International Headache Society (IHS) The International Classification of Headache Disorders, 3rd edition. Cephalalgia 38(1):1–211
Diener H-C, Förderreuther S, Kropp P et al (2022) Therapie der Migräneattacke und Prophylaxe der Migräne, S1-Leitlinie. DGN und DMKG, in: Deutsche Gesellschaft für Neurologie (Hrsg.), Leitlinien für Diagnostik und Therapie in der Neurologie. http://www.dgn.org/leitlinien. Retrieved on August 10, 2023
Hadjikhani N, Sanchez Del Rio M, Wu O, Schwartz D, Bakker D, Fischl B et al (2001) Mechanisms of migraine aura revealed by functional MRI in human visual cortex. Proc Natl Acad Sci U S A 98(8):4687–4692
Kolarova M, García-Sierra F, Bartos A, Ricny J, Ripova D (2012) Structure and pathology of tau protein in Alzheimer disease. Int J Alzheimers Dis 2012:731526
Kmezic I, Samuelsson K, Finn A, Upate Z, Blennow K, Zetterberg H et al (2022) Neurofilament light chain and total tau in the differential diagnosis and prognostic evaluation of acute and chronic inflammatory polyneuropathies. Eur J Neurol 29(9):2810–2822
Sotiropoulos I, Lopes AT, Pinto V, Lopes S, Carlos S, Duarte-Silva S et al (2014) Selective impact of Tau loss on nociceptive primary afferents and pain sensation. Exp Neurol 261:486–493
Goadsby PJ, Edvinsson L, Ekman R (1990) Vasoactive peptide release in the extracerebral circulation of humans during migraine headache. Ann Neurol 28(2):183–187
Pérez-Pereda S, Toriello-Suárez M, Ocejo-Vinyals G, Guiral-Foz S, Castillo-Obeso J, Montes-Gómez S et al (2020) Serum CGRP, VIP, and PACAP usefulness in migraine: a case-control study in chronic migraine patients in real clinical practice. Mol Biol Rep 47(9):7125–7138
Zlotnik Y, Plakht Y, Aven A, Engel Y, Am NB, Ifergane G (2014) Alcohol consumption and hangover patterns among migraine sufferers. J Neurosci Rural Pract 5(2):128–134
Gendron TF, McCartney S, Causevic E, Ko LW, Yen SH (2008) Ethanol enhances tau accumulation in neuroblastoma cells that inducibly express tau. Neurosci Lett 443(2):67–71
Acknowledgements
We greatly acknowledge Dr. M. Steinicke, Dr. I. Laumeier, Dr. M. D. Hofacker, J. Mecklenburg, and Dipl. Psych. I. Hubalek who assisted with the recruitment of subjects.
Funding
Open Access funding enabled and organized by Projekt DEAL. This study was supported by a Novartis AG (Switzerland) research grant to UR.
Author information
Authors and Affiliations
Contributions
LHO, BR, RF, WS, MK, MA, NT, JK, UR contributed to conception and design of the study. LHO, BR, RF, AS, MS, AMM, AP, KR, UR contributed to data acquisition, and analysis of data. LHO, BR, RF, UR drafted the text and prepared figures. All authors have seen the manuscript and approved the submission.
Corresponding author
Ethics declarations
Competing interests
LHO has nothing to declare. BR received speaker fees from Abbvie/Allergan, Lilly, Lundbeck, Novartis, Teva, and grant support for research or education form Novartis, the German Migraine and Headache Society (DMKG) and the German Research Foundation (DFG). BR is member of the junior editorial board of The Journal of Headache and Pain, and editorial board of Frontiers in Neurology. RF received speaker fees from Novartis. MS has nothing to declare. AV has nothing to declare AMM has nothing to declare. KR received speaker fees from Bayer, travel grants from Guthy Jackson Charitable Foundation, and grant support for research or education from Novartis Pharma, Merck Serono, German Ministry of Education and Research, European Union (821283–2), Stiftung Charité (BIH Clinical Fellow Program) and Arthur Arnstein Foundation. WS, MK, MA, and ND are current or former employees of Novartis Pharma. AS received speaker fees from Teva and congress fees from Novartis. JK received speaker fees, research support, or travel support from or served on advisory boards for Swiss MS Society, Swiss National Research Foundation (320030_189140/1), University of Basel, Progressive MS Alliance, Bayer, Biogen, Celgene, Merck, Novartis, Roche, and Sanofi. UR received speaker or consulting fees from Amgen, Allergan, Abbvie, Eli Lilly, Lundbeck, Novartis, Medscape, Pfizer, StreaMedUp, and Teva, and received grant support for research or education from Novartis (CHERUB01) and German Federal Ministry of Education and Research. UR is treasurer of the EHF and serves as associate editor of the Journal of Headache and Pain and Frontiers in Neurology.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Supplementary Table 1.
Clinical and demographic characteristics of the subgroups of episodic and chronic migraine.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Overeem, L.H., Raffaelli, B., Fleischmann, R. et al. Serum tau protein elevation in migraine: a cross-sectional case–control study. J Headache Pain 24, 130 (2023). https://doi.org/10.1186/s10194-023-01663-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s10194-023-01663-5