
Abstract
Background
Dihydroergotamine (DHE) is an antimigraine drug that produces cranial vasoconstriction and inhibits trigeminal CGRP release; furthermore, it inhibits the vasodepressor sensory CGRPergic outflow, but the receptors involved remain unknown. Prejunctional activation of α2A/2C-adrenergic, serotonin 5-HT1B/1F, or dopamine D2-like receptors results in inhibition of this CGRPergic outflow. Since DHE displays affinity for these receptors, this study investigated the pharmacological profile of DHE-induced inhibition of the vasodepressor sensory CGRPergic outflow.
Methods
Pithed rats were pretreated i.v. with hexamethonium (2 mg/kg·min) followed by continuous infusions of methoxamine (20 μg/kg·min) and DHE (3.1 μg/kg·min). Then, stimulus-response curves (spinal electrical stimulation; T9-T12) or dose-response curves (i.v. injections of α-CGRP) resulted in frequency-dependent or dose-dependent decreases in diastolic blood pressure.
Results
DHE inhibited the vasodepressor responses to electrical stimulation (0.56–5.6 Hz), without affecting those to i.v. α-CGRP (0.1–1 μg/kg). This inhibition by DHE (not produced by the methoxamine infusions): (i) was abolished by pretreatment with the combination of the antagonists rauwolscine (α2-adrenoceptor; 310 μg/kg) plus GR127935 (5-HT1B/1D; 31 μg/kg); and (ii) remained unaffected after rauwolscine (310 μg/kg), GR127935 (31 μg/kg) or haloperidol (D2-like; 310 μg/kg) given alone, or after the combination of rauwolscine plus haloperidol or GR127935 plus haloperidol at the aforementioned doses.
Conclusion
DHE-induced inhibition of the vasodepressor sensory CGRPergic outflow is mainly mediated by prejunctional rauwolscine-sensitive α2-adrenoceptors and GR127935-sensitive 5-HT1B/1D receptors, which correlate with α2A/2C-adrenoceptors and 5-HT1B receptors, respectively. These findings suggest that DHE-induced inhibition of the perivascular sensory CGRPergic outflow may facilitate DHE’s vasoconstrictor properties resulting in an increased vascular resistance.
Similar content being viewed by others

Background
Dihydroergotamine (DHE) is a primary drug effective in the acute treatment of migraine [1,2,3] and its therapeutic effect may involve: (i) cranial vasoconstriction via vascular 5-HT1B and α2A/2C-adrenoceptors [4]; (ii) inhibition of neurogenic cranial vasodilatation produced by trigeminal release of calcitonin gene-related peptide (CGRP) [5, 6]; and probably (iii) inhibition of trigeminal nociceptive reflexes [7, 8]. More recently, DHE has been shown to increase diastolic blood pressure (an index of peripheral vascular resistance) by activation of vascular α1 (α1A, α1B and α1D) and α2 (α2A, α2B and α2C)-adrenoceptors [9]. Interestingly, at peripheral level, CGRP released from primary sensory perivascular nerves induces vasodepressor responses [10,11,12], but this neuropeptide does not seem to be involved in the physiological regulation of blood pressure [13]. Notwithstanding, evidence is now growing suggesting that CGRP has a protective role in the generation of hypertension, which is most likely mediated via its effects at peripheral receptors [14]. Thus, the potential side-effects produced by DHE on the systemic CGRPergic transmission via its prejunctional interactions on perivascular sensory CGRPergic nerves deserve special attention [15]. Indeed, DHE is capable of inhibiting the vasodepressor responses induced by spinal stimulation of the perivascular sensory CGRPergic outflow in pithed rats [16]; however, the pharmacological profile of the receptors involved in this inhibitory action remains thus far unclear, probably because DHE displays complex pharmacological properties as it has affinity for an array of receptors [1,2,3, 17]. In this respect, by using selective agonists and antagonists, our group has previously shown that the rat vasodepressor sensory CGRPergic outflow (an index of sensory perivascular CGRP release in resistance blood vessels [12]) can be inhibited by prejunctional activation of receptors coupled to Gi/o proteins, including: (i) α2 (specifically α2A/2C)-adrenoceptors [18]; (ii) serotonin 5-HT1B [19] and 5-HT1F [20] receptors; and (iii) dopamine D2-like receptors [21]. Since DHE displays affinity for these receptors (see Table 1), it is reasonable to hypothesize that these receptors could be involved in DHE-induced inhibition of the vasodepressor sensory CGRPergic outflow. On this basis, the present study in pithed rats was designed to investigate: (a) whether DHE is capable of inhibiting the vasodepressor responses induced by either stimulation of the perivascular sensory CGRPergic outflow or i.v. bolus injections of exogenous α-CGRP; and (b) the pharmacological profile of the receptors involved in DHE-induced inhibition of the vasodepressor sensory CGRPergic outflow by analysing the effects of pre-treatment with the antagonists rauwolscine (α2-adrenoceptors), GR127935 (5-HT1B/1D) and haloperidol (D2-like).
Methods
Animals
Male Wistar normotensive rats (300–350 g) were maintained at a 12/12-h light/dark cycle (with light beginning at 07:00 h) and housed in a special room at constant temperature (22 ± 2 °C) and humidity (50%), with food and water freely available in their home cages. All animal procedures, number of animals and the protocols of the present investigation were approved by our Institutional Ethics Committee on the use of animals in scientific experiments (CICUAL Cinvestav; protocol number 507–12), and followed the regulations established by the Mexican Official Norm (NOM-062-ZOO-1999), in accordance with ARRIVE (Animal Research: Reporting In Vivo Experiments) reporting guidelines for the care and use of laboratory animals.
General methods
Experiments were carried out in a total of 90 rats. After anaesthesia with ether and cannulation of the trachea, the rats were pithed by inserting a stainless-steel rod through the orbit and foramen magnum into the vertebral foramen [22]. Then, the animals were artificially ventilated with room air using a model 7025 Ugo Basile pump (56 strokes per min; stroke volume = 20 ml/kg), as established by Kleinman and Radford [23]. After bilateral vagotomy, catheters were placed in: (i) the left and right femoral and jugular veins, for the continuous infusions of agonists (methoxamine and DHE) and i.v. administration of the antagonists, respectively; and (ii) the left carotid artery, connected to a Grass pressure transducer (P23XL), for the recording of arterial blood pressure. Heart rate was measured with a tachograph (7P4, Grass Instrument Co., Quincy, MA, USA) triggered from the blood pressure signal. Both blood pressure and heart rate were recorded simultaneously by a model 7 Grass polygraph (Grass Instrument Co., Quincy, MA, USA). At this point, the 90 rats were divided into two main sets, so that the effects produced by the continuous infusions of methoxamine and DHE under different treatments could be evaluated on the vasodepressor responses induced by: (i) electrical stimulation of the vasodepressor sensory CGRPergic outflow (set 1; n = 80); and (ii) i.v. bolus injections of exogenous α-CGRP (set 2; n = 10). The vasodepressor stimulus-response curves and dose-response curves by electrical stimulation and exogenous α-CGRP, respectively, were elicited using a sequential schedule at 5–10 min intervals (see below) and were completed in about 50 min. Each response was elicited under unaltered values of resting blood pressure. The body temperature of each pithed rat was maintained at 37 °C by a lamp and monitored with a rectal thermometer.
Experimental protocols
After the animals (n = 90) had been in a stable haemodynamic condition for at least 15 min, baseline values of diastolic blood pressure (a more accurate indicator of peripheral vascular resistance, as previously established [12, 18,19,20,21]) and heart rate were determined.
Protocol 1. Electrical stimulation of the perivascular (vasodepressor) sensory outflow
In the first set of rats (n = 80), the pithing rod was replaced by an electrode enamelled except for 1.5 cm length 9 cm from the tip, so that the uncovered segment was situated at T9-T12 of the spinal cord, and an indifferent electrode was placed dorsally [16, 18,19,20,21,22]. Before electrical stimulation, the animals received (i.v.): (i) a bolus injection of gallamine (25 mg/kg) to avoid electrically-induced muscular twitchings; (ii) ten min later, a continuous infusion of hexamethonium (2 mg/kg·min) to block the electrically-induced vasopressor responses that are produced by stimulation of the preganglionic sympathetic vasopressor outflow; and (iii) ten min later, a continuous infusion of methoxamine (20 μg/kg·min) to produce a sustained increase in diastolic blood pressure that allows us to produce the subsequent induction of vasodepressor responses, as previously described [16, 18,19,20,21]. Ten min later, this set of rats was divided into three groups.
The first group (n = 10) was subdivided into two subgroups (n = 5 each one) that received: (i) nothing (control experiment with no vehicles; see below); and (ii) an i.v. continuous infusion of DHE (3.1 μg/kg·min), a dose that has previously been shown to produce (amongst several doses) a maximal inhibition of the vasodepressor sensory CGRPergic outflow in pithed rats [16]. Twenty minutes later, diastolic blood pressure and heart rate were determined again, and then, the vasodepressor sensory CGRPergic outflow was electrically stimulated during the above treatments to elicit vasodepressor responses by applying 10-s trains of monophasic, rectangular pulses (2 msec, 50 V), at increasing frequencies of stimulation (0.56, 1, 1.8, 3.1 and 5.6 Hz). When diastolic blood pressure had returned to baseline levels, the next frequency was applied. This procedure was systematically performed until the stimulus-response curve had been completed.
The second group (n = 35) received an i.v. continuous infusion of 1% propylene glycol (PPG; vehicle for dissolving DHE) (0.02 ml/min). Ten min later, this group was subdivided into seven subgroups (n = 5 each) comprising i.v. bolus injections of, respectively: (i) saline (1 ml/kg); (ii) rauwolscine (310 μg/kg); (iii) GR127935 (31 μg/kg); (iv) haloperidol (310 μg/kg); (v) rauwolscine+GR127935 (310 and 31 μg/kg, respectively); (vi) rauwolscine+ haloperidol (310 μg/kg, each); and (vii) GR127935 + haloperidol (31 and 310 μg/kg, respectively). After 10 min, a stimulus-response curve was constructed as described above during the infusion of methoxamine to determine the effect of these antagonists per se.
The third group (n = 35) received an i.v. continuous infusion of DHE (3.1 μg/kg·min). Ten min later, this group was subdivided into seven subgroups (n = 5 each) comprising i.v. bolus injections of, respectively: (i) saline (1 ml/kg); (ii) rauwolscine (310 μg/kg); (iii) GR127935 (31 μg/kg); (iv) haloperidol (310 μg/kg); (v) rauwolscine+GR127935 (310 and 31 μg/kg, respectively); (vi) rauwolscine+haloperidol (310 μg/kg, each); and (vii) GR127935 + haloperidol (31 and 310 μg/kg, respectively). Ten minutes later, a stimulus-response curve was constructed as described above, during the infusion of DHE.
Protocol 2. Administration of exogenous α-CGRP
The second set of rats (n = 10) was prepared as describe above, but the pithing rod was left throughout the experiment and the administration of both gallamine and hexamethonium was omitted, as previously described [16, 18,19,20,21]. Then, this set received and i.v. continuous infusion of methoxamine (20 μg/kg·min); after 10 min, this set was divided into two groups (n = 5 each) that received, respectively: (i) nothing (control group); or (ii) an i.v. continuous infusion of DHE (3.1 μg/kg·min). Twenty min later, the values of diastolic blood pressure and heart rate were determined again, and then, the vasodepressor responses elicited by i.v. bolus injections of exogenous α-CGRP (0.1, 0.18, 0.31, 0.56 and 1 μg/kg) were examined during the infusions of methoxamine and DHE.
Other procedures applying to protocols 1 and/or 2
The doses of hexamethonium, methoxamine and DHE were continuously infused at a rate of 0.02 ml/min by a WPI model sp100i pump (World Precision Instruments Inc., Sarasota, FL, USA). The dose of DHE was selected from a previous study [16]. The intervals between the different stimulation frequencies or doses of α-CGRP applied were dependent on the duration of the resulting vasodepressor responses (5–10 min), as we waited until diastolic blood pressure had returned to baseline values.
Drugs
Apart from the anaesthetic (diethyl ether), the compounds used in this study (obtained from the sources indicated) were: gallamine triethiodide, hexamethonium chloride, rat α-CGRP, methoxamine hydrochloride, rauwolscine hydrochloride, (Sigma Chemical Co., St Louis, MO, USA); N-[methoxy-3-(4-methyl-1-piperazinyl)phenyl]-2′-methyl-4′-(5-methyl-1,2,4-oxadiazol-3-yl)[1,1-biphenyl]-4-carboxamidehydrochloride (GR127935) (gift from GlaxoSmithKline, Stevenage, Hertfordshire, UK); and DHE tartrate (gift from Novartis Pharma A.G., Basel, Switzerland). All compounds were dissolved in saline, except: (i) DHE, which was dissolved in propylene glycol and gauged with saline to have a final solution of 1% PPG; and (ii) haloperidol, which was dissolved in some drops of 5% ascorbic acid and the resulting solution was finally diluted with saline. These vehicles had no effect on baseline diastolic blood pressure or heart rate (data not shown). Fresh solutions were prepared for each experiment. The doses of agonists refer to their respective salts, whereas those of the antagonists refer to their free base.
Data presentation and statistical evaluation
All data in the text, tables and figures, unless stated otherwise, are presented as mean ± standard error of the mean (S.E.M). The peak changes in diastolic blood pressure by electrical stimulation or exogenous α-CGRP were expressed as percent change from baseline, as previously reported [16, 18,19,20,21]. The difference in the absolute values of diastolic blood pressure and heart rate within one subgroup of animals before and during the continuous infusions of methoxamine (20 μg/kg·min) and DHE (3.1 μg/kg·min) were evaluated with paired Student’s t-test. Moreover, a one-way analysis of variance was used to compare the absolute values of diastolic blood pressure and heart rate obtained during the continuous infusions of methoxamine (20 μg/kg·min) and DHE (3.1 μg/kg·min) before, immediately after and 10 min after administration of saline or the antagonists used. Finally, the vasodepressor responses induced by electrical stimulation or exogenous α-CGRP in the different subgroups of animals were compared with a two-way analysis of variance. The one- and two-way analyses of variance were followed, if applicable, by the Student-Newman-Keuls’ test. Statistical significance was accepted at P < 0.05. The statistical analysis was performed using the SigmaPlot software (V 12.0; Systat Software, Inc.), whereas the graphs were made with GraphPad Prism® software (V 6.01; GraphPad Software, Inc.).
Results
Systemic haemodynamic effects of the different treatments
The baseline values of diastolic blood pressure and heart rate in the 90 pithed rats were 57 ± 5 mmHg and 243 ± 8 beats per min, respectively; these variables remained unchanged after gallamine or hexamethonium. Twenty min after starting the i.v. continuous infusions of methoxamine, baseline values of diastolic blood pressure and heart rate were significantly (P < 0.05) increased in all animals (i.e. 140 ± 4 mmHg and 273 ± 4 beats per min, respectively). It is noteworthy that during the infusions of methoxamine and/or DHE a transient, but significant, decrease in diastolic blood pressure was produced immediately after administration of an i.v. bolus injections of rauwolscine, haloperidol or the combinations of these antagonists, but not with saline or GR127935 (see Table 2). However, the values of diastolic blood pressure in the different subgroups before and 10 min after administration of saline, or antagonists, were not significantly different (P > 0.05) (Table 2). Furthermore, the increase in diastolic blood pressure produced by the continuous infusion of methoxamine was sustained throughout the experiments, as illustrated in Fig. 1a.

Effect of dihydroergotamine (DHE) on the vasodepressor CGRPergic outflow in pithed rats. a Original experimental tracings illustrating the vasodepressor responses induced by electrical stimulation of the perivascular sensory CGRPergic outflow during continuous infusions of either methoxamine (control; above) or DHE (below). Note that during continuous infusions of DHE (3.1 μg/kg·min) the vasodepressor responses induced by electrical stimulation were attenuated versus control. In both cases, the vasodepressor responses were selective as no changes in heart rate were observed. Panels (b) and (c) show the vasodepressor responses by electrical stimulation or i.v. bolus injections of α-CGRP, respectively, induced during an i.v. continuous infusions of 3.1 μg/kg·min DHE (n = 5 each). For the sake of clarity, control responses (○) were induced during continuous infusions of methoxamine (20 μg/kg·min). * Significantly different responses (P < 0.05) vs. control. BP, blood pressure; HR, heart rate
Vasodepressor responses produced by electrical stimulation or exogenous α-CGRP
Figure 1a shows some representative experimental tracings illustrating that during the infusion of methoxamine the onset of the responses induced by electrical stimulation (0.56–5.6 Hz) of the vasodepressor sensory outflow (T9-T12) were immediate and resulted in frequency-dependent decreases in diastolic blood pressure. It must be emphasized that these vasodepressor responses were due to selective stimulation of the vasodepressor sensory CGRPergic outflow, since only negligible and inconsistent effects in heart rate were observed, as described earlier [16, 18,19,20,21]. In addition, as previously reported by Lozano-Cuenca et al. [16], stimulation of the vasodepressor sensory CGRPergic outflow also resulted in vasodepressor responses during the infusion of DHE (3.1 μg/kg·min), but the magnitude of these responses was clearly smaller than those elicited during the infusion of methoxamine (20 μg/kg·min).
Moreover, during the methoxamine infusion (control; 20 μg/kg·min): (i) electrical stimulation of the perivascular sensory outflow resulted in frequency-dependent vasodepressor responses, which were inhibited during the infusion 3.1 μg/kg·min DHE (see Fig. 1b); and (ii) i.v. bolus injections of exogenous α-CGRP elicited dose-dependent vasodepressor responses, but these responses, unlike those by electrical stimulation, remained unchanged during the infusion of 3.1 μg/kg·min DHE Fig. 1c). In view that 3.1 μg/kg·min DHE inhibited the electrically-induced vasodepressor responses without affecting those by exogenous α-CGRP, we considered this infusion dose of DHE for further pharmacological analysis. In all cases, the vasodepressor responses to electrical stimulation or exogenous α-CGRP: (i) appeared about 10 s after starting each electrical stimulus or dose of α-CGRP, and reached a maximum 1 min after the stimulus had ended; and (ii) returned to baseline levels within 5–10 min after each stimulus/dose, as previously reported [18].
Effect per se of saline, rauwolscine, GR127935 or haloperidol (given separately or in combination) on the neurogenic vasodepressor responses during an infusion of methoxamine
During the methoxamine infusion (control; 20 μg/kg·min), the vasodepressor responses to electrical stimulation in control animals did not significantly differ from those elicited in animals pre-treated (see Additional file 1: Figure S1 [S1]) with an i.v. bolus injection of: (i) vehicle (1 ml/kg; Additional file 1: Figure S1a); (ii) rauwolscine (α2-drenoceptor antagonist, 310 μg/kg; Additional file 1: Figure S1b); (iii) GR127935 (5-HT1B/1D receptor antagonist, 31 μg/kg; Additional file 1: Figure S1a); (iv) haloperidol (D2-like receptor antagonist, 310 μg/kg; Additional file 1: Figure S1d); (v) rauwolscine+GR127935 (310 and 31 μg/kg respectively; Additional file 1: Figure S1e); (vi) rauwolscine+ haloperidol (310 μg/kg each; Additional file 1: Figure S1f); and (vii) GR127935+ haloperidol (31 and 310 μg/kg respectively; Additional file 1: Figure S1g). These results indicate that these compounds, at the doses used and under the present experimental conditions, were essentially devoid of any effect per se on the electrically-induced vasodepressor responses.
Effect of saline, rauwolscine, GR127935 or haloperidol (given separately or in combination) on DHE-induced inhibition of the neurogenic vasodepressor responses
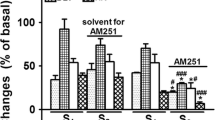
Figure 2 shows that the inhibition induced by DHE (3.1 μg/kg·min) of the electrically-induced vasodepressor responses, which remained unaltered in animals pretreated with vehicle (1 ml/kg; Fig. 2a), was: (i) abolished in animals pretreated with rauwolscine+GR127935 (310 and 31 μg/kg respectively; Fig. 2e); and (ii) resistant to blockade in animals pretreated with rauwolscine (310 μg/kg; Fig. 2b); GR127935 (31 μg/kg; Fig. 2c); haloperidol (310 μg/kg; Fig. 2d); rauwolscine+haloperidol (310 and 310 μg/kg each; Fig. 2f); or GR127935+ haloperidol (31 and 310 μg/kg respectively; Fig. 2g).

Effect of i.v. bolus injections of: (a) saline (1 ml/kg); (b) rauwolscine (310 μg/kg); (c) GR127935 (31 μg/kg); or (d) haloperidol (310 μg/kg) given separately, as well as the combinations (e) rauwolscine plus GR127935 (310 and 31 μg/kg, respectively); (f) rauwolscine plus haloperidol (310 μg/kg each); or (g) GR127935 plus haloperidol (31 and 310 μg/kg, respectively) on the inhibition induced by dihydroergotamine (DHE; 3.1 μg/kg·min; □) of the electrically-induced vasodepressor responses. The control responses (○) represent that of animals receiving an i.v. continuous infusion of methoxamine (20 μg/kg·min) which is shown for comparison. * Significantly different responses (P < 0.05) vs. control
Discussion
General
Apart from the implications discussed below, our study confirms that DHE can inhibit the vasodepressor sensory CGRPergic outflow in pithed rats by prejunctional mechanisms, as previously reported by Lozano-Cuenca et al. [16]. However, these authors made no attempt to identify the pharmacological profile of receptors involved in such inhibition by DHE. Hence, by using antagonists for α2-adrenoceptors (rauwolscine), 5-HT1B/1D receptors (GR127935) and D2-like receptors (haloperidol) (since DHE displays affinity for these receptors; see Table 1), the present study suggests that α2-adrenoceptors and 5-HT1B/1D receptors (but not D2-like receptors) are involved in the prejunctional mechanisms by which DHE inhibits the vasodepressor sensory CGRPergic outflow in pithed rats.
Moreover, it is important to note that we did not measure sensory nerve activity directly, but the electrically-induced CGRP release in the systemic vasculature could be estimated indirectly by measurement of the evoked vasodepressor response, as previously established using the CGRP receptor antagonists CGRP8–37 [12] and olcegepant [24]. Hence, the inhibition by DHE was considered sensory-inhibitory since this ergot inhibited the vasodepressor responses induced by spinal (T9-T12) stimulation of the vasodepressor sensory CGRPergic outflow (Fig. 1b), without affecting those by exogenous α-CGRP (Fig. 1c).
Systemic haemodynamic effects produced by methoxamine and DHE
As previously established in pithed rats [16, 18,19,20,21], the artificial and sustained increase in diastolic blood pressure (at around 140 mmHg) by a continuous infusion of the α1-adrenoceptor agonist methoxamine (20 μg/kg·min; Fig. 1a) is a conditio sine qua non for inducing vasodepressor responses. Otherwise, the basal blood pressure in pithed rats is so low that there is no “window” for eliciting further decreases in this variable. The methoxamine-induced increase in diastolic blood pressure has been attributed to an increase in peripheral vascular resistance [25]. In addition, it is noteworthy that 3.1 μg/kg·min DHE can slightly increase diastolic blood pressure when the methoxamine infusion is not given (i.e. when basal diastolic blood pressure is too low; data not shown). Accordingly, the methoxamine-induced increase in blood pressure, which is maximal [16, 18], could most probably have masked the slight effect of DHE on this variable. In fact, the pressor effect of DHE has been extensively described in humans [26, 27], and its pressor effect in pithed rats has recently been associated with vascular activation of α1 (α1A, α1B and α1D) and α2 (α2A, α2B and α2C)-adrenoceptors [9].
Effects of several antagonists per se on systemic haemodynamic variables and on the sensory-induced vasodepressor responses
To identify the mechanisms involved in the prejunctional inhibition by DHE (Fig. 1b and c), we decided to evaluate the effect of several antagonists per se (Table 1) on systemic haemodynamic conditions and on the vasodepressor responses induced by electrical stimulation. A transient, but significant, decrease in diastolic blood pressure was observed when animals received a bolus injection of rauwolscine and/or haloperidol (Table 2). In the case of haloperidol, this effect could be explained by considering that this compound exhibits high affinity for α1-adrenoceptors (pK i : 8.0; see Table 1). Thus, it is tempting to suggest that haloperidol may have an antagonistic effect on methoxamine (α1-adrenoceptor agonist)-induced increase in blood pressure. In contrast, we have no clear-cut explanation for the decreases in diastolic blood pressure induced by rauwolscine, which does not display affinity for α1-adrenoceptors. Nevertheless, in all cases, 10 min after administration of antagonists the values of diastolic blood pressure had returned to baseline values (Table 2; before and 10 min after). These results, coupled to the lack of effect of the above antagonists (alone or in combination) on the electrically-induced vasodepressor responses (see Additional file 1: Figure S1) indicates that these compounds, at the doses used, were devoid of any effects per se on the above variables. Accordingly, these data suggest that any effect of a given antagonist on DHE-induced sensory inhibition is due to a direct interaction of the antagonist with its respective receptors. It must be emphasized that: (i) our suggestion supporting and/or excluding the role of α2-adrenergic, 5-HT1B/1D or D2-like receptors is based on the assumption that species differences between the binding of agonists and antagonists used do not play a major role (Table 1); and (ii) the doses of antagonists used were high enough to completely block prejunctional α2-adrenoceptors (rauwolscine; [18]), 5-HT1B/1D receptors (GR127935; [19, 20, 28, 29]) and D2-like receptors (haloperidol; [21]) mediating inhibition of neurogenic cardiovascular responses in pithed rats.
Role of α2-adrenergic and 5-HT1B/1D, but not D2-like, receptors in the inhibition by DHE
As previously pointed out, DHE displays affinity for α2-adrenergic, 5-HT1 and D2-like receptors (see Table 1). Activation of these receptors, which are coupled to G i/o proteins, may inhibit adenylyl cyclase activity, inactivate Ca2+ channels and/or activate inwardly rectifying K+ channels [30, 31]. These are signal transduction systems usually associated with inhibition of neurotransmitter release [30, 31]. With this idea in mind and considering our results (Fig. 2), the simplest interpretation of these findings suggests that DHE-induced inhibition mainly involves the activation of prejunctional α2-adrenergic and 5-HT1B/1D receptors, but not of D2-like receptors since the DHE response was: (i) only abolished by rauwolscine plus GR127935 (Fig. 2e); and (ii) resistant to blockade by rauwolscine (Fig. 2b), GR127935 (Fig. 2c), haloperidol (Fig. 2d), rauwolscine plus haloperidol (Fig. 2f) or GR127935 plus haloperidol (Fig. 2g). However, the lack of blockade by some of the above treatments deserves further considerations. For example, the fact that rauwolscine or GR127935 alone failed to block DHE-induced inhibition may reflect the fact that a maximal dose of DHE was used [16]; accordingly, DHE could be stimulating α2-adrenoceptors and 5-HT1B/1D receptors simultaneously; thus, when blocking only one of these receptors, the inhibition produced by the unblocked receptor will overshadow the antagonism produced on the other receptor. In addition, the involvement of D2-like receptors seems unlikely based on the lack of effect of haloperidol, an antagonist with high affinity (pK i ) for the D2-like receptors subtypes (D2: 9.4; D3: 8.5 and D4: 8.8; see Table 1). This suggestion gains weight when considering that DHE-induced inhibition remained unaffected after rauwolscine plus haloperidol (Fig. 2f) or GR127935 plus haloperidol (Fig. 2g).
Having established the main involvement of rauwolscine-sensitive α2-adrenoceptors and GR127935-sensitive 5-HT1B/1D receptors in DHE-induced inhibition, we have to recognize that no attempt was made here to further identify the specific subtypes of these main receptor families. The reason for this omission is based on the fact that we have previously shown (using selective agonists and antagonists) that these receptors correlate with the pharmacological profile of, respectively: (i) α2A/2C (but not α2B)-adrenoceptor subtypes [18]; and (ii) 5-HT1B and 5-HT1F (but not 5-HT1A or 5-HT1D) receptor subtypes [19, 20]. However, the fact that DHE-induced inhibition was abolished by the combination rauwolscine (310 μg/kg) + GR127935 (31 μg/kg), where the latter dose is not enough to block the prejunctional 5-HT1F receptors that inhibit the rat vasodepressor sensory CGRPergic outflow [20], makes the role of these subtypes rather unlikely. Finally, it is noteworthy that DHE also displays moderate affinity for other receptors, including the 5-ht1E (pK i : 6.2) subtype (Table 1). However, the 5-ht1E retains its lower-case appellation as it is not a functional receptor [32].
Conclusion
The above results suggest that DHE-induced inhibition of the vasodepressor sensory CGRPergic outflow is mainly mediated by prejunctional activation of rauwolscine-sensitive α2-adrenoceptors and GR127935-sensitive 5-HT1B/1D receptors which, most likely, correlate with α2A/2C-adrenoceptors [18] and 5-HT1B receptors [19], respectively. These findings may shed further light on the vascular side effects produced by DHE, namely: DHE-induced inhibition of the perivascular sensory CGRPergic outflow may facilitate DHE’s vasoconstrictor properties resulting in an increased vascular resistance.
Abbreviations
- ARRIVE:
-
Animal Research: Reporting In Vivo Experiments
- DHE:
-
Dihydroergotamine
- i.p.:
-
Intraperitoneal
- i.v.:
-
Intravenous
- PPG:
-
Propylene glycol
- α-CGRP:
-
α-Calcitonin gene-related peptide
References
Dahlöf C, MaassenVanDenBrink A (2012) Dihydroergotamine, ergotamine, methysergide and sumatriptan–basic science in relation to migraine treatment. Headache 52:707–714
Saper JR, Silberstein S (2006) Pharmacology of dihydroergotamine and evidence for efficacy and safety in migraine. Headache 46:S171–S181
Silberstein SD, McCrory DC (2003) Ergotamine and dihydroergotamine: history, pharmacology and efficacy. Headache 43:144–166
Villalón CM, Centurión D, Willems EW, Arulmani U, Saxena PR, Valdivia LF (2004) 5-HT1B receptors and alpha2A/2C-adrenoceptors mediate external carotid vasoconstriction to dihydroergotamine. Eur J Pharmacol 484:287–290
Buzzi MG, Carter WB, Shimizu T, Heath H III, Moskowitz MA (1991) Dihydroergotamine and sumatriptan attenuate levels of CGRP in plasma in rat superior sagital sinus during electrical stimulation of the trigeminal ganglion. Neuropharmacology 30:1193–1200
Goadsby PJ, Edvinsson L (1993) The trigeminovascular system and migraine: studies characterizing cerebrovascular and neuropeptide changes seen in humans and cats. Ann Neurol 33:48–56
Marichal-Cancino BA, González-Hernández A, Manrique-Maldonado G, Ruiz-Salinas II, Altamirano-Espinoza AH, MaassenVanDenBrink A, Villalon CM (2012) Intrathecal dihydroergotamine inhibits capsaicin-induced vasodilatation in the canine external carotid circulation via GR127935-and rauwolscine-sensitive receptors. Eur J Pharmacol 692:69–77
Price TJ, Hargreaves KM, Cervero F (2006) Protein expression and mRNA cellular distribution of the NKCC1 cotransporter in the dorsal root and trigeminal ganglia of the rat. Brain Res 1112:146–158
Rivera-Mancilla E, Avilés-Rosas VH, Manrique-Maldonado G, Altamirano-Espinoza AH, Villanueva-Castillo B, MaassenVanDenBrink A, Villalón CM (2017) The role of α1- and α2-adrenoceptor subtypes in the vasopressor responses induced by dihydroergotamine in ritanserin-pretreated pithed rats. J Headache Pain 18:104
Deng PY, Li YJ (2005) Calcitonin gene-related peptide and hypertension. Peptides 26:1676–1685
González-Hernández A, Marichal-Cancino BA, Lozano-Cuenca J, López-Canales JS, Muñoz-Islas E, Ramírez-Rosas MB, Villalón CM (2016) Heteroreceptors modulating CGRP release at neurovascular junction: potential therapeutic implications on some vascular related diseases. Biomed Res Int 2016:2056786
Taguchi T, Kawasaki H, Imamura T, Takasaki K (1992) Endogenous calcitonin gene-related peptide mediates nonadrenergic noncholinergic depressor response to spinal cord stimulation in the pithed rats. Circ Res 71:357–364
Smillie SJ, Brain SD (2011) Calcitonin gene-related peptide (CGRP) and its role in hypertension. Neuropeptides 45:93–104
Smillie SJ, King R, Kodji X, Outzen E, Pozsgai G, Fernandes E, Marshall N, De Winter P, Heads RJ, Dessapt-Baradez C, Gnudi L (2014) An ongoing role of α-calcitonin gene–related peptide as part of a protective network against hypertension, vascular hypertrophy, and oxidative stress. Hypertension 63:1056–1062
González-Hernández A, Marichal-Cancino BA, MaassenVanDenBrink A, Villalón CM (2018) Side effects associated with current and prospective antimigraine pharmacotherapies. Exp Opin Drug Metab Toxicol 14:25–41
Lozano-Cuenca J, González-Hernández A, Muñoz-Islas E, Sánchez-López A, Centurión D, Cobos-Puc LE, Villalón CM (2009) Effect of some acute and prophylactic antimigraine drugs on the vasodepressor sensory CGRPergic outflow in pithed rats. Life Sci 84:125–131
Sanders-Bush E, Mayer SE (2006) 5-hydroxytryptamine (serotonin): receptor agonists and antagonists. In: Brunton LL, Lazo JS, Parker KL (eds) Goodman & Gilman’s the pharmacological basis of therapeutics, 11th edn. Mc Graw Hill, New York, pp 297–316
Villalón CM, Albarrán-Juárez JA, Lozano-Cuenca J, Pertz HH, Görnemann T, Centurión D (2008) Pharmacological profile of the clonidine-induced inhibition of vasodepressor sensory outflow in pithed rats: correlation with α2A/2C-adrenoceptors. Br J Pharmacol 154:51–59
González-Hernández A, Muñoz-Islas E, Lozano-Cuenca J, Ramírez-Rosas MB, Sánchez-López A, Centurión D, Ramírez-San Juan E, Villalón CM (2010) Activation of 5-HT1B receptors inhibits the vasodepressor sensory CGRPergic outflow in pithed rats. Eur J Pharmacol 637:131–137
González-Hernández A, Manrique-Maldonado G, Lozano-Cuenca J, Muñoz-Islas E, Centurión D, MaassenVanDenBrink A, Villalón CM (2011) The 5-HT1 receptors inhibiting the rat vasodepressor sensory CGRPergic outflow: further involvement of 5-HT1F, but not 5-HT1A or 5-HT1D, subtypes. Eur J Pharmacol 659:233–243
Manrique-Maldonado G, González-Hernández A, Altamirano-Espinoza AH, Marichal-Cancino BA, Ruiz-Salinas I, Villalón CM (2014) The role of prejunctional D2-like receptors mediating quinpirole-induced inhibition of the vasodepressor sensory CGRPergic out-flow in pithed rats. Basic Clin Pharmacol Toxicol 114:174–180
Gillespie JS, MacLaren A, Pollock D (1970) A method of stimulating different segments of the autonomic outflow from the spinal column to various organs in the pithed cat and rat. Br J Pharmacol 40:257–267
Kleinman L, Radford E (1964) Ventilation standards for small mammals. J Appl Physiol 19:360–362
Avilés-Rosas VH, Rivera-Mancilla E, Marichal-Cancino BA, Manrique-Maldonado G, Altamirano-Espinoza AH, MaassenVanDenBrink A, Villalón CM (2017) Olcegepant blocks neurogenic and non-neurogenic CGRPergic vasodepressor responses and facilitates noradrenergic vasopressor responses in pithed rats. Br J Pharmacol 174:2001–2014
Decker N, Ehrhardt JD, Leclerc G, Schwartz J (1984) Postjunctional α-adrenoceptors: α1 and α2 subtypes in rat vasculature in vitro and in vivo. Naunyn Schmiedeberg’s Arch Pharmacol 326:1–6
Saxena PR, Den Boer MO (1991) Pharmacology of antimigraine drugs. J Neurol 238:S28–S35
Tfelt-Hansen PC, Koehler PJ (2008) History of the use of ergotamine and dihydroergotamine in migraine from 1906 and orward. Cephalalgia 28:877–886
Cobos-Puc LE, Villalón CM, Sánchez-López A, Ramírez-Rosas MB, Lozano-Cuenca J, Pertz HH, Gömermann T, Centurión D (2009) Pharmacological characterization of ergotamine-induced inhibition of the cardioaccelerator sympathetic outflow in pithed rats. Naunyn Schmiedeberg's Arch Pharmacol 379:137–148
Sánchez-López A, Centurión D, Vázquez E, Arulmani U, Saxena PR, Villalón CM (2004) Further characterization of the 5-HT1 receptors mediating cardiac sympatho-inhibition in pithed rats: pharmacological correlation with the 5-HT1B and 5-HT1D subtypes. Naunyn Schmiedeberg's Arch Pharmacol 369:220–227
Boehm S, Kubista H (2002) Fine tuning of sympathetic transmitter release via ionotropic and metabotropic presynaptic receptors. Pharmacol Rev 54:43–99
De Jong AP, Verhage M (2009) Presynaptic signal transduction pathways that modulate synaptic transmission. Curr Opin Neurobiol 19:245–253
Alexander SPH, Christopoulos A, Davenport AP, Kelly E, Marrion NV, Peters JA, Faccenda E, Harding SD, Pawson AJ, Sharman JL, Southan C, Davies JA (2017) CGTP collaborators - the concise guide to PHARMACOLOGY 2017/18: G protein-coupled receptors. Br J Pharmacol 174(S1):S17–S129
Leysen JE, Gommeren W, Heylen L, Luyten WH, Van de Weyer I, Vanhoenacker P, Schotte A, Van Gompel P, Wouters R, Lesage AS (1996) Alniditan, a new 5-hydroxytryptamine1D agonist and migraine-abortive agent: ligand-binding properties of human 5-hydroxytryptamine1D alpha, human 5-hydroxytryptamine1D beta, and calf 5-hydroxytryptamine1D receptors investigated with [3H]5-hydroxytryptamine and [3H]alniditan. Mol Pharmacol 50:1567–1580
Jasper JR, Lesnick JD, Chang LK, Yamanishi SS, Chang TK, Hsu SA, Daunt DA, Bonhaus DW, Eglen RM (1998) Ligand efficacy and potency at recombinant alpha2 adrenergic receptors: agonist-mediated [35S]GTPgammaS binding. Biochem Pharmacol 55:1035–1043
Uhlén S, Porter AC, Neubig RR (1994) The novel alpha-2 adrenergic radioligand [3H]-MK912 is α2C selective among human α2A, α2B and α2C adrenoceptors. J Pharmacol Exp Ther 271:1558–1565
Pauwels PJ (1996) Pharmacological properties of a putative 5-HT1B/1D receptor antagonist GR127935. CNS Drug Rev 2:415–428
Adham N, Kao HT, Schechter LE, Bard J, Olsen M, Urquhart D, Durkin M, Hartig PR, Weinshank RL, Branchek TA (1993) Cloning of another human serotonin receptor (5-HT1F): a fifth 5-HT1 receptor subtype coupled to the inhibition of adenylate cyclase. Proc Natl Acad Sci U S A 90:408–412
Beer MS, Heald MA, McAllister G, Stanton JA (1998) Pharmacological characterisation of a cloned dog 5-HT1B receptor cell line. Eur J Pharmacol 360:117–121
Price GW, Burton MJ, Collin LJ, Duckworth M, Gaster L, Göthert M, Jones BJ, Roberts C, Watson JM, Middlemiss DN (1997) SB-216641 and BRL-15572-compounds to pharmacologically discriminate h5-HT1B and h5-HT1D receptors. Naunyn Schmiedeberg's Arch Pharmacol 356:312–320
Arnt J, Skarsfeldt T (1998) Do novel antipsychotics have similar pharmacological characteristics? A review of the evidence. Neurophsychopharmacol 18:63–101
Millan MJ, Brocco M, Rivet JM, Audinot V, Newman-Tancredi A, Maiofiss L, Queriaux S, Despaux N, Peglion JL, Dekeyne A (2000) S18327 (1-[2-[4-(6-fluoro-1, 2-benzisoxazol-3-yl)piperid-1-yl]ethyl]3-phenyl imidazolin-2-one), a novel, potential antipsychotic displaying marked antagonist properties at α1- and α2-adrenergic receptors: II. Functional profile and a multiparametric comparison with haloperidol, clozapine, and 11 other antipsychotic agents. J Pharmacol Exp Ther 292:54–66
Acknowledgements
The authors would like to thank Mr. Arturo Contreras, Mr. Mauricio Villasana and Engr. José Rodolfo Fernández Calderón for their assistance. We are also indebted to the pharmaceutical companies for their generous gifts (see Drugs Section).
Funding
This work was sponsored by Consejo Nacional de Ciencia y Tecnología (CONACyT, Mexico City, Grant No. 219707 for CMV) and the Netherlands Organization for Scientific Research (NWO; VIDI 917.11.349 for AMVDB).
Availability of data and materials
All relevant data are within the paper. Moreover, a Figure with control results can be found as supplemental material.
Author information
Authors and Affiliations
Contributions
AGH – Performed the experiments, analysed the data and drafted the manuscript. JLC – Technical assistance during the experiments, analysed the data and drafted the manuscript. BAMC – Revised and approved the final manuscript. AMVDB – Revised and approved the final manuscript. CMV – Supervised the experiments and data analysis, drafted and revised the final manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval
The present investigation was approved by our Institutional Ethics Committee (CICUAL Cinvestav; protocol no. 507–12), and followed the regulations established by the Mexican Official Norm (NOM-062-ZOO-1999), in accordance with ARRIVE reporting guidelines for the care and use of laboratory animals.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1:
Figure S1. Effect per se of i.v. bolus injections of: (a) saline (1 ml/kg); (b) rauwolscine (310 μg/kg); (c) GR127935 (31 μg/kg); or (d) haloperidol (310 μg/kg) given separately; as well as the combinations (e) rauwolscine plus GR127935 (310 and 31 μg/kg, respectively); (f) rauwolscine plus haloperidol (310 μg/kg each); or (g) GR127935 plus haloperidol (31 and 310 μg/kg, respectively) on the electrically-induced vasodepressor responses produced during an i.v. continuous infusion of methoxamine (20 μg/kg. min) (n = 5 for each group). No significant effects were produced after administration of compounds (P > 0.05). (PDF 881 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
González-Hernández, A., Lozano-Cuenca, J., Marichal-Cancino, B.A. et al. Dihydroergotamine inhibits the vasodepressor sensory CGRPergic outflow by prejunctional activation of α2-adrenoceptors and 5-HT1 receptors. J Headache Pain 19, 40 (2018). https://doi.org/10.1186/s10194-018-0869-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s10194-018-0869-8