
Abstract
Ensifer medicae Di28 is an aerobic, motile, Gram-negative, non-spore-forming rod that can exist as a soil saprophyte or as a legume microsymbiont of Medicago spp. Di28 was isolated in 1998 from a nodule recovered from the roots of M. polymorpha growing in the south east of Sardinia (Italy). Di28 is an effective microsymbiont of the annual forage legumes M. polymorpha and M. murex and is capable of establishing a partially effective symbiotic association with the perennial M. sativa. Here we describe the features of E. medicae Di28, together with genome sequence information and its annotation. The 6,553,624 bp standard draft genome is arranged into 104 scaffolds of 104 contigs containing 6,394 protein-coding genes and 75 RNA-only encoding genes. This rhizobial genome is one of 100 sequenced as part of the DOE Joint Genome Institute 2010 Genomic Encyclopedia for Bacteria and Archaea-Root Nodule Bacteria (GEBA-RNB) project.
Similar content being viewed by others

Introduction
Legumes are key components of sustainable agricultural systems owing to their ability to form nitrogen (N2)-fixing symbioses with specific soil bacteria referred to as rhizobia (or root nodule bacteria). These rhizobia are housed within legume root nodules, where they receive a source of carbon from the legume and in return supply the host with reduced nitrogen (N) in the form of ammonia [1]. The provision of this bioavailable N to the host fuels legume growth and development without the requirement for supplementation with industrially synthesized N-based fertilizers. Furthermore, some of this biologically fixed N remains in the soil after plant harvest or senescence, resulting in an increase in soil fertility. Growing legumes in rotation with a cereal crop or as a source of forage or fodder is therefore an environmentally sustainable way of improving soil fertility and boosting agricultural productivity [2].
The legume genus Medicago is of prime importance globally as a source of forage or fodder. The perennial M. sativa (alfalfa or lucerne) is the most widely cultivated member of this genus, with over 35 million hectares grown annually. Other important species include the annuals M. polymorpha (burr medic), M. truncatula, (barrel medic) and M. murex [2–4]. In order to maximise the agronomic success of these forage legumes, it is imperative that they are well-matched with an effective N2-fixing microsymbiont [5, 6]. While Ensifer meliloti and E. medicae are two species of rhizobia both able to nodulate and fix N2 with Medicago spp., differences exist between these species with regard to their host range and effectiveness. Specifically, E. medicae is an effective N2-fixing symbiont of the acid tolerant annual Medicago spp. (e.g. M. polymorpha, M. murex and M. arabica), whereas E. meliloti adapted to nodulate and fix N2 with the neutral or slightly alkaline-favoring M. truncatula, M. littoralis and M. tornata [7–9].
The strain E. medicae Di28 was isolated in 1998 from a nodule collected from M. polymorpha growing in the south east of Sardinia (Italy) [10]. In common with many E. medicae strains, E. medicae Di28 is only moderately effective as a microsymbiont with M. sativa [8]. However, Di28 is capable of effective N2 fixation with M. murex and M. polymorpha [8]. Therefore, this strain is a valuable resource in improving our understanding of the genetic determinants of highly efficient N2-fixing symbioses and host range, which would complement information already gained from the sequencing of the genomes of other Medicago-nodulating microsymbionts [11–13]. Here we present a summary classification and a set of general features for this microsymbiont together with a description of its genome sequence and annotation.
Organism information
Classification and general features
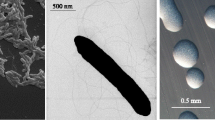
E. medicae Di28 is a motile, Gram-negative rod (Figure 1 Left and Center) in the order Rhizobiales of the class Alphaproteobacteria. It is fast growing, forming colonies within 3–4 days when grown on half strength Lupin Agar (½LA) [14] at 28°C. Colonies on ½LA are white-opaque, slightly domed and moderately mucoid with smooth margins (Figure 1 Right).

Images of Ensifer medicae Di28 using scanning (Left) and transmission (Center) electron microscopy and the appearance of colony morphology on ½LA solid medium (Right).
Minimum Information about the Genome Sequence (MIGS) is provided in Table 1. Figure 2 shows the phylogenetic neighborhood of E. medicae Di28 in a 16S rRNA sequence based tree. This strain shares 100% sequence identity (over 1,290 bp) to the 16S rRNA of the E. medicae A321 type strain and the fully sequenced E. medicae WSM419 [11].

Phylogenetic tree showing the relationship of Ensifer medicae Di28 (shown in bold print) to other Ensifer spp. in the order Rhizobiales based on aligned sequences of the 16S rRNA gene (1,290 bp internal region). All sites were informative and there were no gap-containing sites. Phylogenetic analyses were performed using MEGA, version 5 [27]. The tree was built using the Maximum-Likelihood method with the General Time Reversible model [28]. Bootstrap analysis [29] with 500 replicates was performed to assess the support of the clusters. Type strains are indicated with a superscript T. Brackets after the strain name contain a DNA database accession number and/or a GOLD ID (beginning with the prefix G) for a sequencing project registered in GOLD [30]. Published genomes are indicated with an asterisk.
Evidence codes – IDA: Inferred from Direct Assay; TAS: Traceable Author Statement (i.e., a direct report exists in the literature); NAS: Non-traceable Author Statement (i.e., not directly observed for the living, isolated sample, but based on a generally accepted property for the species, or anecdotal evidence). These evidence codes are from the Gene Ontology project [31].
Symbiotaxonomy
E. medicae strain Di28 was isolated during a germplasm collection carried out in 1998 [10] from a nodule collected from the annual M. polymorpha growing near Villasimius, South East Sardinia (Italy). The site of collection contained ruderal plant species, with soil properties of 1.69% (w/w) organic matter, 0.09% (w/w) total nitrogen and a near-neutral pH. Along with M. polymorpha, other Medicago spp. present at the sampling site were M. rugosa, M. littoralis and M. rigidula. Di28 forms nodules and fixes N2 with M. sativa, M. polymorpha and M. murex. However, while Di28 is fully effective for N2 fixation with M. murex and M. polymorpha, it is only partially effective as a microsymbiont of M. sativa.
Genome sequencing and annotation information
Genome project history
This organism was selected for sequencing on the basis of its environmental and agricultural relevance to issues in global carbon cycling, alternative energy production, and biogeochemical importance, and is part of the Community Sequencing Program at the U.S. Department of Energy, Joint Genome Institute (JGI) for projects of relevance to agency missions. The genome project is deposited in the Genomes OnLine Database [30] and a standard draft genome sequence in IMG. Sequencing, finishing and annotation were performed by the JGI. A summary of the project information is shown in Table 2.
Growth conditions and genomic DNA preparation
E. medicae Di28 was cultured to mid logarithmic phase in 60 ml of TY rich media [32] on a gyratory shaker at 28°C at 250 rpm. DNA was isolated from the cells using a CTAB (Cetyl trimethyl ammonium bromide) bacterial genomic DNA isolation method [33].
Genome sequencing and assembly
The genome of Ensifer medicae Di28 was sequenced at the Joint Genome Institute (JGI) using Illumina technology [34]. An Illumina standard paired-end library was constructed and sequenced using the Illumina HiSeq 2000 platform which generated 16,333,536 reads totaling 2,450 Mbp.
All general aspects of library construction and sequencing performed at the JGI can be found at DOE Joint Genome Institute user homepage. All raw Illumina sequence data was passed through DUK, a filtering program developed at JGI, which removes known Illumina sequencing and library preparation artifacts (Mingkun, L., Copeland, A. and Han, J., unpublished). The following steps were then performed for assembly: (1) filtered Illumina reads were assembled using Velvet [35] (version 1.1.04), (2) 1–3 Kb simulated paired end reads were created from Velvet contigs using wgsim [36], (3) Illumina reads were assembled with simulated read pairs using Allpaths–LG [37] (version r39750). Parameters for assembly steps were: 1) Velvet (velveth: 63 –shortPaired and velvetg: -veryclean yes –exportFiltered yes –mincontiglgth 500 –scaffolding no–covcutoff 10) 2) wgsim (-e 0–1 76–2 76 -r 0 -R 0 -X 0) 3) Allpaths–LG (PrepareAllpathsInputs:PHRED64 = 1 PLOIDY = 1 FRAGCOVERAGE = 125 JUMPCOVERAGE = 25 LONGJUMPCOV = 50, RunAllpath-sLG: THREADS = 8 RUN = stdshredpairs TARGETS = standard VAPIWARNONLY = True OVERWRITE = True). The final draft assembly contained 104 contigs in 104 scaffolds. The total size of the genome is 6.5 Mbp and the final assembly is based on 2,450 Mbp of Illumina data, which provides an average 374× coverage of the genome.
Genome annotation
Genes were identified using Prodigal [38] as part of the DOE-JGI annotation pipeline [39]. The predicted CDSs were translated and used to search the National Center for Biotechnology Information (NCBI) nonredundant database, UniProt, TIGRFam, Pfam, PRIAM, KEGG, COG, and InterPro databases. The tRNAScanSE tool [40] was used to find tRNA genes, whereas ribosomal RNA genes were found by searches against models of the ribosomal RNA genes built from SILVA [41]. Other non–coding RNAs such as the RNA components of the protein secretion complex and the RNase P were identified by searching the genome for the corresponding Rfam profiles using INFERNAL [42]. Additional gene prediction analysis and manual functional annotation was performed within the Integrated Microbial Genomes (IMG-ER) platform [43].
Genome properties
The genome is 6,553,624 nucleotides with 61.15% GC content (Table 3) and comprised of 104 scaffolds (Figure 3) of 104 contigs. From a total of 6,469 genes, 6,394 were protein encoding and 75 RNA only encoding genes. The majority of genes (78.65%) were assigned a putative function whilst the remaining genes were annotated as hypothetical. The distribution of genes into COGs functional categories is presented in Table 4.

Graphical map of the genome of Ensifer medicae Di28 showing the seven largest scaffolds. From bottom to the top of each scaffold: Genes on forward strand (color by COG categories as denoted by the IMG platform), Genes on reverse strand (color by COG categories), RNA genes (tRNAs green, sRNAs red, other RNAs black), GC content, GC skew.
Conclusions
Di28 was isolated from a nodule of M. polymorpha found in Sardinian soil of near-neutral pH. The genome size, gene count, GC content and COG profile of Di28 is comparable to that of the sequenced E. medicae strains WSM244, WSM419, WSM1115, WSM1369 and WSM4191. Of particular interest is the finding that Di28, WSM244 and WSM1369 have a relatively low pseudogene percentage (0.03-0.06%) in comparison to the other strains (4.29-6.83%). One stand-out feature from the genome of Di28 is the absence of the acid-activated lpiA gene (11,32), which is found in all other E. meliloti and E. medicae strains sequenced to date. Furthermore, the regulatory genes tcsA, tcrA and fsrR, which are required for the full acid-activated expression of lpiA, are present in all other sequenced E. medicae strains, but are absent in Di28. The unique attributes of Di28 in comparison to other Ensifer strains, make this an ideal candidate for future work.
References
Terpolilli JJ, Hood GA, Poole PS: What determines the efficiency of N 2 -fixing Rhizobium -Legume symbioses? Adv Microb Physiol 2012, 60: 325–89. PubMed http://dx.doi.org/10.1016/B978–0-12–398264–3.00005-X
Howieson JG, O’Hara GW, Carr SJ: Changing roles for legumes in Mediterranean agriculture: developments from an Australian perspective. Field Crops Res 2000, 65: 107–22. http://dx.doi.org/10.1016/S0378–4290(99)00081–7 10.1016/S0378-4290(99)00081-7
Radović J, Sokolović D, Marković J: Alfalfa-most important perennial forage legume in animal husbandry. Biotech Anim Husbandry 2009, 25: 465–75. http://dx.doi.org/10.2298/BAH0906465R 10.2298/BAH0906465R
Howieson JG, Ewing MA: Acid tolerance in the Rhizobium meliloti-Medicago symbiosis. Aust J Agric Res 1986, 37: 55–64. http://dx.doi.org/10.1071/AR9860055 10.1071/AR9860055
Sessitsch A, Howieson JG, Perret X, Antoun H, Martinez-Romero E: Advances in Rhizobium research. Crit Rev Plant Sci 2002, 21: 323–78. http://dx.doi.org/10.1080/0735–260291044278 10.1080/0735-260291044278
Evans PM, Howieson JG, Nutt BJ: Increased yield and persistence of several annual medic species and Medicago sativa by inoculation with selected strains of Sinorhizobium meliloti and S. medicae . Aust J Exp Agric 2005, 45: 217–24. http://dx.doi.org/10.1071/EA03128 10.1071/EA03128
Rome S, Brunel B, Normand P, Fernandez M, Cleyet-Marel JC: Evidence that two genomic species of Rhizobium are associated with Medicago truncatula . Arch Microbiol 1996, 165: 285–8. PubMed http://dx.doi.org/10.1007/s002030050328 10.1007/s002030050328
Garau G, Reeve WG, Brau L, Yates RJ, James D, Tiwari R, O’Hara GW, Howieson JG: The symbiotic requirements of different Medicago spp. suggest the evolution of Sinorhizobium meliloti and S. medicae with hosts differentially adapted to soil pH. Plant Soil 2005, 276: 263–77. http://dx.doi.org/10.1007/s11104–005–0374–0 10.1007/s11104-005-0374-0
Terpolilli JJ, O’Hara GW, Tiwari RP, Dilworth MJ, Howieson JG: The model legume Medicago truncatula A17 is poorly matched for N 2 fixation with the sequenced microsymbiont Sinorhizobium meliloti 1021. New Phytol 2008, 179: 62–6. PubMed http://dx.doi.org/10.1111/j.1469–8137.2008.02464.x 10.1111/j.1469-8137.2008.02464.x
Brundu G, Camarda I, Caredda M, Garau G, Maltoni S, Deiana P: A contribution to the study of the distribtion of Medicago-Sinorhizobium symbiosis in Sardinia (Italy). Agricoltura Mediterranea 2004, 134: 33–48.
Reeve W, Chain P, O’Hara G, Ardley J, Nandesena K, Brau L, Tiwari R, Malfatti S, Kiss H, Lapidus A, et al.: Complete genome sequence of the Medicago microsymbiont Ensifer ( Sinorhizobium ) medicae strain WSM419. Stand Genomic Sci 2010, 2: 77–86. PubMed http://dx.doi.org/10.4056/sigs.43526 10.4056/sigs.43526
Terpolilli J, Hill Y, Tian R, Howieson J, Bräu L, Goodwin L, Han J, Liolios K, Huntemann M, Pati A, et al.: Genome sequence of Ensifer meliloti strain WSM1022; a highly effective microsymbiont of the model legume Medicago truncatula A17. Stand Genomic Sci 2013, 9: 315–324. 10.4056/sigs.4608286
Terpolilli J, Garau G, Hill Y, Tian R, Howieson J, Bräu L, Goodwin L, Han J, Liolios K, Huntemann M, et al.: Genome sequence of Ensifer medicae strain WSM1369; an effective microsymbiont of the annual legume Medicago sphaerocarpos . Stand Genomic Sci 2013, 9: 420–430. 10.4056/sigs.4838624
Howieson JG, Ewing MA, D’antuono MF: Selection for acid tolerance in Rhizobium meliloti . Plant Soil 1988, 105: 179–88. http://dx.doi.org/10.1007/BF02376781 10.1007/BF02376781
Field D, Garrity G, Gray T, Morrison N, Selengut J, Sterk P, Tatusova T, Thomson N, Allen MJ, Angiuoli SV, et al.: Towards a richer description of our complete collection of genomes and metagenomes “Minimum Information about a Genome Sequence” (MIGS) specification. Nat Biotechnol 2008, 26: 541–7. PubMed http://dx.doi.org/10.1038/nbt1360 10.1038/nbt1360
Woese CR, Kandler O, Wheelis ML: Towards a natural system of organisms: proposal for the domains Archaea , Bacteria, and Eucarya. Proc Natl Acad Sci USA 1990, 87: 4576–9. PubMed http://dx.doi.org/10.1073/pnas.87.12.4576 10.1073/pnas.87.12.4576
Garrity GM, Bell JA, Lilburn T: Phylum XIV. Proteobacteria phyl. nov. In Bergey’s Manual of Systematic Bacteriology, Second Edition, Volume 2, Part B. Edited by: Garrity GM, Brenner DJ, Krieg NR, Staley JT. New York: Springer; 2005:1.
Garrity GM, Bell JA, Lilburn T: Class I. Alphaproteobacteria class. In Bergey’s Manual of Systematic Bacteriology. Second ed. Edited by: Garrity GM, Brenner DJ, Kreig NR, Staley JT. New York: Springer - Verlag; 2005.
Validation List No. 107. List of new names and new combinations previously effectively, but not validly, published Int J Syst Evol Microbiol 2006, 56: 1–6. PubMed http://dx.doi.org/10.1099/ijs.0.64188–0
Kuykendall LD: Order VI: Rhizobiales ord. nov. In Bergey’s Manual of Systematic Bacteriology. Second ed. Edited by: Garrity GM, Brenner DJ, Kreig NR, Staley JT. New York: Springer - Verlag; 2005:324.
Skerman VBD, McGowan V, Sneath PHA: Approved lists of bacterial names. Int J Syst Bacteriol 1980, 30: 225–420. http://dx.doi.org/10.1099/00207713–30–1-225 10.1099/00207713-30-1-225
Conn HJ: Taxonomic relationships of certain non-sporeforming rods in soil. J Bacteriol 1938, 36: 320–1.
Casida LE: Ensifer adhaerens gen. nov., sp. nov.: a bacterial predator of bacteria in soil. Int J Syst Bacteriol 1982, 32: 339–45. http://dx.doi.org/10.1099/00207713–32–3-339 10.1099/00207713-32-3-339
Young JM: The genus name Ensifer Casida 1982 takes priority over Sinorhizobium Chen et al. 1988, and Sinorhizobium morelense Wang et al. 2002 is a later synonym of Ensifer adhaerens Casida 1982. Is the combination Sinorhizobium adhaerens (Casida 1982) Willems et al. 2003 legitimate? Request for an Opinion. Int J Syst Evol Microbiol 2003, 53: 2107–10. PubMed http://dx.doi.org/10.1099/ijs.0.02665–0 10.1099/ijs.0.02665-0
Judicial Commission of the International Committee on Systematics of Prokaryotes: The genus name Sinorhizobium Chen et al. 1988 is a later synonym of Ensifer Casida 1982 and is not conserved over the latter genus name, and the species name ‘ Sinorhizobium adhaerens ’ is not validly published. Opinion 84. Int J Syst Evol Microbiol 2008, 58: 1973. PubMed http://dx.doi.org/10.1099/ijs.0.2008/005991–0
Biological Agents: Technical rules for biological agents. TRBA; ( ):466 http://www.baua.de
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S: MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 2011, 28: 2731–9. PubMed http://dx.doi.org/10.1093/molbev/msr121 10.1093/molbev/msr121
Nei M, Kumar S: Molecular Evolution and Phylogenetics. New York: Oxford University Press; 2000.
Felsenstein J: Confidence limits on phylogenies: an approach using the bootstrap. Evolution 1985, 39: 783–91. http://dx.doi.org/10.2307/2408678 10.2307/2408678
Liolios K, Mavromatis K, Tavernarakis N, Kyrpides NC: The Genomes On Line Database (GOLD) in 2007: status of genomic and metagenomic projects and their associated metadata. Nucleic Acids Res 2008, 36: D475–9. PubMed http://dx.doi.org/10.1093/nar/gkm884 10.1093/nar/gkn240
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al.: Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 2000, 25: 25–9. PubMed http://dx.doi.org/10.1038/75556 10.1038/75556
Reeve WG, Tiwari RP, Worsley PS, Dilworth MJ, Glenn AR, Howieson JG: Constructs for insertional mutagenesis, transcriptional signal localization and gene regulation studies in root nodule and other bacteria. Microbiology 1999, 145: 1307–16. PubMed http://dx.doi.org/10.1099/13500872–145–6-1307 10.1099/13500872-145-6-1307
DOE Joint Genome Institute user homepage http://my.jgi.doe.gov/general/index.html
Bennett S: Solexa Ltd. Pharmacogenomics 2004, 5: 433–8. PubMed http://dx.doi.org/10.1517/14622416.5.4.433 10.1517/14622416.5.4.433
Zerbino DR: Using the Velvet de novo assembler for short-read sequencing technologies. Current Protocols in Bioinformatics 2010, Chapter 11: Unit 11 5.
Gnerre S, MacCallum I, Przybylski D, Ribeiro FJ, Burton JN, Walker BJ, Sharpe T, Hall G, Shea TP, Sykes S, et al.: High-quality draft assemblies of mammalian genomes from massively parallel sequence data. Proc Natl Acad Sci USA 2011, 108: 1513–8. PubMed http://dx.doi.org/10.1073/pnas.1017351108 10.1073/pnas.1017351108
Hyatt D, Chen GL, Locascio PF, Land ML, Larimer FW, Hauser LJ: Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 2010, 11: 119. PubMed http://dx.doi.org/10.1186/1471–2105–11–119 10.1186/1471-2105-11-119
Mavromatis K, Ivanova NN, Chen IM, Szeto E, Markowitz VM, Kyrpides NC: The DOE-JGI Standard operating procedure for the annotations of microbial genomes. Stand Genomic Sci 2009, 1: 63–7. PubMed http://dx.doi.org/10.4056/sigs.632 10.4056/sigs.632
Lowe TM, Eddy SR: tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res 1997, 25: 955–64. PubMed http://dx.doi.org/10.1093/nar/25.5.0955 10.1093/nar/25.5.0955
Pruesse E, Quast C, Knittel K, Fuchs BM, Ludwig W, Peplies J, Glöckner FO: SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res 2007, 35: 7188–96. PubMed http://dx.doi.org/10.1093/nar/gkm864 10.1093/nar/gkm864
INFERNAL http://infernal.janelia.org
Markowitz VM, Mavromatis K, Ivanova NN, Chen IM, Chu K, Kyrpides NC: IMG ER: a system for microbial genome annotation expert review and curation. Bioinformatics 2009, 25: 2271–8. PubMed http://dx.doi.org/10.1093/bioinformatics/btp393 10.1093/bioinformatics/btp393
Acknowledgements
This work was performed under the auspices of the US Department of Energy Office of Science, Biological and Environmental Research Program, and by the University of California, Lawrence Berkeley National Laboratory under contract No. DE-AC02-05CH11231, Lawrence Livermore National Laboratory under Contract No. DE-AC52-07NA27344, and Los Alamos National Laboratory under contract No. DE-AC02-06NA25396. We gratefully acknowledge Early Career Research funding for J. Terpolilli awarded by the School of Veterinary and Life Sciences at Murdoch University.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
GG and JH supplied the strain and background information for this project, TR supplied DNA to JGI and performed all imaging, GG and JT drafted the paper, YH performed phylogenetic analysis, WR coordinated the project and all other authors were involved in either sequencing the genome and/or editing the paper. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Garau, G., Terpolilli, J., Hill, Y. et al. Genome sequence of Ensifer medicae Di28; an effective N2-fixing microsymbiont of Medicago murex and M. polymorpha . Stand in Genomic Sci 9, 4 (2014). https://doi.org/10.1186/1944-3277-9-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1944-3277-9-4