
Abstract
Pituitary adenylate cyclase activating polypeptide (PACAP) is an ubiquitous peptide involved, among others, in neurodevelopment, neuromodulation, neuroprotection, neurogenic inflammation and nociception. Presence of PACAP and its specific receptor, PAC1, in the trigeminocervical complex, changes of PACAP levels in migraine patients and the migraine-inducing effect of PACAP injection strongly support the involvement of PACAP/PAC1 receptor in migraine pathogenesis. While antagonizing PAC1 receptor is a promising therapeutic target in migraine, the diverse array of PACAP’s functions, including protection in ischemic events, requires that the cost-benefit of such an intervention is well investigated by taking all the beneficial effects of PACAP into account. In the present review we summarize the protective effects of PACAP in ischemia, especially in neuronal ischemic injuries, and discuss possible points to consider when developing strategies in migraine therapy interfering with the PACAP/PAC1 receptor system.
Similar content being viewed by others

Introduction
PACAP is an ubiquitous peptide discovered almost three decades ago [1], and it has been described to be involved in neurodevelopment, neuromodulation, neuroprotection, neurogenic inflammation and nociception [2]. It belongs to the vasoactive intestinal peptide (VIP)/glucagon/growth hormone releasing factor/secretin superfamily [2] and is encoded by the ADCYAP1 gene, located on chromosome 18, which expresses a proprotein that is further processed into multiple mature peptides. Alternative splicing results in multiple transcript variants, including two forms that contain either 27 or 38 amino acids (PACAP27 and PACAP38). Since in mammals PACAP38 is the most prevalent form [3], in this review PACAP38 will be referred to simply as PACAP unless stated otherwise.
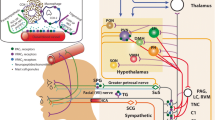
PACAP exerts its functions through the activation of three different G-protein coupled receptors (GPCRs): VPAC1, VPAC2 and PAC1. While VPAC1 and VPAC2 receptors are coupled to Gs proteins and show similar affinity for VIP, the PAC1 receptor has a 100-fold selectivity for PACAP27 and PACAP38 over VIP, leading to the activation of adenylate-cyclase and phospholipase C signaling transduction pathways [4].
In the central nervous system (CNS), PACAP has been described in the pituitary, thalamus, hypothalamus, hippocampus, locus coeruleus, periaqueductal grey area, the dorsal horn of the spinal cord and in astrocytes [5,6,7,8,9,10,11,12,13,14]. Of special interest, PACAP is expressed in the trigeminal nucleus caudalis (TNC) and trigeminal ganglia [15], which could suggest a possible role for PACAP in migraine pathogenesis. In rats, injection of PACAP into the paraventricular nucleus of the hypothalamus increases the activity of the TNC, which can be reverted by administration of the PAC1 receptor antagonist [16], and intrathecal injection of PACAP induces hyperalgesia [8]. PACAP plasma levels in migraineurs are elevated during a migraine attack, in comparison with the interictal levels [17]. Most importantly, if injected peripherally to migraineurs, PACAP is able to induce an immediate headache in 90% of the cases, that is followed by a delayed migraine-like headache in almost 60% of the subjects; conversely, only 15% of the healthy controls experience the delayed migraine-like headache [15]. These findings are similar to those obtained after peripheral administration of calcitonin gene-related peptide (CGRP) [18]. Interestingly, PACAP is a weaker dilator of the human meningeal artery when compared to VIP [19]. Since VIP was earlier reported not to induce migraine-like headaches [20], this could suggest that the role of PACAP in migraine is probably through modulation of the trigeminocervical complex via the PAC1 receptor.
In view of the suggested role of PACAP, but not VIP, in migraine, an antibody against the PAC1 receptor (AMG 301) has been developed for the treatment of migraine (Clinical trials identifier: NCT03238781). In preclinical studies, AMG 301 has been shown to inhibit stimulus-evoked nociceptive activity in the TNC and the results are comparable to the inhibition observed with sumatriptan, supporting the role of PAC1 receptor in migraine pathophysiology. However, it is important to consider the ubiquitous nature of PACAP and its receptors, since they have also been described to be widely expressed in the periphery, such as in the thyroid and parathyroid glands, lungs, pancreas, liver, colon, stomach and blood vessels [3, 11, 21,22,23,24,25]; thus they participate in several respiratory, gastrointestinal, reproductive and cardiovascular (patho) physiological processes [2] and, as it will be discussed, play a significant role in the homeostatic responses to ischemic events [26,27,28,29,30,31,32], Table 1.
Review
PACAP in brain ischemia
PACAP has been shown to be neuroprotective in vitro in different neuronal cultures against various toxic insults and in models of neuronal injuries in vivo [33, 34]. Numerous in vivo data have been published showing its protective actions in cerebral ischemia [33, 35]. The first proof for the in vivo neuroprotective effect came from a rat global ischemia study, where intravenous or intracerebroventricular (icv) PACAP administration reduced hippocampal neuronal loss [36]. This was achieved via suppression of JNK and p38, while stimulation of ERK activity [37,38,39]. These observations were followed by studies demonstrating that PACAP was also effective in transient and permanent focal ischemia in rats and mice induced by middle cerebral artery occlusion (MCAO) [27, 40,41,42,43,44].
Subsequent studies provided further details on the neuroprotective mechanisms. Anti-apoptotic and anti-inflammatory actions seem to be the main protective mechanisms in PACAP’s actions in rat and mouse models of cerebral ischemia. PACAP decreased apoptosis in the ischemic penumbra [45], inhibited expression of bcl-2-associated death promoter, caspase-3, macrophage inflammatory protein-1alpha, inducible nitric oxide synthase2, tumor necrosis factor-(TNF) alpha mRNAs and increased ERK2, bcl-2 and IL-6 [40, 41, 46]. Decreased inflammatory response was also found after post-stroke PACAP-producing stem cell transplantation, where numerous chemokines as well as TNF, NFkappaB and IL-1 decreased [47]. In brain cortical neurons subjected to oxygen-glucose deprivation and reoxygenation, PACAP induced neuronal protection by both direct actions through PAC1 receptor, and indirect pathways via neurotrophin release, activation of trkB receptors and attenuation of neuronal growth inhibitory signaling molecules p75NTR and Nogo receptor [41]. In addition, PACAP induced apurinic/apyrimidinic endonuclease APE1 in hippocampal neurons that can be an additional factor reducing DNA stress and hippocampal CA1 neuronal death in global ischemia [48]. In mouse MCAO, several genes were affected in the ischemic core and penumbra after PACAP treatment [49,50,51,52]. Among the upregulated genes was IL-6, which was strongly induced during the critical first 24 h, suggesting a relationship between PACAP and IL-6 in accordance with previous findings by Ohtaki and co-workers [40]. Several other cytokines and growth factors were altered in a region-specific and time-dependent fashion after post-ischemic PACAP treatment, such as brain derived neurotrophic factor [50, 51]. Whether alterations of these factors are consequences of PACAP reducing infarct volume by other mechanisms or represent a causative factor is not known at the moment. Only in case of IL-6, it has been proven that PACAP failed to improve ischemic lesion in IL-6-deficient mice, showing the causative role of IL-6 in PACAP-mediated neuroprotection in mice [40]. Numerous further factors playing a role in neuronal defense, axonal growth and development were also modified after ischemia [52]. A relationship between hypoxia inducible factor (HIF) and PACAP was described in several studies in different experimental paradigms [53,54,55]. Under in vitro and in vivo hypoxic conditions, HIF1-alpha activation upregulated PACAP, which in turn activated PAC1 receptor [56]. Although PACAP reduced HIF1-alpha expression in a model of diabetic retinopathy 2 weeks after the treatment, bone marrow-derived stem cells homing into the ischemic brain was also facilitated by a recently described HIF1-alpha-activated PACAP38-PAC1 signaling process [55]. A detailed time-dependent analysis of PACAP’s effect on cerebral HIF1 expression could clarify the role of this pathway in PACAP-induced neuroprotection in ischemia. Analogs of PACAP were also tested in focal ischemic models. In a study of ischemia/reperfusion injury, a potent metabolically stable PACAP38 analog [acetyl-(Ala15, Ala20) PACAP38-propylamide] led to the same degree of protection as native PACAP38 [46]. This is an important finding, as one of the limitations of PACAP’s therapeutic use is its poor stability. However, according to these data enhancing its plasmatic half-life did not lead to an increase of its neuroprotective potential [46], but analogs might have less vasomotor side effects, as described in another study [57].
As far as functional recovery is concerned, PACAP is able to improve functional deficits in association with the morphological amelioration in stroke models. In rat permanent focal cerebral ischemia, PACAP improved certain sensorimotor deficits, such as reaction times to body surface touch [27]. Another study further supported this in a transient MCAO, evaluating neurological impairment by degree of limb flexion, grasping and symmetry of movements [46]. In a permanent focal ischemia model, PACAP-producing stem cells transplanted icv 3 days after stroke promoted functional recovery even when given beyond the therapeutic window for structural recovery [47].
PACAP is known to cross the blood-brain barrier (BBB), but it is still questionable whether the rate is sufficient to lead to effects in the brain under physiological or pathological conditions [2, 38]. Although ischemic conditions change region-specific crossing, it is suggested that the passage is sufficient enough to induce neuroprotection in ischemic brains [58]. Antisenses inhibit efflux pumps of the BBB, and could inhibit PACAP27 efflux and reduce the infarct size in mouse focal ischemia [59]. Regarding changes in cerebral blood flow, in some studies PACAP increased cerebral blood flow in ischemic conditions, while in others no change or even decrease was found [27, 46, 60]. PACAP has potent vasodilatory effects, which can also be included in the pathomechanism of migraine [61,62,63]. However, given the contradictory data on cerebral blood flow after PACAP treatment, it remains unknown at the moment whether this effect plays a role in post-ischemic neuroprotection.
The role of endogenous PACAP was suggested by upregulation of PACAP signaling in different ischemia models and from knockout studies (Table 1). In a gerbil model of global ischemia, decrease in PACAP expression was followed by an increase 5 days later. This was accompanied by increases in PAC1 receptor expression in the vulnerable CA1 region, in contrast to the more resistant CA3 area, where PACAP expression did not change [36, 64]. Upregulation of PAC1 receptor could also be observed after focal ischemia [65, 66]. A massive upregulation of PACAP was found in peri-infarct regions [67]. In a rat global ischemia model, moderate PAC1 mRNA decrease was observed throughout the hippocampus, while granule cells showed increased PACAP expression [68]. It was suggested that the altered PACAP and PAC1 receptor expression might play a role in regulated neurogenesis after stroke [68]. In mouse hippocampal astrocytes, PAC1 receptor expression was increased 7 days after stroke, suggesting an important role of PACAP in reactive astrocytes [69, 70]. Further evidence for the endogenous protection by PACAP came from studies using PACAP deficient mice. Hetero- and homozygous PACAP knockout animals had increased infarct volume with increased edema formation and more severe neurological deficits after MCAO, and these could be ameliorated by PACAP injection [40, 71]. Furthermore, cytochrome-c release was higher, while mitochondrial bcl-2 was lower in mice lacking PACAP. It was also suggested that these protective effects could be mediated in part by IL-6 [40]. Endogenous PACAP also promotes hippocampal neurogenesis after stroke, as proliferation of neuronal stem cells in the subgranular zone of the hippocampus was found to be increased in wild type mice, but not in PACAP heterozygous animals [72].
The few available human data also support that PACAP might play a role in ischemic neuronal conditions. It was hypothesized that the elevated blood PACAP levels may reflect an increased leakage into circulation or an overproduction of PACAP as a pathological response to the loss of neural tissue in the CNS and it might be associated with the neuroprotective effects of the neuropeptide [73]. Plasma PACAP concentrations were higher in patients after acute spontaneous basal ganglia and aneurysmal subarachnoid hemorrhages than in healthy control subjects [73, 74]. Positive association was shown between PACAP levels and neurological score, as well as with hematoma volume. Patients, who died within the first week after admission, had higher PACAP levels and overall survival times were shorter in individuals with high PACAP concentrations [73, 74]. It is suggested that PACAP could be a good prognostic predictor in hemorrhage patients. These studies suggest that PACAP can be an independent predictor of survival and a potential prognostic biomarker of brain hemorrhage.
PACAP in retinal ischemia
PACAP is considered to be a potent neuroprotective peptide with potential therapeutic use also in retinal diseases [34, 75,76,77,78,79]. Similarly to models of cerebral ischemia, protective effects have been described in animal models of retinal ischemia. Intravitreal injection of PACAP38 or PACAP27 following bilateral common carotid artery occlusion in rats preserved the thickness of all retinal layers and reduced cell loss in the ganglionic layer. Immunohistochemistry demonstrated that PACAP rescued fully or partially several retinal cell types from ischemia-induced damage. The PACAP antagonist PACAP6–38 could block these protective effects [76, 80]. Electroretinography showed that ischemia caused functional loss in the retina, whereas PACAP treatment resulted a preserved retinal function [81]. Endogenous PACAP had similar protective effects, as knockout mice were more susceptible to retinal ischemic injury [82]. Efficacy of PACAP was also shown in another retinal ischemia model induced by high intraocular pressure, which could be blocked by a cAMP antagonist [83]. Testing possible therapeutic effects of various PACAP fragments and analogues, and three related peptides (VIP, secretin, glucagon) revealed that the most effective forms were PACAP38 and PACAP27, while the other fragments had either no effects or slight antagonistic effects [84, 85]. Related peptides had no effect except for VIP, which was retinoprotective at concentrations ten times higher than it is required for PACAP [84, 86]. Recent results have shown that PACAP38 and 27 are able to cross the ocular barriers and exert retinoprotective effects in ischemia even when given in form of eye drops [87, 88], providing the basis for an easy route of future therapeutic use.
Examining the protective mechanisms in retinal hypoperfusion, several studies have revealed possible signaling pathways resulting in neuroprotection. Another study investigated possible receptorial mechanisms. All three PACAP receptors (PAC1, VPAC1, VPAC2) are expressed in the retina, with PAC1 receptor showing dominant role in the retinoprotective effects [34]. Our research group confirmed the involvement of PAC1 receptors in the PACAP-induced retinoprotection using a selective PAC1 receptor agonist maxadilan in permanent common carotid artery ligation. Maxadilan rescued retinal layers from ischemia-induced degeneration and decreased expression of cytokines such as CINC-1, IL-1α, and L-selectin [89]. In another study, intravitreal PACAP increased the activation of the protective Akt and ERK1/2, while decreased both p38MAPK and JNK activation in hypoperfused retinas. After ischemia several cytokines were overexpressed (CINC, CNTF, fractalkine, sICAM, IL-1, LIX, Selectin, MIP-1, RANTES and TIMP-1), but attenuated by PACAP38 [90]. Moreover, the neuropeptide further increased vascular endothelial growth factor and thymus chemokine levels. These results suggest that PACAP can ameliorate hypoperfusion injury involving Akt, MAPK pathways and anti-inflammatory actions.
PACAP in cardiac and other peripheral ischemic conditions
The cytoprotective effects of PACAP in ischemic conditions have also been observed outside the nervous system in various peripheral organs. Expression of PACAP mRNA increased after myocardial infarction in mice, and immunohistochemistry revealed a gp130-dependent elevation in PACAP38 in the stellate ganglion [91]. PACAP38 immunoreactivity was not detected in sham hearts, but was high in the infarct 3 days after infarction, suggesting an important role in cardiac and neuronal remodeling after ischemia-reperfusion [91]. Human data also propose the involvement of PACAP in cardiac ischemia: PACAP38- and PACAP27-like immunoreactivity was higher in ischemic heart diseases than in valve disorders [92]. Differences were also observed between ischemic and non-ischemic heart failure patient plasma, suggesting that PACAP might play an important role in the pathomechanism and progression of ischemic heart failure and it might be a potential biomarker of cardiac diseases [93]. A few available reports showed that PACAP was protective in cardiomyocyte ischemia in vitro [94, 95]. Cultured cardiomyocytes, exposed to ischemia/reperfusion, reacted to PACAP with increased cell viability and decreased apoptosis. PACAP induced the phosphorylation of Akt and protein kinaseA, while inactivated Bad, a pro-apoptotic member of the Bcl-2 family. Furthermore, PACAP increased the levels of Bcl-xL and 14–3-3 proteins, both of which promote cell survival, and decreased the apoptosis executor caspase-3 cleavage [94]. In another study, cardiomyocytes were exposed to brief preconditioning ischemia followed by 2 h ischemia and 4 h reperfusion. PACAP treatment could again increase cell viability and decrease cell death, and further reduced the level of cleaved caspase-8 under in preconditioning [95].
Numerous studies have provided evidence for the protective effects of PACAP in several other peripheral organs, like small intestine, kidney and liver. Liver ischemia/reperfusion injury triggered the expression of intrinsic PACAP and its receptors, whereas the hepatocellular damage was exacerbated in PACAP deficient mice [96]. Both PACAP27 and PACAP38 protected against hepatic ischemia, accompanied by decreased serum alanine aminotransferase levels, more preserved hepatic morphology with less cell death signs and reduced inflammation [96]. In small intestinal ischemia/reperfusion injury PACAP was protective both exogenously and endogenously. PACAP deficient mice reacted with more severe tissue damage than wild types [97, 98]. Preservation of morphological structure of small intestine after ligation of mesenteric artery followed by reperfusion was accompanied by decreased oxidative stress and increased anti-oxidant capacity in PACAP-treated animals [97, 98]. Similar results have been obtained in the kidney [99]. Both homo- and heterozygous PACAP knockout mice showed increased injury after renal artery clamping [100, 101]. Cell cultures isolated from wild type and PACAP deficient mice showed that cells from PACAP deficient mice had higher vulnerability to in vitro hypoxia [102]. In vivo, knockout mice also displayed increased tissue damage accompanied by increased inflammatory cytokine expression, decreased anti-oxidant capacity and increased expression of apoptotic markers [100, 101]. When PACAP was given as an exogenous treatment in rat renal ischemia/reperfusion injury, PACAP-treated animals had decreased mortality and inflammatory status, better preserved morphological structure in all tested histological parameters and decreased apoptotic and cytokine activity [103, 104]. All these results show that PACAP has protective effects in ischemic injuries not only in the nervous system, but also in several peripheral organs suggesting a general anti-ischemic protective role of this neuropeptide.
Discussion
As discussed above, several in vitro and in vivo studies have shown that PACAP has protective effects in the CNS, as well as in peripheral organs during ischemic injuries [26, 31, 33, 34, 40, 41, 43, 45, 46, 89, 91, 95, 97, 101]. These actions are thought to be mediated via anti-apoptotic and anti-inflammatory mechanisms through direct activation of PAC1 receptors and indirect pathways [34, 41, 89]. Therefore, PACAP and the PAC1 receptor seem to be a promising therapeutic target for ischemic conditions [46], as well as for several neurodegenerative disorders [28, 30, 33].
Conversely, studies have shown expression of PACAP and PAC1 receptor in the TNC [15] and elevated PACAP plasma levels during migraine attacks [17]. Furthermore, the peripheral injection of PACAP induces migraine-like headaches to migraineurs [15]. This has led to the development of AMG 301, an antibody against the PAC1 receptor for the treatment of migraine, that is currently in Phase II (Clinical trials identifier: NCT03238781). As mentioned above, the PAC1 receptor was proposed as the most relevant PACAP receptor in migraine partly because this is stimulated exclusively by PACAP and not, as the VPAC1 and VPAC2 receptor, also by VIP. This latter peptide failed to induce migraine-like attacks in migraine patients [20]. Notwithstanding the evidence supporting a role for the PAC1 receptor in migraine, it cannot be completely ruled out that the differences in migraine-generation properties of PACAP and VIP are rather due to their pharmacokinetic characteristics (difference in half-life) than due to differences in their pharmacodynamic action. Thus, we feel that it is too early to exclude VPAC1 and VPAC2 as additional potential antimigraine targets.
Certainly the prophylactic treatment of migraine with AMG 301 seems promising; however, it is important to have in mind that migraineurs present an increased risk of ischemic stroke [105,106,107,108,109] and that PACAP and PAC1 play a key role in the homeostatic responses to ischemic conditions. Therefore, the question remains whether a mild ischemic event could transform into a full-blown infarct when PACAP’s actions are blocked; similar concerns have been raised with the novel CGRP (receptor)-antibodies [109, 110]. Although the benefits of blocking CGRP seem greater than the drawbacks, more research is warranted. Similarly, concerning blockade of the PAC1 receptor, further studies are required to determine the possible side effects of long-term blockade of PAC1 signaling, and to study whether the activation of indirect pathways involved in the protective actions of PACAP is sufficient during ischemic events [111].
Further, it remains to be established whether the same patients that show a positive therapeutic response to CGRP (receptor)-antibodies will have a positive response to PAC1 antibodies, or that both types of medications are most effective in a separate population of migraine patients, depending on the peptide that is most predominant in their individual migraine pathophysiology. In view of the role of both CGRP and PACAP in preserving homeostasis under ischemic conditions, it remains of particular interest whether these antimigraine drugs could be combined, or whether simultaneous use would augment their side-effect potential.
Conclusions
In conclusion, PAC1 antibodies may present a valuable new tool in the treatment of migraine. Larger clinical studies will shed more light on the efficacy of these antibodies in migraine. The cardiovascular safety should be investigated in both preclinical models as well as in relevant patient populations.
Abbreviations
- BBB:
-
Blood-brain barrier
- BCCAO:
-
Bilateral common carotid artery occlusion
- CGRP:
-
Calcitonin gene-related peptide
- CNS:
-
Central nervous system
- GPCRs:
-
G-protein coupled receptors
- HIF:
-
Hypoxia inducible factor
- icv:
-
Intracerebroventricular
- IL-6:
-
Interleukin-6
- MCAO:
-
Middle cerebral artery occlusion
- PACAP:
-
Pituitary adenylate cyclase activating polypeptide
- TNC:
-
Trigeminal nucleus caudalis
- TNF:
-
Tumor necrosis factor
- VIP:
-
Vasoactive intestinal peptide
- 4VO:
-
4 vessel occlusion
References
Miyata A, Arimura A, Dahl RR, Minamino N, Uehara A, Jiang L, Culler MD, Coy DH (1989) Isolation of a novel 38 residue-hypothalamic polypeptide which stimulates adenylate cyclase in pituitary cells. Biochem Biophys Res Commun 164(1):567–574
Vaudry D, Gonzalez BJ, Basille M, Yon L, Fournier A, Vaudry H (2000) Pituitary adenylate ayclase-activating polypeptide and its receptors: from structure to functions. Pharmacol Rev 52(2):269–324
Arimura A (1992) Receptors for pituitary adenylate cyclase-activating polypeptide comparison with vasoactive intestinal peptide receptors. Trends Endocrinol Metab 3(8):288–294
Laburthe M, Couvineau A, Marie JC (2002) VPAC receptors for VIP and PACAP. Receptors Channels 8(3–4):137–153
Masuo Y, Ohtaki T, Masuda Y, Nagai Y, Suno M, Tsuda M, Fujino M (1991) Autoradiographic distribution of pituitary adenylate cyclase activating polypeptide (PACAP) binding sites in the rat brain. Neurosci Lett 126(2):103–106
Masuo Y, Suzuki N, Matsumoto H, Tokito F, Matsumoto Y, Tsuda M, Fujino M (1993) Regional distribution of pituitary adenylate cyclase activating polypeptide (PACAP) in the rat central nervous system as determined by sandwich-enzyme immunoassay. Brain Res 602(1):57–63
Uddman R, Tajti J, Hou M, Sundler F, Edvinsson L (2002) Neuropeptide expression in the human trigeminal nucleus caudalis and in the cervical spinal cord C1 and C2. Cephalalgia 22(2):112–116
Narita M, Dun SL, Dun NJ, Tseng LF (1996) Hyperalgesia induced by pituitary adenylate cyclase-activating polypeptide in the mouse spinal cord. Eur J Pharmacol 311(2–3):121–126
Tajti J, Uddman R, Edvinsson L (2001) Neuropeptide localization in the ‘migraine generator’ region of the human brainstem. Cephalalgia 21(2):96–101
Vigh S, Arimura A, Koves K, Somogyvari-Vigh A, Sitton J, Fermin CD (1991) Immunohistochemical localization of the neuropeptide, pituitary adenylate cyclase activating polypeptide (PACAP), in human and primate hypothalamus. Peptides 12(2):313–318
Gottschall PE, Tatsuno I, Miyata A, Arimura A (1990) Characterization and distribution of binding sites for the hypothalamic peptide, pituitary adenylate cyclase-activating polypeptide. Endocrinology 127(1):272–277
Koves K, Arimura A, Somogyvari-Vigh A, Vigh S, Miller JIM (1990) Immunohistochemical demonstration of a novel hypothalamic peptide, pituitary adenylate cyclase-activating polypeptide, in the ovine hypothalamus. Endocrinology 127(1):264–271
Gourlet P, Woussen-Colle MC, Robberecht P, Neef P, Cauvin A, Vandermeers-Piret MC, Vandermeers A, Christophe J (1991) Structural requirements for the binding of the pituitary adenylate-cyclase-activating peptide to receptors and adenylate-cyclase activation in pancreatic and neuronal membranes. Eur J Biochem 195(2):535–541
Tatsuno I, Gottschall PE, Koves K, Arimura A (1990) Demonstration of specific binding sites for pituitary adenylate cyclase activating polypeptide (PACAP) in rat astrocytes. Biochem Biophys Res Commun 168(3):1027–1033
Schytz HW, Birk S, Wienecke T, Kruuse C, Olesen J, Ashina M (2009) PACAP38 induces migraine-like attacks in patients with migraine without aura. Brain 132(Pt1):16–25
Robert C, Bourgeais L, Arreto CD, Condes-Lara M, Noseda R, Jay T, Villanueva L (2013) Paraventricular hypothalamic regulation of trigeminovascular mechanisms involved in headaches. J Neurosci 33(20):8827–8840
Tuka B, Helyes Z, Markovics A, Bagoly T, Szolcsanyi J, Szabo N, Toth E, Kincses ZT, Vecsei L, Tajti J (2013) Alterations in PACAP-38-like immunoreactivity in the plasma during ictal and interictal periods of migraine patients. Cephalalgia 33(13):1085–1095
Lassen LH, Haderslev PA, Jacobsen VB, Iversen HK, Sperling B, Olesen J (2002) CGRP may play a causative role in migraine. Cephalalgia 22(1):54–61
Chan KY, Baun M, de Vries R, van den Bogaerdt AJ, Dirven CM, Danser AH, Jansen-Olesen I, Olesen J, Villalon CM, Maassen Van DenBrink A, Gupta S (2011) Pharmacological characterization of VIP and PACAP receptors in the human meningeal and coronary artery. Cephalalgia 31(2):181–189
Rahmann A, Wienecke T, Hansen JM, Fahrenkrug J, Olesen J, Ashina M (2008) Vasoactive intestinal peptide causes marked cephalic vasodilation, but does not induce migraine. Cephalalgia 28(3):226–236
Lam HC, Takahashi K, Ghatei MA, Kanse SM, Polak JM, Bloom SR (1990) Binding sites of a novel neuropeptide pituitary-adenylate-cyclase-activating polypeptide in the rat brain and lung. Eur J Biochem 193(3):725–729
Uddman R, Luts A, Arimura A, Sundler F (1991) Pituitary adenylate cyclase-activating peptide (PACAP), a new vasoactive intestinal peptide (VIP)-like peptide in the respiratory tract. Cell Tissue Res 265(1):197–201
Fahrenkrug J, Hannibal J (2011) Localisation of the neuropeptide PACAP and its receptors in the rat parathyroid and thyroid glands. Gen Comp Endocrinol 171(1):105–113
Lauffer JM, Modlin IM, Tang LH (1999) Biological relevance of pituitary adenylate cyclase-activating polypeptide (PACAP) in the gastrointestinal tract. Regul Pept 84(1–3):1–12
Lenti L, Domoki F, Kis D, Hegyi O, Toth GK, Busija DW, Bari F (2007) Pituitary adenylate cyclase-activating polypeptide induces pial arteriolar vasodilation through cyclooxygenase-dependent and independent mechanisms in newborn pigs. Brain Res 1165:81–88
Atlasz T, Babai N, Kiss P, Reglodi D, Tamas A, Szabadfi K, Toth G, Hegyi O, Lubics A, Gabriel R (2007) Pituitary adenylate cyclase activating polypeptide is protective in bilateral carotid occlusion-induced retinal lesion in rats. Gen Comp Endocrinol 153:108–114
Reglodi D, Tamas A, Somogyvari-Vigh A, Szanto Z, Kertes E, Lenard L, Arimura A, Lengvari I (2002) Effects of pretreatment with PACAP on the infarct size and functional outcome in rat permanent focal cerebral ischemia. Peptides 23(12):2227–2234
Reglodi D, Tamas A, Lubics A, Szalontay L, Lengvari I (2004) Morphological and functional effects of PACAP in 6-hydroxydopamine-induced lesion of the substantia nigra in rats. Regul Pept 123(1–3):85–94
Babai N, Atlasz T, Tamas A, Reglodi D, Toth G, Kiss P, Gabriel R (2005) Degree of damage compensation by various PACAP treatments in monosodium glutamate-induced retinal degeneration. Neurotox Res 8(3–4):227–233
Reglodi D, Lubics A, Kiss P, Lengvari I, Gaszner B, Toth G, Hegyi O, Tamas A (2006) Effect of PACAP in 6-OHDA-induced injury of the substantia nigra in intact young and ovariectomized female rats. Neuropeptides 40(4):265–274
Racz B, Gallyas F, Kiss P, Toth G, Hegyi O, Gasz B, Borsiczky B, Ferencz A, Roth E, Tamas A, Lengvari I, Lubics A, Reglodi D (2006) The neuroprotective effects of PACAP in monosodium glutamate-induced retinal lesion involve inhibition of proapoptotic signaling pathways. Regul Pept 137(1–2):20–26
Tamas A, Lubics A, Lengvari I, Reglodi D (2006) Protective effects of PACAP in excitotoxic striatal lesion. Ann N Y Acad Sci 1070:570–574
Somogyvari-Vigh A, Reglodi D (2004) Pituitary adenylate cyclase activating polypeptide: a potential neuroprotective peptide. Curr Pharm Des 10(23):2861–2889
Atlasz T, Vaczy A, Werling D, Kiss P, Tamas A, Kovacs K, Fabian E, Kvarik T, Mammel B, Danyadi B, Lokos E, Reglodi D (2016) Neuroprotective effects of PACAP in the retina. In: Reglodi D, Tamas A (eds) Pituitary adenylate cyclase activating polypeptide-PACAP, vol 11. Springer Nature, New York, pp 501–527
Ohtaki H, Nakamachi T, Dohi K, Shioda S (2008) Role of PACAP in ischemic neural death. J Mol Neurosci 36:16–25
Uchida D, Arimura A, Somogyvári-Vigh A, Shioda S, Banks WA (1996) Prevention of ischemia-induced death of hippocampal neurons by pituitary adenylate cyclase activating polypeptide. Brain Res 736(1–2):280–286
Dohi K, Mizushima H, Nakajo S, Ohtaki H, Matsunaga S, Aruga T, Shioda S (2002) Pituitary adenylate cyclase-activating polypeptide (PACAP) prevents hippocampal neurons from apoptosis by inhibiting JNK/SAPK and p38 signal transduction pathways. Regul Pept 109(1–3):83–88
Banks WA, Uchida D, Arimura A, Somogyvari-Vigh A, Shioda S (1996) Transport of pituitary adenylate cyclase-activating polypeptide across the blood-brain barrier and the prevention of ischemia-induced death of hippocampal neurons. Ann N Y Acad Sci 805:270–277
Shioda S, Ozawa H, Dohi K, Mizushima H, Matsumoto K, Nakajo S, Takaki A, Zhou CJ, Nakai Y, Arimura A (1998) PACAP protects hippocampal neurons against apoptosis: involvement of JNK/SAPK signaling pathway. Ann N Y Acad Sci 865:111–117
Ohtaki H, Nakamachi T, Dohi K, Aizawa Y, Takaki A, Hodoyama K, Yofu S, Hashimoto H, Shintani N, Baba A, Kopf M, Iwakura Y, Matsuda K, Arimura A, Shioda S (2006) Pituitary adenylate cyclase-activating polypeptide (PACAP) decreases ischemic neuronal cell death in association with IL-6. Proc Natl Acad Sci 103(19):7488–7493
Lazarovici P, Cohen G, Arien-Zakay H, Chen J, Zhang C, Chopp M, Jiang H (2012) Multimodal neuroprotection induced by PACAP38 in oxygen–glucose deprivation and middle cerebral artery occlusion stroke models. J Mol Neurosci 48(3):526–540
Tamas A, Reglodi D, Szanto Z, Borsiczky B, Nemeth J, Lengvari I (2002) Comparative neuroprotective effects of preischemic PACAP and VIP administration in permanent occlusion of the middle cerebral artery in rats. Neuro Endocrinol Lett 23(3):249–254
Reglodi D, Somogyvari-Vigh A, Vigh S, Kozicz T, Arimura A (2000) Delayed systemic administration of PACAP38 is neuroprotective in transient middle cerebral artery occlusion in the rat. Stroke 31(6):1411–1417
Reglodi D, Somogyvari-Vigh A, Vigh S, Maderdrut JL, Arimura A (2000) Neuroprotective effects of PACAP38 in a rat model of transient focal ischemia under various experimental conditions. Ann N Y Acad Sci 921:119–128
Reglodi D, Fabian Z, Tamas A, Lubics A, Szeberenyi J, Alexy T, Toth K, Marton Z, Borsiczky B, Roth E, Szalontay L, Lengvari I (2004) Effects of PACAP on in vitro and in vivo neuronal cell death, platelet aggregation, and production of reactive oxygen radicals. Regul Pept 123(1–3):51–59
Dejda A, Seaborn T, Bourgault S, Touzani O, Fournier A, Vaudry H, Vaudry D (2011) PACAP and a novel stable analog protect rat brain from ischemia: insight into the mechanisms of action. Peptides 32(6):1207–1216
Brifault C, Gras M, Liot D, May V, Vaudry D, Wurtz O (2015) Delayed pituitary adenylate cyclase-activating polypeptide delivery after brain stroke improves functional recovery by inducing m2 microglia/macrophage polarization. Stroke 46(2):520–528
Stetler RA, Gao Y, Zukin RS, Vosler PS, Zhang L, Zhang F, Cao G, Bennett MV, Chen J (2010) Apurinic/apyrimidinic endonuclease APE1 is required for PACAP-induced neuroprotection against global cerebral ischemia. Proc Natl Acad Sci U S A 107(7):3204–3209
Hori M, Nakamachi T, Shibato J, Rakwal R, Shioda S, Numazawa S (2015) Unraveling the specific ischemic core and penumbra transcriptome in the permanent middle cerebral artery occlusion mouse model brain treated with the neuropeptide PACAP38. Microarrays (Basel) 4(1):2–24
Hori M, Shibato J, Nakamachi T, Rakwal R, Ogawa T, Shioda S, Numazawa S (2015) Two-color dye-swap DNA microarray approach toward confident gene expression profiling in PMCAO mouse model for ischemia-related and PACAP38-influenced genes. Genom Data 3:148–154
Hori M, Nakamachi T, Shibato J, Rakwal R, Tsuchida M, Shioda S, Numazawa S (2014) PACAP38 differentially effects genes and CRMP2 protein expression in ischemic core and penumbra regions of permanent middle cerebral artery occlusion model mice brain. Int J Mol Sci 15(9):17014–17034
Hori M, Nakamachi T, Rakwal R, Shibato J, Ogawa T, Aiuchi T, Tsuruyama T, Tamaki K, Shioda S (2012) Transcriptomics and proteomics analyses of the PACAP38 influenced ischemic brain in permanent middle cerebral artery occlusion model mice. J Neuroinflammation 9:256
Maugeri G, D'Amico AG, Saccone S, Federico C, Cavallaro S, D'Agata V (2017) PACAP and VIP inhibit HIF-1α-mediated VEGF expression in a model of diabetic macular edema. J Cell Physiol 232(5):1209–1215
D'Amico AG, Maugeri G, Reitano R, Bucolo C, Saccone S, Drago F, D'Agata V (2015) PACAP modulates expression of hypoxia-inducible factors in streptozotocin-induced diabetic rat retina. J Mol Neurosci 57(4):501–509
Lin CH, Chiu L, Lee HT, Chiang CW, Liu SP, Hsu YH, Lin SZ, Hsu CY, Hsieh CH, Shyu WC (2015) PACAP38/PAC1 signaling induces bone marrow-derived cells homing to ischemic brain. Stem Cells 33(4):1153–1172
Doeppner TR, Kaltwasser B, Schlechter J, Jaschke J, Kilic E, Bahr M, Hermann DM, Weise J (2015) Cellular prion protein promotes post-ischemic neuronal survival, angioneurogenesis and enhances neural progenitor cell homing via proteasome inhibition. Cell Death Dis 6:e2024
Lamine A, Letourneau M, Doan ND, Maucotel J, Couvineau A, Vaudry H, Chatenet D, Vaudry D, Fournier A (2016) Characterizations of a synthetic pituitary adenylate cyclase-activating polypeptide analog displaying potent neuroprotective activity and reduced in vivo cardiovascular side effects in a Parkinson's disease model. Neuropharmacology 108:440–450
Somogyvari-Vigh A, Pan W, Reglodi D, Kastin AJ, Arimura A (2000) Effect of middle cerebral artery occlusion on the passage of pituitary adenylate cyclase activating polypeptide across the blood-brain barrier in the rat. Regul Pept 91(1–3):89–95
Dogrukol-Ak D, Kumar VB, Ryerse JS, Farr SA, Verma S, Nonaka N, Nakamachi T, Ohtaki H, Niehoff ML, Edwards JC, Shioda S, Morley JE, Banks WA (2009) Isolation of peptide transport system-6 from brain endothelial cells: therapeutic effects with antisense inhibition in Alzheimer and stroke models. J Cereb Blood Flow Metab 29(2):411–422
Ohtaki H, Dohi K, Yofu S, Nakamachi T, Kudo Y, Endo S, Aruga T, Goto N, Watanabe J, Kikuyama S, Shioda S (2004) Effect of pituitary adenylate cyclase-activating polypeptide 38 (PACAP38) on tissue oxygen content--treatment in central nervous system of mice. Regul Pept 123(1–3):61–67
Ivic I, Fulop BD, Juhasz T, Reglodi D, Toth G, Hashimoto H, Tamas A, Koller A (2017) Backup Mechanisms Maintain PACAP/VIP-Induced Arterial Relaxations in Pituitary Adenylate Cyclase-Activating Polypeptide-Deficient Mice. J Vasc Res 54:180–192
Amin FM, Asghar MS, Guo S, Hougaard A, Hansen AE, Schytz HW, van der Geest RJ, de Koning PJ, Larsson HB, Olesen J, Ashina M (2012) Headache and prolonged dilatation of the middle meningeal artery by PACAP38 in healthy volunteers. Cephalalgia 32:140–149
Edvinsson L (2016) Pituitary adenylate cyclase activating polypeptide (PACAP) in migraine pathophysiology. In: Pituitary adenylate cyclase activating polypeptide-PACAP, volume 11. Springer Nature, New York, pp 583–606
Shin CM, Chung YH, Kim MJ, Cha CI (2001) Spatial and temporal distribution of pituitary adenylate cyclase activating polypeptide in gerbil global cerebral ischemia. Neurosci Lett 309(1):53–56
Gillardon F, Hata R, Hossmann KA (1998) Delayed up-regulation of Zac1 and PACAP type I receptor after transient focal cerebral ischemia in mice. Brain Res Mol Brain Res 61(1–2):207–210
Ciani E, Hoffmann A, Schmidt P, Journot L, Spengler D (1999) Induction of the PAC1-R (PACAP-type I receptor) gene by p53 and Zac. Brain Res Mol Brain Res 69(2):290–294
Stumm R, Kolodziej A, Prinz V, Endres M, Wu DF, Hollt V (2007) Pituitary adenylate cyclase-activating polypeptide is up-regulated in cortical pyramidal cells after focal ischemia and protects neurons from mild hypoxic/ischemic damage. J Neurochem 103(4):1666–1681
Riek-Burchardt M, Kolodziej A, Henrich-Noack P, Reymann KG, Hollt V, Stumm R (2010) Differential regulation of CXCL12 and PACAP mRNA expression after focal and global ischemia. Neuropharmacology 58(1):199–207
Nakamachi T, Farkas J, Kagami N, Wada Y, Hori M, Tsuchikawa D, Tsuchida M, Yoshikawa A, Imai N, Hosono T, Atrata S, Shioda S (2013) Expression and distribution of pituitary adenylate cyclase-activating polypeptide receptor in reactive astrocytes induced by global brain ischemia in mice. Acta Neurochir Suppl 118:55–59
Nakamachi T, Tsuchida M, Kagami N, Yofu S, Wada Y, Hori M, Tsuchikawa D, Yoshikawa A, Imai N, Nakamura K, Arata S, Shioda S (2012) IL-6 and PACAP receptor expression and localization after global brain ischemia in mice. J Mol Neurosci 48(3):518–525
Nakamachi T, Ohtaki H, Yofu S, Dohi K, Watanabe J, Mori H, Sato A, Hashimoto H, Shintani N, Baba A, Shioda S (2010) Endogenous pituitary adenylate cyclase activating polypeptide is involved in suppression of edema in the ischemic brain. Acta Neurochir Suppl 106:43–46
Matsumoto M, Nakamachi T, Watanabe J, Sugiyama K, Ohtaki H, Murai N, Sasaki S, Xu Z, Hashimoto H, Seki T, Miyazaki A, Shioda S (2016) Pituitary adenylate cyclase-activating polypeptide (PACAP) is involved in adult mouse hippocampal neurogenesis after stroke. J Mol Neurosci 59(2):270–279
Ma BQ, Zhang M, Ba L (2015) Plasma pituitary adenylate cyclase-activating polypeptide concentrations and mortality after acute spontaneous basal ganglia hemorrhage. Clin Chim Acta 439:102–106
Jiang L, Wang WH, Dong XQ, Yu WH, Du Q, Yang DB, Wang H, Shen YF (2016) The change of plasma pituitary adenylate cyclase-activating polypeptide levels after aneurysmal subarachnoid hemorrhage. Acta Neurol Scand 134(2):131–139
Atlasz T, Szabadfi K, Kiss P, Racz B, Gallyas F, Tamas A, Gaal V, Marton Z, Gabriel R, Reglodi D (2010) Pituitary adenylate cyclase activating polypeptide in the retina: focus on the retinoprotective effects. Ann N Y Acad Sci 1200:128–139
Atlasz T, Szabadfi K, Kiss P, Tamas A, Toth G, Reglodi D, Gabriel R (2010) Evaluation of the protective effects of PACAP with cell-specific markers in ischemia-induced retinal degeneration. Brain Res Bull 81:497–504
Cervia D, Casini G (2013) The neuropeptide systems and their potential role in the treatment of mammalian retinal ischemia: a developing story. Curr Neuropharmacol 11:95–101
Shioda S, Takenoya F, Wada N, Hirabayashi T, Seki T, Nakamachi T (2016) Pleiotropic and retinoprotective functions of PACAP. Anat Sci Int 4:313–324
Nakamachi T, Matkovits A, Seki T, Shioda S (2012) Distribution and protective function of pituitary adenylate cyclase-activating polypeptide in the retina. Front Endocrinol (Lausanne) 3:145
Szabadfi K, Mester L, Reglodi D, Kiss P, Babai N, Racz B, Kovacs K, Szabo A, Tamas A, Gabriel R, Atlasz T (2010) Novel neuroprotective strategies in ischemic retinal lesions. Int J Mol Sci 11:544–561
Danyadi B, Szabadfi K, Reglodi D, Mihalik A, Danyadi T, Kovacs Zs BI, Tamas A, Kiss P, Toth G, Gabriel R (2014) PACAP application improves functional outcome of chronic retinal ischemic injury in rats – evidence from electroretinographic measurements. J Mol Neurosci 54:293–299
Szabadfi K, Atlasz T, Kiss P, Danyadi B, Tamas A, Helyes Zs HH, Shintani N, Baba A, Toth G, Gabriel R, Reglodi D (2012a) Mice deficient in pituitary adenylate cyclase activating polypeptide (PACAP) are more susceptible to retinal ischemic injury in vivo. Neurotox Res 21:41–48
Seki T, Itoh H, Nakamachi T, Endo K, Wada Y, Nakamura K, Shioda S (2011) Suppression of rat retinal ganglion cell death by PACAP following transient ischemia induced by high intraocular pressure. J Mol Neurosci 43:30–34
Werling D, Reglodi D, Kiss P, Toth G, Szabadfi K, Tamas A, Biro Z, Atlasz T (2014) Investigation of PACAP fragments and related peptides in chronic retinal hypoperfusion. J Ophthalmol 2014:563812
Atlasz T, Szabadfi K, Reglodi D, Kiss P, Tamas A, Toth G, Molnar A, Szabo K, Gabriel R (2009) Effects of pituitary adenylate cyclase activating polypeptide and its fragments on retinal degeneration induced by neonatal monosodium glutamate treatment. Ann N Y Acad Sci 1163:348–352
Szabadfi K, Danyadi B, Kiss P, Tamas A, Fabian E, Gabriel R, Reglodi D (2012) Protective effects of vasoactive intestinal peptide (VIP) in ischemic retinal degeneration. J Mol Neurosci 48:501–507
Werling D, Banks WA, Salameh TS, Kvarik T, Kovacs LA, Vaczy A, Szabo E, Mayer F, Varga R, Tamas A, Toth G, Biro Z, Atlasz T, Reglodi D (2017) Passage through the ocular barriers and beneficial effects in retinal ischemia of topical application of PACAP1-38 in rodents. Int J Mol Sci 18(3)
Werling D, Reglodi D, Banks WA, Salameh TS, Kovacs K, Kvarik T, Vaczy A, Kovacs L, Mayer F, Danyadi B, Lokos E, Tamas A, Toth G, Biro Z, Atlasz T (2016) Ocular delivery of PACAP1-27 protects the retina from ischemic damage in rodents. Invest Ophthalmol Vis Sci 57(15):6683–6691
Vaczy A, Reglodi D, Somoskeoy T, Kovacs K, Lokos E, Szabo E, Tamas A, Atlasz T (2016) The protective role of PAC1-receptor agonist maxadilan in BCCAO-induced retinal degeneration. J Mol Neurosci 60(2):186–194
Szabo A, Danyadi B, Bognar E, Szabadfi K, Fabian E, Kiss P, Mester L, Manavalan S, Atlasz T, Gabriel R, Toth G, Tamas A, Reglodi D, Kovacs K (2012) Effect of PACAP on MAP kinases, Akt and cytokine expressions in rat retinal hypoperfusion. Neurosci Lett 523:93–98
Alston EN, Parrish DC, Hasan W, Tharp K, Pahlmeyer L, Habecker BA (2011) Cardiac ischemia-reperfusion regulates sympathetic neuropeptide expression through gp130-dependent and independent mechanisms. Neuropeptides 45(1):33–42
Szanto Z, Sarszegi Z, Reglodi D, Nemeth J, Szabadfi K, Kiss P, Varga A, Banki E, Csanaky K, Gaszner B, Pinter O, Szalai Z, Tamas A (2012) PACAP immunoreactivity in human malignant tumor samples and cardiac diseases. J Mol Neurosci 48(3):667–673
Sarszegi Z, Szabo D, Gaszner B, Konyi A, Reglodi D, Nemeth J, Lelesz B, Polgar B, Jungling A, Tamas A (2018) Examination of pituitary adenylate cyclase-activating polypeptide (PACAP) as a potential biomarker in heart failure patients. J Mol Neurosci in press. https://doi.org/10.1007/s12031-017-1025-7
Racz B, Gasz B, Gallyas F Jr, Kiss P, Tamas A, Szanto Z, Lubics A, Lengvari I, Toth G, Hegyi O, Roth E, Reglodi D (2008) PKA-Bad-14-3-3 and Akt-Bad-14-3-3 signaling pathways are involved in the protective effects of PACAP against ischemia/reperfusion-induced cardiomyocyte apoptosis. Regul Pept 145(1–3):105–115
Roth E, Weber G, Kiss P, Horvath G, Toth G, Gasz B, Ferencz A, Gallyas F Jr, Reglodi D, Racz B (2009) Effects of PACAP and preconditioning against ischemia/reperfusion-induced cardiomyocyte apoptosis in vitro. Ann N Y Acad Sci 1163:512–516
Ji H, Zhang Y, Shen XD, Gao F, Huang CY, Abad C, Busuttil RW, Waschek JA, Kupiec-Weglinski JW (2013) Neuropeptide PACAP in mouse liver ischemia and reperfusion injury: immunomodulation by the cAMP-PKA pathway. Hepatology 57(3):1225–1237
Ferencz A, Racz B, Tamas A, Reglodi D, Lubics A, Nemeth J, Nedvig K, Kalmar-Nagy K, Horvath OP, Weber G, Roth E (2009) Influence of PACAP on oxidative stress and tissue injury following small bowel autotransplantation. J Mol Neurosci 37:168–176
Ferencz A, Kiss P, Weber G, Helyes Z, Shintani N, Baba A, Reglodi D (2010) Comparison of intestinal warm ischemic injury in PACAP knock-out and wild-type mice. J Mol Neurosci 42:435–442
Laszlo E, Kiss P, Horvath G, Szakaly P, Tamas A, Reglodi D (2014) The effects of pituitary adenylate cyclase activating polypeptide in renal ischemia/reperfusion. Acta Biol Hung 65(4):369–378
Laszlo E, Varga A, Kovacs K, Jancso G, Kiss P, Tamas A, Szakaly P, Fulop B, Reglodi D (2015) Ischemia/reperfusion-induced kidney injury in heterozygous PACAP deficient mice. Transplant Proc 47:2210–2215
Szakaly P, Laszlo E, Kovacs K, Racz B, Horvath G, Ferencz A, Lubics A, Kiss P, Tamas A, Brubel R, Opper B, Baba A, Hashimoto H, Farkas J, Matkovits A, Magyarlaki T, Helyes Zs RD (2011) Mice deficient in pituitary adenylate cyclase activating polypeptide (PACAP) show increased susceptibility to in vivo renal ischemia/reperfusion injury. Neuropeptides 45:113–121
Horvath G, Racz B, Szakaly P, Kiss P, Laszlo E, Hau L, Tamas A, Helyes Zs LA, Hashimoto H, Baba A, Reglodi D (2010) Mice deficient in neuropeptide PACAP demonstrate increased sensitivity to in vitro kidney hypoxia. Transplant Proc 42:2293–2295
Horvath G, Racz B, Reglodi D, Kovacs K, Kiss P, Gallyas F Jr, Bognar Z, Szabo A, Magyarlaki T, Laszlo E, Lubics A, Tamas A, Toth G, Szakaly P (2010) Effects of PACAP on mitochondrial apoptotic pathways and cytokine expression in rats subjected to renal ischemia-reperfusion. J Mol Neurosci 42: 411–418
Szakaly P, Kiss P, Lubics A, Magyarlaki T, Tamas A, Racz B, Lengvari I, Toth G, Reglodi D (2008) Effects of PACAP on survival and renal morphology in rats subjected to renal ischemia-reperfusion. J Mol Neurosci 36:89–96
Chang CL, Donaghy M, Poulter N (1999) Migraine and stroke in young women: case-control study. BMJ 318(7175):13–18
Etminan M, Takkouche B, Isorna FC, Samii A (2005) Risk of ischaemic stroke in people with migraine: systematic review and meta-analysis of observational studies. BMJ 330(7482):63
Schurks M, Rist PM, Bigal ME, Buring JE, Lipton RB, Kurth T (2009) Migraine and cardiovascular disease: systematic review and meta-analysis. BMJ 339:b3914
Spector JT, Kahn SR, Jones MR, Jayakumar M, Dalal D, Nazarian S (2010) Migraine headache and ischemic stroke risk: an updated meta-analysis. Am J Med 123(7):612–624
Maassen Van DenBrink A, Meijer J, Villalon CM, Ferrari MD (2016) Wiping out CGRP: potential cardiovascular risks. Trends Pharmacol Sci 37(9):779–788
Deen M, Correnti E, Kamm K, Kelderman T, Papetti L, Rubio-Beltran E, Vigneri S, Edvinsson L, Maassen Van Den Brink A (2017) Blocking CGRP in migraine patients - a review of pros and cons. J Headache Pain 18(1):96
Brifault C, Vaudry D, Wurtz O (2016) The neuropeptide PACAP, a potent disease modifier candidate for brain stroke treatment. In: Pituitary adenylate cyclase activating polypeptide-PACAP, volume 11. Springer Nature, New York, pp 583–606
Acknowledgements
N/A
Funding
2017–1.2.1-NKP-2017-00002, PTE ÁOK KA Research Grant; GINOP-2.3.2–15–2016-00050 “PEPSYS”, MTA-TKI 14016, NKFIH K119759, 115874; EFOP-3.6.2–16–2017-00008. “The role of neuro-inflammation in neurodegeneration: from molecules to clinics”, Neuroscience Centre of Pecs.
Availability of data and materials
N/A
Author information
Authors and Affiliations
Contributions
DR, AV, ERB, AM wrote the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Authors’ information
N/A
Ethics approval and consent to participate
N/A
Consent for publication
N/A
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Reglodi, D., Vaczy, A., Rubio-Beltran, E. et al. Protective effects of PACAP in ischemia. J Headache Pain 19, 19 (2018). https://doi.org/10.1186/s10194-018-0845-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s10194-018-0845-3