
Abstract
Background
Due to the effect of midgut bacteria on proliferation of parasites and their potential as paratransgenesis tools, their identification in malaria vector mosquitoes is important. Anopheles culicifacies s.l. is one of the main malaria vectors in Asia; however, its midgut microbiota remains un-studied. This work was primarily designed to isolate potential candidates for use in a paratransgenesis approach, but also to give a picture of the midgut microbiota of wild-caught An. culicifacies larvae and adults from the southeast corner of Iran, which has the highest malaria endemicity in the country.
Methods
A total of 68 larvae and 34 adult females (newly eclosed and older) from three different biotopes in Iran were analyzed for their midgut microflora. The mosquitoes had their midgut bacterial contents plated on three different culture media (brain heart agar, nutrient agar and blood agar) yielding 57 bacterial isolates. The 16S rRNA genes of the isolates were sequence analyzed for species designation, which then was confirmed by biochemical analysis.
Results
A total of twelve bacterial genera were identified: Acinetobacter, Aeromonas, Bacillus, Chryseobacterium, Delftia, Exiguobacterium, Kurthia, Microbacterium, Pseudomonas, Staphylococcus, Thorsellia and Variovorax. In older females, only Gram-negative bacteria were found, whereas larvae and newly-eclosed adults also harbored Gram-positive bacteria. The diversity of isolates also varied between sampling sites and mosquito stages, with the largest number of genera found in the Anguri district and in larvae, respectively. Pseudomonas was the most common genus retrieved from all sampling sites, and in both larvae and adults, suggesting a potential transstadial passage of these bacteria. Interestingly, identical 16S sequences of Pseudomonas were found in mosquitoes originating from different habitats at least 45 km apart, which could suggest that these bacteria have been adapted to the mosquitoes.
Conclusions
The study of vector mosquito microbiota has recently gathered increased interest because of the potential influence on vector competence. By adding data from a hitherto uncharacterized malaria mosquito, a better picture of gut flora in vector mosquitoes was obtained. Furthermore, some species of the predominant genus Pseudomonas will be evaluated for the selection of a paratransgenesis candidate.
Similar content being viewed by others


Background
Vector-borne diseases cause health problems in several parts of the world and among them malaria is the most important with almost 700,000 deaths annually [1]. Malaria control programs focused on mosquito vectors have reduced mortality and morbidity, but emerging insecticide-resistant vectors and environmental issues related to application of pesticides have necessitated development of new control strategies with less environmental impacts/damage and higher efficacy [2, 3].
One of the control approaches under development is paratransgenesis, which has been defined as using symbiotic organisms (such as bacteria) to deliver anti-parasitic effector molecules to wild vector populations [4]. Initial steps of developing paratransgenesis against malaria have been taken in laboratory experiments on An. gambiae mosquitoes. In an early study, a single-chain antibody targeting Plasmodium berghei ookinete Pbs21 was linked to the lytic peptide Shiva-1 and expressed in Escherichia coli, which resulted in 95.6% transmission blockage [5]. More recently, it was shown that engineered Pantoea agglomerans bacteria isolated from mosquitoes inhibited development of the human malaria parasite Plasmodium falciparum and the rodent malaria parasite Plasmodium berghei by up to 98% [6].
In mosquitoes, obligate symbionts are yet to be found; therefore, a first step in paratransgenesis is to identify the normal midgut microbiota of mosquitoes and to isolate candidates for further modification [7, 8]. So far, a limited number of studies have been carried out on the microbiota of Anopheles mosquitoes [9]. The studied species include: laboratory-reared Anopheles stephensi, An. gambiae, and An. albimanus[10], field-collected An. albimanus from southern Mexico [11], field-collected An. gambiae s.s., An. arabiensis and An. funestus from western Kenya [12], field-collected An. darlingi from Brazil [13], laboratory-reared and wild-caught An. stephensi from India [14], semi-field collected An. gambiae from Kenya [15], field-collected An. maculipennis and An. stephensi from Iran [16], and field-collected An. stephensi from Iran [17].
An. culicifacies Giles is one of the main malaria vectors in the tropical parts of South and Southwest Asia [18–23], where it transmits both Plasmodium falciparum and P. vivax[24]. In this study, culturable midgut bacteria isolated from wild-caught An. culicifacies in southeastern Iran are identified using 16S rRNA sequence analysis with the aim of selecting potential candidates for paratransgenesis.
Methods
Ethics statement
Prior to the approval of all projects by the Tehran University of Medical Sciences (TUMS), they are reviewed and endorsed by the ethical committee of the TUMS. Mosquito collection was carried out from private dwellings. At least one week prior to any mosquito collection, the owners were informed by the Local Health System officers. The research and its objectives were explained by Ali Reza Chavshin (ARC), to residents. Owners of the land (for larval collection) and dwellings (for adult mosquito collection) gave permission to conduct the study on these sites. After their permission, the samples were collected at an agreed date and time. The whole process was coordinated, managed and documented by the “Local manager of malaria control program” and “Local Health System officer” in the study areas and signed by ARC. Also it is declared that the collected species is not endangered or protected in the area of investigation.
Field collection of An. culicifacies and isolation of midgut bacteria
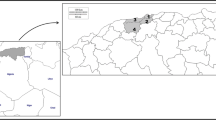
Wild caught mosquito samples were collected during July-September 2010, from 1) the Iranshahr district, an urban region; 2) the Anguri district, a mountainous or hilly rural region with temporal rivers; and 3) the Saraydan district, a rural plain region (Table 1). The distances between the three sites are 45–90 km (Iranshahr to Saraydan 45 km, Saraydan to Anguri 50 km, and Iranshahr to Anguri 90 km). All three areas are parts of the Sistan and Balouchistan Province, which borders to Pakistan and comprises the most important urban and rural malaria foci in southeastern Iran (Figure 1).

Map of Iran indicating the locations of the Sistan-va-Baluchistan Province, and the districts/villages where the specimens were collected. 1) Iranshahr, 2) Anguri, and 3) Saraydan.
A standard dipping technique was used for larval sample collection (350 ml dipper). Adult mosquitoes were collected by aspiration from the walls and the roofs of dwellings and pit shelters where adults normally rest after feeding [25]. The live specimens were transferred to the laboratory of Iranshahr, National Institute of Health Research (NIHR). All collected samples were identified to species level using standard morphological keys [26]. In this part of Iran, An. culicifacies subspecies A is the most abundant [27]. Among the identified species, the An. culicifacies s.l. were separated and analyzed for midgut microbiota. Specimens analyzed for midgut microbiota were from three sources: 1) wild-caught females, 2) wild-caught fourth instar larvae, and 3) newly emerged female adults (immediately dissected after emergence from pupa reared from a subset of the larvae in 2).
Preparation, sterilization and dissection of specimens were done according to a previously described method [28]. Obtained midguts were mashed and suspended in 500 μL of Brain Heart Infusion (BHI). A 100 μL aliquot of the contents was serially diluted up to 10-6 and plated onto different media: 1) Brain Heart agar (BHA), 2) Nutrient agar (NA), and 3) Blood agar (BA) (Merck, Germany) and incubated at 28 ± 2°C for 24-48 hours. The sterility of all reagents was checked and controls for the efficiency of sterilization were treated like the other samples. Continuous sub-culture of every grown bacterial colony was performed in order to isolate single purified colonies of the bacteria. The single colonies of the bacteria were later used for DNA extraction and PCR, and biochemical investigations.
DNA extraction, 16S rRNA gene amplification and sequencing
Each purified bacterial colony was subjected to genomic DNA extraction using the QIAGEN DNeasy Kit (Qiagen, Germany) according to the manufacturer’s instructions. The 16S rRNA universal primers 16suF 5′-GAGTTTGATCCTGGCTCAG-3′ and 16suR 5′-GTTACCTTGTTACGACTT-3′ [29] were used to amplify about 1500 bp long sequences. The PCR program had an initial denaturation at 94°C for 10 min, followed by 35 cycles of denaturation at 95°C for 30 s, annealing at 56.5°C for 40 s, and extension at 72°C for 30 s, followed by a final extension at 72°C for 8 min. All amplicons were sent for sequencing to SeqLab (Germany). The Mallard program (http://www.bioinformatics-toolkit.org) was used for all acquired sequences to check the presence of probable chimeric sequences and the specimens with suspicious sequences were removed from the data set. The resultant sequences were compared to the data-bases of the Ribosomal Database Project (RDP II; Michigan State University, http://rdp.cme.msu.edu) and the GenBank (http//:http://www.ncbi.nlm.nih.gov/BLAST) for confident sequence analysis and their seq-match and sequences similarity check tools were used.
Based on sequence comparison with the GenBank and RDPII entries, identification of isolates and their classification at genus and species level was done; 99 percent or higher sequence identities with the GenBank entries were assumed for species delineation [30]. All isolates were also identified using classical phenotyping and biochemical methods such as Gram-staining, oxi/ferm tests and using selective cultivation media [31]. The results of biochemical and phenotyping methods were compared to the sequencing results and only those bacteria that confirmed the sequencing results are presented. All confirmed sequences were submitted to GenBank. All GenBank reference numbers are displayed in Figure 2.

Phylogenetic analysis of 16S rRNA sequences (~1500 bp) of all bacteria isolated from An. culicifacies using Maximum Likelihood (ML) based on the Tamura 3-parameter model[33]. The first part of the sequence ID indicates from which life stage the bacteria were isolated, adult (A), newly emerged adult (NEA), or larva (L). The second part of the sequence ID denotes the sampling locality, Anguri (A), Iranshahr (I), or Saraydan (S). The third part of the sequence ID is the GenBank accession number. Only the significant bootstrap values (>50%) from 1000 replicates are shown on the nodes.
The MEGA5.05 [32] software was used for phylogenetic analysis and tree construction. Phylogenetic trees were built using Maximum Likelihood (ML) based on the Tamura 3-parameter model [33] (1000 bootstrap replicates) analyses.
Results
In this study, midguts of a total of 102 specimens of An. culicifacies (68 larvae and 34 adults) from three different sampling sites were analyzed (Table 1). The specimens were dissected and screened for cell-free cultivable bacteria on different media resulting in a total of 57 bacterial isolates (Table 2). Forty-one isolates were members of seven genera of Gram-negative bacteria and the remaining 16 isolates belonged to Gram-positive bacteria. Gram-negative bacteria were isolated from larvae and both adult stages, but Gram-positive bacteria were only isolated from larvae and newly emerged adults. The domination of Gram-negative bacteria was statistically significant in larvae and in mosquitoes from the Iranshahr area (Table 3). Sequence data of the bacteria that were found in the midgut of larval and adult stages were used for phylogenetic analysis.
A total of 19 bacterial species were isolated and identified from the different stages of the collected An. culicifacies specimens. In Figure 2, all non-identical 16S sequences are represented with information on stage and sampling locality. According to the number of isolated and identified species, it was shown that the larval stage of the wild-caught samples had a higher diversity with 17 species divided in 12 genera of bacteria: Acinetobacter, Aeromonas, Bacillus, Chryseobacterium, Delftia, Exiguobacterium, Kurthia, Microbacterium, Pseudomonas, Staphylococcus, Thorsellia and Variovorax. In contrast, in wild-caught adults only three species from three genera were identified: Brachybacterium, Pseudomonas and Variovorax. In the newly emerged adults from wild caught larvae four species from three genera of bacteria were identified: Acinetobacter, Bacillus, and Pseudomonas (Table 4).
The genus Pseudomonas was the most frequently isolated bacteria in this study (20/57, 57%) and members of Pseudomonas have been also identified in other malaria vectors [10, 13, 14, 16, 17]. To show the relationship between sequence data and origin of samples, all the sequences belonging to the genus Pseudomonas from An. darlingi, An. gambiae, An. maculipennis and An. stephensi available in GenBank were retrieved and analyzed together with the ones found for An. culicifacies in this study (Figure 3).

Phylogenetic analysis of 16S rRNA sequences (~1500 bp) of Pseudomonas strains using Maximum Likelihood (ML) based on the Tamura 3-parameter model[33]from different Anopheles mosquitoes: An. culicifacies from Iran (Ir_Cu; this study), An. darlingi from Brazil (Br_DA; [[13]]), An. gambiae from Kenya (Ke_GA; [[12]]), An. stephensi from India (In_St; [[14]]), An. stephensi from Iran (Ir_St; [[17]]) and An. maculipennis from Iran (Ir_Maculi; [[16]]). Only the significant bootstrap values ( >50%) are shown on the nodes. Scale of genetic distance is shown underneath.
Discussion
Identification of gut bacteria in vector mosquitoes has received notable attention since recent studies suggest that the composition of the vector gut microbiota affects the outcome of mosquito infection with Plasmodium parasites [34, 35]. This corroborates earlier results, for example that some bacteria have a natural anti-Plasmodium effect [10, 11, 36]. Although a number of studies have been carried out to identify the bacterial microbiota of Anopheles mosquitoes, several important vectors remain un-studied. Among those was An. culicifacies, which has been investigated in the current study, and from which aerobic midgut bacteria have been isolated and identified. Strikingly, as seen by the lack of branch length differences between the isolates in Figure 2, many are very similar despite coming from different stages and different areas. This suggests that the bacteria acquired by An. culicifacies is not a random selection, but instead points to possible co-adaptation between bacteria and mosquitoes and/or their breeding waters.
Albeit the number of isolates in this study is moderate, the data suggest that Gram-negative bacteria dominate the flora of An. culicifacies (Table 3). This agrees with earlier results from culture-based studies on An. stephensi and An. maculipennis in Iran [16] and An. gambiae and An. funestus in Kenya and Mali [37], and also data from sequence-based studies on An. darlingi in Brazil [13] and An. gambiae in Kenya [15] all showing predominance for Gram-negative bacteria. In Wang et al. [15], the proportion of Gram-negative bacteria increases from larvae to adults, similar to what we see for An. culicifacies.
Comparing the results of this study with other similar studies showed that some bacteria are common among several important vectors. For instance the genus Thorsellia, which was isolated from An. arabiensis[12] and An. stephensi[14] and also found in An. gambiae s.l. [15, 38], has now been identified in An. culicifacies (this study). In two different Kenyan populations of An. gambiae mosquitoes, Thorsellia was the dominant genus [15, 38]. Briones et al. [38] consistently isolated T. anophelis from the water surface micro-layer (SML), i.e., where Anopheles larvae feed, as well as in 40% of the adults, while Wang et al. [15] found that almost 70% of the bacteria in young adults belonged to the Thorsellia genus. Thorsellia is adapted to a life in mosquitoes with high tolerance for the alkaline conditions found in the larvae and with increased growth rates in blood medium [38]. Thorsellia has only been found in vector mosquitoes and their breeding waters, and appears to be a unique genus of bacteria only distantly related to other members of gamma-proteobacteria [39].
Another common bacterial genus is Pseudomonas, which has been found among several mosquito vectors in Asia and the Americas [11, 13, 14, 17, 36]; however, a recent study found Pseudomonas only at a low level in Kenyan mosquitoes [40]. In this study, we identified Pseudomonas as the most frequently isolated bacteria from An. culicifacies. The same finding was made previously in An. stephensi midguts [17]. It should be noted that although Pseudomonas is the most frequently isolated species, other non-culturable species may be important constituents of the midgut microbiota in An. culicifacies. However, even in studies on mosquito gut flora mainly using PCR-based methods for identification, those bacteria possible to grow under laboratory conditions dominate the gut flora [13, 15, 40]. The high frequency of Pseudomonas isolates is promising for a paratransgenesis approach also because of its possibility to grow in cell-free and ordinary culture media and suitability for genetic transformation [41]. However, characteristics such as transstadial transmission, non-pathogenicity, immunological and physiological adaptation to mosquito midgut conditions, colonization in the mosquito midgut including effective competition with resident bacteria and persistency in the gut for a reasonable time, should be studied before selection of the isolates for paratransgenesis. A note of caution is that among the more than 100 Pseudomonas species some are pathogenic; for example, Pseudomonas aeruginosa is an opportunistic human pathogen in severely immunocompromised patients [42]. Therefore, care must be taken so that for a paratransgenesis approach only Pseudomonas species that are non-pathogenic should be selected.
In a paratransgenesis strategy against mosquito-borne diseases, it would be an advantage if the transformed bacteria expressing effector molecules could remain in the vector population. Some kind of transstadial transmission would therefore be an essential characteristic for an ideal paratransgenesis candidate. However, it was suggested that most bacteria are lost during metamorphosis in the pupal stage [43]. In this study, the sequence and phylogenetic analysis showed that some isolates of the genus Pseudomonas were common and present in field-collected larvae, adults, and newly emerged adults from field-collected larvae. Some of the Pseudomonas isolates found in different stages of An. culicifacies from the same area have 16S sequences with 100% sequence identity suggesting transstadial transmission. This finding is similar to a possible transstadial transmission of some Pseudomonas isolates in An. stephensi[17], but needs to be further investigated.
Conclusions
This is the first study on microbiota in An. culicifacies, an important vector of malaria in Asia. Further studies are needed regarding the biological characteristics of the bacteria and interactions between the gut microbiota and the host. The fact that Pseudomonas bacteria with identical 16S sequences are present both in different locations and different stages could suggest that this species has adapted to a life in An. culicifacies. The potential symbiotic relationship between An. culicifacies and Pseudomonas makes it a good candidate for paratransgenesis.
References
WHO: World Malaria Report. 2012, Geneva: World Health Organization
Coutinho-Abreu IV, Zhu KY, Ramalho-Ortigao M: Transgenesis and paratransgenesis to control insect-borne diseases: current status and future challenges. Parasitol Int. 2010, 59: 1-8. 10.1016/j.parint.2009.10.002.
Hill CA, Kafatos FC, Stansfield SK, Collins FH: Arthropod-borne diseases: vector control in the genomics era. Nat Rev Microbiol. 2005, 3: 262-268. 10.1038/nrmicro1101.
Beard CB, Cordon-Rosales C, Durvasula RV: Bacterial symbionts of the triatominae and their potential use in control of Chagas disease transmission. Annu Rev Entomol. 2002, 47: 123-141. 10.1146/annurev.ento.47.091201.145144.
Yoshida S, Ioka D, Matsuoka H, Endo H, Ishii A: Bacteria expressing single-chain immunotoxin inhibit malaria parasite development in mosquitoes. Mol Biochem Parasitol. 2001, 113: 89-96. 10.1016/S0166-6851(00)00387-X.
Wang S, Ghosh AK, Bongio N, Stebbings KA, Lampe DJ, Jacobs-Lorena M: Fighting malaria with engineered symbiotic bacteria from vector mosquitoes. Proc Natl Acad Sci U S A. 2012, 109: 12734-12739. 10.1073/pnas.1204158109.
Ricci I, Damiani C, Rossi P, Capone A, Scuppa P, Cappelli A, Ulissi U, Mosca M, Valzano M, Epis S: Mosquito symbioses: from basic research to the paratransgenic control of mosquito-borne diseases. J Appl Entomol. 2011, 135: 487-493. 10.1111/j.1439-0418.2011.01613.x.
Riehle MA, Jacobs-Lorena M: Using bacteria to express and display anti-parasite molecules in mosquitoes: current and future strategies. Insect Biochem Mol Biol. 2005, 35: 699-707. 10.1016/j.ibmb.2005.02.008.
Minard G, Mavingui P, Moro CV: Diversity and function of bacterial microbiota in the mosquito holobiont. Parasit Vectors. 2013, 6: 146-10.1186/1756-3305-6-146.
Pumpuni CB, Demaio J, Kent M, Davis JR, Beier JC: Bacterial population dynamics in three anopheline species: the impact on Plasmodium sporogonic development. Am J Trop Med Hyg. 1996, 54: 214-218.
Gonzalez-Ceron L, Santillan F, Rodriguez MH, Mendez D, Hernandez-Avila JE: Bacteria in midguts of field-collected Anopheles albimanus block Plasmodium vivax sporogonic development. J Med Entomol. 2003, 40: 371-374. 10.1603/0022-2585-40.3.371.
Lindh JM, Terenius O, Faye I: 16S rRNA gene-based identification of midgut bacteria from field-caught Anopheles gambiae sensu lato and A. funestus mosquitoes reveals new species related to known insect symbionts. Appl Environ Microbiol. 2005, 71: 7217-7223. 10.1128/AEM.71.11.7217-7223.2005.
Terenius O, De Oliveira CD, Pinheiro WD, Tadei WP, James AA, Marinotti O: 16S rRNA gene sequences from bacteria associated with adult Anopheles darlingi (Diptera: Culicidae) mosquitoes. J Med Entomol. 2008, 45: 172-175. 10.1603/0022-2585(2008)45[172:SRGSFB]2.0.CO;2.
Rani A, Sharma A, Rajagopal R, Adak T, Bhatnagar R: Bacterial diversity analysis of larvae and adult midgut microflora using culture-dependent and culture-independent methods in lab-reared and field-collected Anopheles stephensi-an Asian malarial vector. BMC Microbiol. 2009, 9: 96-10.1186/1471-2180-9-96.
Wang Y, Gilbreath TM, Kukutla P, Yan G, Xu J: Dynamic gut microbiome across life history of the malaria mosquito Anopheles gambiae in Kenya. PLoS One. 2011, 6: e24767-10.1371/journal.pone.0024767.
Djadid ND, Jazayeri H, Raz A, Favia G, Ricci I, Zakeri S: Identification of the midgut microbiota of An. stephensi and An. maculipennis for their application as a paratransgenic tool against malaria. PLoS One. 2011, 6: e28484-10.1371/journal.pone.0028484.
Chavshin AR, Oshaghi MA, Vatandoost H, Pourmand MR, Raeisi A, Enayati AA, Mardani N, Ghoorchian S: Identification of bacterial microflora in the midgut of the larvae and adult of wild caught Anopheles stephensi: A step toward finding suitable paratransgenesis candidates. Acta Trop. 2012, 121: 129-134. 10.1016/j.actatropica.2011.10.015.
Vatandoost H, Emami SN, Oshaghi MA, Abai MR, Raeisi A, Piazzak N, Mahmoodi M, Akbarzadeh K, Sartipi M: Ecology of malaria vector Anopheles culicifacies in a malarious area of Sistan va Baluchestan province, south-east Islamic Republic of Iran. East Mediterr Health J. 2011, 17: 439-445.
Zaim M, Subbarao SK, Manouchehri AV, Cochrane AH: Role of Anopheles culicifacies s.l. and An. pulcherrimus in malaria transmission in Ghassreghand (Baluchistan), Iran. J Am Mosq Control Assoc. 1993, 9: 23-26.
Subbarao SK, Adak T, Sharma VP: Anopheles culicifacies: sibling species distribution and vector incrimination studies. J Commun Dis. 1980, 12: 102-104.
Mahmood F, Sakai RK, Akhtar K: Vector incrimination studies and observations on species A and B of the taxon Anopheles culicifacies in Pakistan. Trans R Soc Trop Med Hyg. 1984, 78: 607-616. 10.1016/0035-9203(84)90219-0.
Subbarao SK, Vasantha K, Raghavendra K, Sharma VP, Sharma GK: Anopheles culicifacies: siblings species composition and its relationship to malaria incidence. J Am Mosq Control Assoc. 1988, 4: 29-33.
Hanafi-Bojd AA, Azari-Hamidian S, Vatandoost H, Charrahy Z: Spatio-temporal distribution of malaria vectors (Diptera: Culicidae) across different climatic zones of Iran. Asian Pac J Trop Med. 2011, 4: 498-504. 10.1016/S1995-7645(11)60134-X.
Jude P, Dharshini S, Vinobaba M, Surendran S, Ramasamy R: Anopheles culicifacies breeding in brackish waters in Sri Lanka and implications for malaria control. Malar J. 2010, 9: 106-10.1186/1475-2875-9-106.
Service M, Townson H: The Anopheles vector. Essential Malariology. Edited by: Warrell D, Gilles H. 2002, Florida, USA: Arnold Publishers, 85-106. 4
Azari-Hamidian S, Harbach RE: Keys to the adult females and fourth-instar larvae of the mosquitoes of Iran (Diptera: Culicidae). Zootaxa:. 2009, 2078: 1-33.
Zaim M, Javaherian Z: Occurrence of Anopheles culicifacies species A in Iran. J Am Mosq Control Assoc. 1991, 7: 324-326.
Pidiyar VJ, Jangid K, Patole MS, Shouche YS: Studies on cultured and uncultured microbiota of wild Culex quinquefasciatus mosquito midgut based on 16 s ribosomal RNA gene analysis. Am J Trop Med Hyg. 2004, 70: 597-603.
Weisburg WG, Barns SM, Pelletier DA, Lane DJ: 16S ribosomal DNA amplification for phylogenetic study. J Bacteriol. 1991, 173: 697-703.
Drancourt M, Raoult D: Sequence-based identification of new bacteria: a proposition for creation of an orphan bacterium repository. J Clin Microbiol. 2005, 43: 4311-4315. 10.1128/JCM.43.9.4311-4315.2005.
Staley JT, Brenner DJ, Goodfellow M, Krieg NR, Rainey FA, Schleifer K-H: Bergey's Manual of Systematic Bacteriology. 2001, New York, USA: Springer, 2
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S: MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011, 28: 2731-2739. 10.1093/molbev/msr121.
Tamura K: Estimation of the number of nucleotide substitutions when there are strong transition-transversion and G + C-content biases. Mol Biol Evol. 1992, 9: 678-687.
Boissière A, Tchioffo MT, Bachar D, Abate L, Marie A, Nsango SE, Shahbazkia HR, Awono-Ambene PH, Levashina EA, Christen R: Midgut microbiota of the malaria mosquito vector Anopheles gambiae and interactions with Plasmodium falciparum infection. PLoS Path. 2012, 8: e1002742-10.1371/journal.ppat.1002742.
Cirimotich CM, Dong Y, Clayton AM, Sandiford SL, Souza-Neto JA, Mulenga M, Dimopoulos G: Natural microbe-mediated refractoriness to Plasmodium infection in Anopheles gambiae. Science. 2011, 332: 855-858. 10.1126/science.1201618.
Pumpuni CB, Beier MS, Nataro JP, Guers LD, Davis JR: Plasmodium falciparum: inhibition of sporogonic development in Anopheles stephensi by gram-negative bacteria. Exp Parasitol. 1993, 77: 195-199. 10.1006/expr.1993.1076.
Straif SC, Mbogo CN, Toure AM, Walker ED, Kaufman M, Toure YT, Beier JC: Midgut bacteria in Anopheles gambiae and An. funestus (Diptera: Culicidae) from Kenya and Mali. J Med Entomol. 1998, 35: 222-226.
Briones AM, Shililu J, Githure J, Novak R, Raskin L: Thorsellia anophelis is the dominant bacterium in a Kenyan population of adult Anopheles gambiae mosquitoes. ISME J. 2008, 2: 74-82. 10.1038/ismej.2007.95.
Kämpfer P, Lindh JM, Terenius O, Haghdoost S, Falsen E, Busse H-J, Faye I: Thorsellia anophelis gen. nov., sp. nov., a new member of the Gammaproteobacteria. Int J Syst Evol Microbiol. 2006, 56: 335-338. 10.1099/ijs.0.63999-0.
Osei-Poku J, Mbogo C, Palmer W, Jiggins F: Deep sequencing reveals extensive variation in the gut microbiota of wild mosquitoes from Kenya. Mol Ecol. 2012, 21: 5138-5150. 10.1111/j.1365-294X.2012.05759.x.
Choi KH, Kumar A, Schweizer HP: A 10-min method for preparation of highly electrocompetent Pseudomonas aeruginosa cells: Application for DNA fragment transfer between chromosomes and plasmid transformation. J Microbiol Meth. 2006, 64: 391-397. 10.1016/j.mimet.2005.06.001.
Gellatly SL, Hancock REW: Pseudomonas aeruginosa: new insights into pathogenesis and host defenses. Pathog Dis. 2013, 67: 159-173. 10.1111/2049-632X.12033.
Moll RM, Romoser WS, Modrakowski MC, Moncayo AC, Lerdthusnee K: Meconial peritrophic membranes and the fate of midgut bacteria during mosquito (Diptera: Culicidae) metamorphosis. J Med Entomol. 2001, 38: 29-32. 10.1603/0022-2585-38.1.29.
Acknowledgment
The authors would like to thank the staff of the Iranshahr research center and National Institute of Health Research (NIHR-TUMS) for their valuable help and support. This work was supported by the Tehran University of Medical Sciences (TUMS) and the Swedish University of Agricultural Sciences (SLU).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
ARC, AR, HV and MAO designed the study. ARC collected the samples, carried out the laboratory process, analyzed the results and wrote the draft of the manuscript. MRP participated in the species designation of bacteria and related biochemical tests. AR facilitated field work. ARC and OT carried out the phylogenetic analysis. HV, OT and MAO supervised the project and finalized the manuscript. All authors read the manuscript and approved its contents.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Chavshin, A.R., Oshaghi, M.A., Vatandoost, H. et al. Isolation and identification of culturable bacteria from wild Anopheles culicifacies, a first step in a paratransgenesis approach. Parasites Vectors 7, 419 (2014). https://doi.org/10.1186/1756-3305-7-419
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1756-3305-7-419