
Abstract
Strains of a pink-pigmented Methylobacterium sp. are effective nitrogen- (N2) fixing microsymbionts of species of the African crotalarioid genus Listia. Strain WSM2598 is an aerobic, motile, Gram-negative, non-spore-forming rod isolated in 2002 from a Listia bainesii root nodule collected at Estcourt Research Station in South Africa. Here we describe the features of Methylobacterium sp. WSM2598, together with information and annotation of a high-quality draft genome sequence. The 7,669,765 bp draft genome is arranged in 5 scaffolds of 83 contigs, contains 7,236 protein-coding genes and 18 RNA-only encoding genes. This rhizobial genome is one of 100 sequenced as part of the DOE Joint Genome Institute 2010 G enomic E ncyclopedia for B acteria and A rchaea-R oot N odule B acteria (GEBA-RNB) project.
Similar content being viewed by others


Introduction
Nodulated legumes are important and established components of Australian agricultural systems: the value of atmospheric nitrogen (N2) fixed by rhizobia in symbiotic association with these legumes is estimated to be worth more than $2 billion annually [1, 2]. The major agricultural region of south-western Australia has a Mediterranean climate, with soils that are often acid, have a low clay content and low organic matter, and tend to be inherently infertile [3, 4]. The last forty years, however, have seen a sharp decrease in average winter rainfall by about 15–20% [5]. This, together with the development of dryland salinity [6], has challenged the sustainability of using the commonly sown subterranean clover and annual medics as pasture legumes in these systems. Alternative perennial legume species (and their associated rhizobia) are therefore being sought [2]. We have identified a suite of South African perennial, herbaceous forage legumes, including several species in the crotalarioid genus Listia (previously Lotononis) [7], that are potentially well-adapted to the arid climate and acid, infertile soils of the target agricultural areas.
Listia species are found in seasonally wet habitats throughout southern and tropical Africa [8]. They produce stoloniferous roots [8, 9] and form lupinoid nodules rather than the indeterminate type found in other crotalarioid species [7, 10]. Rhizobial infection occurs by epidermal entry rather than via root hair curling [7]. Listia-rhizobia symbioses are highly specific. The tropically distributed L. angolensis forms effective (i.e. N2-fixing) nodules with newly described species of Microvirga [11], while all other studied Listia species are only nodulated by strains of pigmented methylobacteria [7, 10, 12]. Unlike the methylotrophic Methylobacterium nodulans, which specifically nodulates some species of Crotalaria [13], the Listia methylobacteria are unable to utilize methanol as a sole carbon source [14]. In Australia, strains of pigmented methylobacteria have been used as commercial inoculants for Listia bainesii and are able to persist in acidic, sandy, infertile soils, while remaining symbiotically and serologically stable [10, 15].
A pigmented Methylobacterium strain, WSM2598, isolated from a root nodule of L. bainesii cv “Miles” in South Africa in 2002, was found to be a highly effective nitrogen fixing microsymbiont of both L. bainesii and Listia heterophylla (previously Lotononis listii) [10]. Here we present a set of preliminary classification and general features for Methylobacterium sp. strain WSM2598, together with the description of the genome sequence and annotation.
Organism information
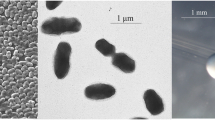
Methylobacterium sp. strain WSM2598 is a motile, non-sporulating, non-encapsulated, Gram-negative rod with one to several flagella. It is a member of the family Methylobacteriaceae in the class Alphaproteobacteria. The rod-shaped form varies in size with dimensions of approximately 0.5 μm in width and 1.0-1.5 μm in length (Figure 1 Left and 1 Center). WSM2598 is medium to slow growing, forming 0.5-1.5 mm diameter colonies within 6–7 days at 28°C. WSM2598 is pigmented, an unusual property for rhizobia. When grown on half strength Lupin Agar (½LA) [10], WSM2598 forms dark pink pigmented, opaque, slightly domed colonies with smooth margins (Figure 1 Right).

Images of Methylobacterium sp. strain WSM2598 using scanning (Left) and transmission (Center) electron microscopy as well as light microscopy to visualize colony morphology on solid ½LA [10] (Right).
WSM2598 alkalinizes ½LA containing universal indicator (BDH Laboratory Supplies). WSM2598 cultured in minimal medium [16] is unable to utilize arabinose, galactose, glucose, mannitol, methanol, methylamine or formaldehyde as sole carbon sources, but grows poorly on formate and well on succinate and glutamate [14]. Minimum Information about the Genome Sequence (MIGS) is provided in Table 1 and Additional file 1: Table S1.
Figure 2 shows the phylogenetic neighborhood of Methylobacterium sp. WSM2598 in a 16S rRNA sequence based tree. The 16S rDNA sequence of WSM2598 has 99% (1,358/1,364 bp) and 98% (1,334/1,365 bp) sequence identity to the 16S rRNA of the fully sequenced strains Methylobacterium sp. 4–46 (Gc00857) and M. nodulans ORS2060 (Gc00935), respectively.

Phylogenetic tree showing the relationships of Methylobacterium sp. WSM2598 (shown in blue print) with some of the root nodule bacteria in the order Rhizobiales based on aligned sequences of the 16S rRNA gene (1,340 bp internal region). All sites were informative and there were no gap-containing sites. Phylogenetic analyses were performed using MEGA, version 5 [28]. The tree was built using the maximum likelihood method with the General Time Reversible model. Bootstrap analysis [29] with 500 replicates was performed to assess the support of the clusters. Type strains are indicated with a superscript T. Brackets after the strain name contain an accession number. Strains with a genome sequencing project registered in GOLD [30] are in bold print and the GOLD ID is mentioned after the accession number. Published genomes are designated with an asterisk.
Symbiotaxonomy
Methylobacterium sp. WSM2598 forms nodules on (Nod+), and fixes N2 (Fix+), with southern African species of Listia. On Listia angolensis, some species of the crotalarioid genus Leobordea and the promiscuous legume Macroptilium atropurpureum, WSM2598 forms white, ineffective (Fix-) nodules. It does not form nodules on other tested legumes [7], [Table 2].
Genome sequencing and annotation information
Genome project history
This organism was selected for sequencing on the basis of its environmental and agricultural relevance to issues in global carbon cycling, alternative energy production, and biogeochemical importance, and is part of the Community Sequencing Program at the U.S. Department of Energy, Joint Genome Institute (JGI) for projects of relevance to agency missions. The genome project is deposited in the Genomes OnLine Database [30] and an improved-high-quality-draft genome sequence in IMG. Sequencing, finishing and annotation were performed by the JGI. A summary of the project information is shown in Table 3.
Growth conditions and DNA isolation
Methylobacterium sp. WSM2598 was grown to mid-logarithmic phase in TY rich media on a gyratory shaker at 28°C [32]. DNA was isolated from 60 mL of cells using a CTAB (Cetyl trimethyl ammonium bromide) bacterial genomic DNA isolation method [33].
Genome sequencing and assembly
The draft genome of Methylobacterium sp. WSM2598 was generated at the DOE Joint Genome Institute (JGI) using Illumina technology [34, 35]. For this genome, we constructed and sequenced an Illumina short-insert paired-end library with an average insert size of 270 bp which generated 19,048,548 reads and an Illumina long-insert paired-end library with an average insert size of 6354.14 +/− 3100.07 bp which generated 18,876,864 reads totaling 5,689 Mbp of Illumina data. (unpublished, Feng Chen). All general aspects of library construction and sequencing performed at the JGI can be found at the JGI website. The initial draft assembly contained 141 contigs in 41 scaffold(s). The initial draft data was assembled with Allpaths, version 39750, and the consensus was computationally shredded into 10 Kbp overlapping fake reads (shreds). The Illumina draft data was also assembled with Velvet, version 1.1.05 [36] and the consensus sequences were computationally shredded into 1.5 Kbp overlapping fake reads (shreds). The Illumina draft data was assembled again with Velvet using the shreds from the first Velvet assembly to guide the next assembly. The consensus from the second VELVET assembly was shredded into 1.5 Kbp overlapping fake reads. The fake reads from the Allpaths assembly and both Velvet assemblies and a subset of the Illumina CLIP paired-end reads were assembled using parallel phrap, version 4.24 (High Performance Software, LLC). Possible mis-assemblies were corrected with manual editing in Consed [37–39]. Gap closure was accomplished using repeat resolution software (Wei Gu, unpublished), and sequencing of bridging PCR fragments with Sanger and/or PacBio (unpublished, Cliff Han) technologies. One round of manual/wet lab finishing was also completed. 17 PCR PacBio consensus sequences were completed to close gaps and to raise the quality of the final sequence. The total (“estimated size” for the unfinished) size of the genome is 8.3 Mbp and the final assembly is based on 5,689 Mbp of Illumina draft data, which provides an average 685× coverage of the genome.
Genome annotation
Genes were identified using Prodigal [40] as part of the DOE-JGI Annotation pipeline [41], followed by a round of manual curation using the JGI GenePRIMP pipeline [42]. Within the Integrated Microbial Genomes (IMG-ER) system [43], predicted CDSs were translated and used to search the National Center for Biotechnology Information (NCBI) nonredundant database, UniProt, TIGRFam, Pfam, PRIAM, KEGG, COG, and InterPro databases. These data sources were combined to assert a product description for each predicted protein. Non-coding genes and miscellaneous features were predicted using tRNAscan-SE [44], RNAMMer [45], Rfam [46], TMHMM [47], and SignalP [48]. Additional gene prediction analyses and functional annotation were performed within IMG.
Genome properties
The genome is 7,669,765 nucleotides with 71.17% GC content (Table 4) and comprised of 5 scaffolds (Figure 3) of 83 contigs. From a total of 7,349 genes, 7,236 were protein encoding and 18 RNA only encoding genes. The majority of genes (71.22%) were assigned a putative function whilst the remaining genes were annotated as hypothetical. The distribution of genes into COGs functional categories is presented in Table 5.

Graphical map of the 5 scaffolds assembled for the genome of Methylobacterium sp. WSM2598. From top to bottom, the scaffolds are: WSM2598: MET2598DRAFT _scaffold1.1, WSM2598: MET2598DRAFT_scaffold2.2, WSM2598: MET2598DRAFT _scaffold3.3, WSM2598: MET2598DRAFT _scaffold4.4, and WSM2598: MET2598DRAFT _scaffold5.5. From the bottom to the top of each scaffold: Genes on forward strand (color by COG categories as denoted by the IMG platform), Genes on reverse strand (color by COG categories), RNA genes (tRNAs green, sRNAs red, other RNAs black), GC content, GC skew.
Conclusion
WSM2598 was sequenced as part of the DOE Joint Genome Institute GEBA-RNB project. In common with other sequenced rhizobial strains, WSM2598 has a comparatively large genome of around 7.69 Mbp, with a high proportion of genes assigned to the COG functional categories associated with transcription control and signal transduction (14.69%), transport and metabolism (29.38%) and secondary metabolite biosynthesis (3.12%). These features are characteristic of soil bacteria, which inhabit oligotrophic environments with typically diverse but scarce nutrient sources. Rhizobial methylobacteria are unusual, however, in that they form symbiotic associations exclusively with African crotalarioid legume hosts, several species of which are well-adapted to arid climates and acid, infertile soils and are therefore potentially useful pasture plants in marginal agricultural systems. The molecular basis for this symbiotic specificity has yet to be determined. As WSM2598 is highly effective for N2-fixation on several of these hosts, its sequenced genome is a valuable resource for gaining an understanding of symbiotic specificity and N2-fixation in a currently understudied group of legumes and rhizobia.
References
Herridge DF, Peoples MB, Boddey RM: Global inputs of biological nitrogen fixation in agricultural systems. Plant Soil 2008, 311: 1–18. http://dx.doi.org/10.1007/s11104–008–9668–3 10.1007/s11104-008-9668-3
Howieson JG, Yates RJ, Foster K, Real D, Besier B: Prospects for the future use of legumes. In Leguminous Nitrogen-Fixing Symbioses. Edited by: Dilworth MJ, James EK, Sprent JI, Newton WE. London, UK: Elsevier; 2008:363–394.
Howieson JG, O’Hara GW, Carr SJ: Changing roles for legumes in Mediterranean agriculture: developments from an Australian perspective. Field Crops Res 2000, 65: 107–122. http://dx.doi.org/10.1016/S0378–4290(99)00081–7 10.1016/S0378-4290(99)00081-7
Nichols P, Loi A, Nutt B, Snowball R, Revell C: Domestication of new Mediterranean annual pasture legumes. Edited by: Huyghe C. Dordrecht, Netherlands: Sustainable Use of Genetic Diversity in Forage and Turf Breeding: Springer; 2010:137–142.
Initiative IOC: Climate variability and change in southwest Western Australia. 2002, 1–34.
Sy A, Giraud E, Jourand P, Garcia N, Willems A, de Lajudie P, Prin Y, Neyra M, Gillis M, Boivin-Masson C, Dreyfus B: Long-term groundwater trends and their impact on the future extent of dryland salinity in Western Australia in a varible climate. University of Western Australia: Crawley, Western Australia: Salinity Forum; 2008.
Ardley JK, Reeve WG, O’Hara GW, Yates RJ, Dilworth MJ, Howieson JG: Nodule morphology, symbiotic specificity and association with unusual rhizobia are distinguishing features of the genus Listia within the southern African crotalarioid clade Lotononis s.l. Ann Bot (Lond) 2013, 112: 1–15. PubMed http://dx.doi.org/10.1093/aob/mct095 10.1093/aob/mct095
van Wyk BE: A Synopsis of the Genus Lotononis (Fabaceae: Crotalarieae). Cape Town, South Africa: Rustica Press; 1991.
Boatwright JS, Wink M, van Wyk BE: The generic concept of Lotononis (Crotalarieae, Fabaceae): Reinstatement of the genera Euchlora, Leobordea and Listia and the new genus Ezoloba . Taxon 2011, 60: 161–177.
Yates RJ, Howieson JG, Reeve WG, Nandasena KG, Law IJ, Bräu L, Ardley JK, Nistelberger HM, Real D, O’Hara GW: Lotononis angolensis forms nitrogen fixing, lupinoid nodules with phylogenetically unique, fast-growing, pink-pigmented bacteria, which do not nodulate L. bainesii or L. listii . Soil Biol Biochem 2007, 39: 1680–1688. http://dx.doi.org/10.1016/j.soilbio.2007.01.025 10.1016/j.soilbio.2007.01.025
Ardley JK, Parker MA, De Meyer SE, Trengove RD, O’Hara GW, Reeve WG, Yates RJ, Dilworth MJ, Willems A, Howieson JG: Microvirga lupini sp. nov., Microvirga lotononidis sp. nov. and Microvirga zambiensis sp. nov. are alphaproteobacterial root-nodule bacteria that specifically nodulate and fix nitrogen with geographically and taxonomically separate legume hosts. Int J Syst Evol Microbiol 2012, 62: 2579–2588. PubMed http://dx.doi.org/10.1099/ijs.0.035097–0 10.1099/ijs.0.035097-0
Norris DO: A red strain of Rhizobium from Lotononis bainesii Baker. Aust J Agric Res 1958, 9: 629–632. http://dx.doi.org/10.1071/AR9580629 10.1071/AR9580629
Sy A, Giraud E, Jourand P, Garcia N, Willems A, de Lajudie P, Prin Y, Neyra M, Gillis M, Boivin-Masson C, et al.: Methylotrophic Methylobacterium bacteria nodulate and fix nitrogen in symbiosis with legumes. J Bacteriol 2001, 183: 214–220. PubMed http://dx.doi.org/10.1128/JB.183.1.214–220.2001 10.1128/JB.183.1.214-220.2001
Ardley JK, O’Hara GW, Reeve WG, Yates RJ, Dilworth MJ, Tiwari RP, Howieson JG: Root nodule bacteria isolated from South African Lotononis bainesii, L. listii and L. solitudinis are species of Methylobacterium that are unable to utilize methanol. Arch Microbiol 2009, 191: 311–318. PubMed http://dx.doi.org/10.1007/s00203–009–0456–0 10.1007/s00203-009-0456-0
Diatloff A: Ecological studies of root-nodule bacteria introduced into field environments—6. Antigenic and symbiotic stability in Lotononis rhizobia over a 12-year period. Soil Biol Biochem 1977, 9: 85–88. http://dx.doi.org/10.1016/0038–0717(77)90042–6 10.1016/0038-0717(77)90042-6
O’Hara GW, Goss TJ, Dilworth MJ, Glenn AR: Maintenance of intracellular pH and acid tolerance in Rhizobium meliloti . Appl Environ Microbiol 1989, 55: 1870–1876. PubMed
Field D, Garrity G, Gray T, Morrison N, Selengut J, Sterk P, Tatusova T, Thomson N, Allen M, Angiuoli SV, Ashburner M, Axelrod N, Baldauf S, Ballard S, Boore J, Cochrane G, Cole J, Dawyndt P, De Vos P, De Pamphilis C, Edwards R, Faruque N, Feldman R, Gilbert J, Gilna P, Glöckner FO, Goldstein P, Guralnick R, Haft D, Hancock D: Towards a richer description of our complete collection of genomes and metagenomes “Minimum Information about a Genome Sequence ” (MIGS) specification. Nat Biotechnol 2008, 26: 541–547. PubMed http://dx.doi.org/10.1038/nbt1360 10.1038/nbt1360
Woese CR, Kandler O, Wheelis ML: Towards a natural system of organisms: proposal for the domains Archaea , Bacteria, and Eucarya. Proc Natl Acad Sci USA 1990, 87: 4576–4579. PubMed http://dx.doi.org/10.1073/pnas.87.12.4576 10.1073/pnas.87.12.4576
Garrity GM, Bell JA, Phylum LT, XIV: Proteobacteria phyl. nov. In Bergey’s Manual of Systematic Bacteriology. Edited by: Garrity GM, Brenner DJ, Krieg NR, Staley JT. Springer, New York: Part B; 2005:1. Second Edition, Volume 2
Garrity GM, Bell JA, Lilburn TG: Class I. Alphaproteobacteria . In Bergey’s Manual of Systematic Bacteriology. Volume Volume 2. Second edition. Edited by: Garrity GM, Brenner DJ, Krieg NR, Staley JT. New York: Springer - Verlag; 2005.
Validation List No. 107: List of new names and new combinations previously effectively, but not validly, published. Int J Syst Evol Microbiol 2006, 56: 1–6. PubMed http://dx.doi.org/10.1099/ijs.0.64188–0
Kuykendall LD: Order VI. Rhizobiales ord. nov. In Bergey’s Manual of Systematic Bacteriology. Second edition. Edited by: Garrity GM, Brenner DJ, Kreig NR, Staley JT. New York: Springer - Verlag; 2005:324.
Garrity GM, Bell JA, Lilburn TG: Family IX. Methylobacteriaceae . In Bergey’s Manual of Systematic Bacteriology. Volume Volume 2. Second edition. Edited by: Garrity GM, Brenner DJ, Krieg NR, Staley JT. New York: Springer - Verlag; 2005.
Skerman VBD, McGowan V, Sneath PHA: Approved Lists of Bacterial Names. Int J Syst Bacteriol 1980, 30: 225–420. http://dx.doi.org/10.1099/00207713–30–1-225 10.1099/00207713-30-1-225
Patt TE, Cole GC, Hanson RS: Methylobacterium , a new genus of facultatively methylotrophic bacteria. Int J Syst Bacteriol 1976, 26: 226–229. http://dx.doi.org/10.1099/00207713–26–2-226 10.1099/00207713-26-2-226
Green PN, Bousfield IJ: Emendation of Methylobacterium Patt, Cole and Hanson 1976; Methylobacterium rhodinum (Heumann 1962) comb. nov. corrig.; Methylobacterium radiotolerans , (Ito and Iizuka 1971) comb. nov. corrig.; and Methylobacterium mesophilicum , (Austin and Goodfellow 1979) comb. nov. Int J Syst Bacteriol 1983, 33: 875–877. http://dx.doi.org/10.1099/00207713–33–4-875 10.1099/00207713-33-4-875
Biological Agents: Technical rules for biological agents. TRBA. ( ):466 http://www.baua.de
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S: MEGA5: Molecular evolutionary genetics analysis using Maximum Likelihood, evolutionary distance, and Maximum Parismony methods. Mol Biol Evol 2011, 28: 2731–2739. PubMed http://dx.doi.org/10.1093/molbev/msr121 10.1093/molbev/msr121
Felsenstein J: Confidence limits on phylogenies: an approach using the bootstrap. Evolution 1985, 39: 783–791. http://dx.doi.org/10.2307/2408678 10.2307/2408678
Liolios K, Mavromatis K, Tavernarakis N, Kyrpides NC: The Genomes On Line Database (GOLD) in 2007: status of genomic and metagenomic projects and their associated metadata. Nucleic Acids Res 2008, 36: D475-D479. PubMed http://dx.doi.org/10.1093/nar/gkm884 10.1093/nar/gkn240
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al.: Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 2000, 25: 25–29. PubMed http://dx.doi.org/10.1038/75556 10.1038/75556
Reeve WG, Tiwari RP, Worsley PS, Dilworth MJ, Glenn AR, Howieson JG: Constructs for insertional mutagenesis, transcriptional signal localization and gene regulation studies in root nodule and other bacteria. Microbiology 1999, 145: 1307–1316. PubMed http://dx.doi.org/10.1099/13500872–145–6-1307 10.1099/13500872-145-6-1307
DOE Joint Genome Institute user home http://my.jgi.doe.gov/general/index.html
Bennett S: Solexa Ltd. Pharmacogenomics 2004, 5: 433–438. PubMed http://dx.doi.org/10.1517/14622416.5.4.433 10.1517/14622416.5.4.433
Doe Joint Genome Institute. http://www.jgi.doe.gov
Zerbino DR: Using the Velvet de novo assembler for short-read sequencing technologies. Current Protocols in Bioinformatics 2010, Chapter 11: Unit 11 5.
Ewing B, Green P: Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res 1998, 8: 186–194. PubMed http://dx.doi.org/10.1101/gr.8.3.175
Ewing B, Hillier L, Wendl MC, Green P: Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res 1998, 8: 175–185. PubMed http://dx.doi.org/10.1101/gr.8.3.175 10.1101/gr.8.3.175
Gordon D, Abajian C, Green P: Consed: a graphical tool for sequence finishing. Genome Res 1998, 8: 195–202. PubMed http://dx.doi.org/10.1101/gr.8.3.195 10.1101/gr.8.3.195
Hyatt D, Chen GL, Locascio PF, Land ML, Larimer FW, Hauser LJ: Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 2010, 11: 119. PubMed http://dx.doi.org/10.1186/1471–2105–11–119 10.1186/1471-2105-11-119
Mavromatis K, Ivanova NN, Chen IM, Szeto E, Markowitz VM, Kyrpides NC: The DOE-JGI Standard operating procedure for the annotations of microbial genomes. Stand Genomic Sci 2009, 1: 63–67. PubMed http://dx.doi.org/10.4056/sigs.632 10.4056/sigs.632
Pati A, Ivanova NN, Mikhailova N, Ovchinnikova G, Hooper SD, Lykidis A, Kyrpides NC: GenePRIMP: a gene prediction improvement pipeline for prokaryotic genomes. Nat Methods 2010, 7: 455–457. PubMed http://dx.doi.org/10.1038/nmeth.1457 10.1038/nmeth.1457
Markowitz VM, Mavromatis K, Ivanova NN, Chen IM, Chu K, Kyrpides NC: IMG ER: a system for microbial genome annotation expert review and curation. Bioinformatics 2009, 25: 2271–2278. PubMed http://dx.doi.org/10.1093/bioinformatics/btp393 10.1093/bioinformatics/btp393
Lowe TM, Eddy SR: tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res 1997, 25: 955–964. PubMed http://dx.doi.org/10.1093/nar/25.5.0955 10.1093/nar/25.5.0955
Lagesen K, Hallin P, Rodland EA, Staerfeldt HH, Rognes T, Ussery DW: RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res 2007, 35: 3100–3108. PubMed http://dx.doi.org/10.1093/nar/gkm160 10.1093/nar/gkm160
Griffiths-Jones S, Bateman A, Marshall M, Khanna A, Eddy SR: Rfam: an RNA family database. Nucleic Acids Res 2003, 31: 439–441. PubMed http://dx.doi.org/10.1093/nar/gkg006 10.1093/nar/gkg006
Krogh A, Larsson B, von Heijne G, Sonnhammer ELL: Predicting transmembrane protein topology with a hidden Markov model: Application to complete genomes. J Mol Biol 2001, 305: 567–580. PubMed http://dx.doi.org/10.1006/jmbi.2000.4315 10.1006/jmbi.2000.4315
Bendtsen JD, Nielsen H, von Heijne G, Brunak S: Improved prediction of signal peptides: SignalP 3.0. J Mol Biol 2004, 340: 783–795. PubMed http://dx.doi.org/10.1016/j.jmb.2004.05.028 10.1016/j.jmb.2004.05.028
Acknowledgements
This work was performed under the auspices of the US Department of Energy Office of Science, Biological and Environmental Research Program, and by the University of California, Lawrence Berkeley National Laboratory under contract No. DE-AC02-05CH11231, Lawrence Livermore National Laboratory under Contract No. DE-AC52-07NA27344, and Los Alamos National Laboratory under contract No. DE-AC02-06NA25396. We gratefully acknowledge Strategic Research Funds allocated by Murdoch University to support this project.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
JA, JH and RY supplied the strain and background information for this project and contributed to the assembly of the manuscript with WR, TR supplied DNA to JGI and performed all imaging, WR coordinated the project and all other authors were involved in either sequencing the genome and/or editing the paper. All authors read and approved the final manuscript.
Electronic supplementary material
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Ardley, J., Tian, R., Howieson, J. et al. Genome sequence of the dark pink pigmented Listia bainesii microsymbiont Methylobacterium sp. WSM2598. Stand in Genomic Sci 9, 5 (2014). https://doi.org/10.1186/1944-3277-9-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1944-3277-9-5