
Abstract
Background
Interstitial deletions of chromosome bands 14q24.1q24.3 are very rare with only three reported cases.
Results
We describe a 7-year-old boy with a 5.345 Mb de novo interstitial deletion at 14q24.1q24.3 band detected by array-CGH who had a complex phenotype characterized by seizures, congenital heart defects, dysmorphisms, psychomotor delay, and bronchopulmonary, skeletal, and brain anomalies.
Conclusion
The deleted region contains numerous genes, but we focused our attention on three of them (C14orf169, NUMB, and PSEN1), which could account, at least partially, for the phenotype of the boy. We therefore discuss the involvement of these genes and the observed phenotype compared to that of previously described patients.
Similar content being viewed by others

Background
Interstitial deletions involving chromosome band 14q24.1q24.3 are very rare. The use of array-CGH in routine cytogenetic diagnostics allowed the detection of pathogenic copy number variants (CNVs), which contribute to the delineation of new genomic disorders. However, there are chromosome regions in which no well-characterized aberrations were found, such as 14q24.1q24.3. Recently, Oehl-Jaschkowitz et al.,[1] described the first three unrelated patients with overlapping de novo interstitial 14q23.1q24.3 deletion characterized by array-CGH. All three patients had mild intellectual disability, congenital heart defect, brachydactyly, hypertelorism, broad nasal bridge and thin upper lip.
Here, we report on a de novo 14q24.1q24.3 deletion in a boy with complex phenotype characterized by seizures, congenital heart defects, dysmorphisms, psychomotor delay, and bronchopulmonary, skeletal, and brain anomalies. We compare the phenotype of our patient with that of previously reported patients and discuss the role of the deleted genes in order to investigate the possibility of a genotype-phenotype correlation.
Case presentation
The proband is a 7-year-old Italian male born to non consanguineous, healthy parents with another older healthy child. The child was born at 37 weeks of gestation by caesarean section after an uneventful pregnancy. Birth weight was 3050 g (90th centile), length 54 cm (90th centile), and head circumference (OFC) 36 cm (50th centile). Hypotonia, poor feeding, and mild dysmorphisms were observed from the first days of life. He was referred to the Istituto Giannina Gaslini at one month of life because of bronchiolitis and pneumonia complicated by respiratory arrest treated with intubation, ventilatory support, and oxygen supplementation for 2 months. Chest CT scans showed areas of parenchymal consolidation caused by ventilation defects characterized by thick mucous secretions in the upper right lobe and hilar areas bilaterally. During hospitalisation, the patient had seizures documented by abnormal areas in temporal lobes on EEG and was started on phenobarbital. Metabolic screening, abdominal ultrasound, and routine blood biochemical tests were normal. Cardiac ultrasound showed atrial septal defect with spontaneous closure at follow-up. Fundus oculi and visually evoked potentials (VEP) were normal. Because of gastroesophageal reflux and antral gastritis, PPI therapy was started and Nissen fundoplication was necessary at the age of three years. During the first years of life, the child experienced recurrent episodes of wheezing and upper and lower respiratory tract infections complicated by respiratory insufficiency with desaturation requiring admission to emergency care. Sweat test and genetic testing for cystic fibrosis were normal. Serum immunoglobulin levels showed transient childhood hypogammaglobulinemia. Lymphocyte counts, lymphocyte proliferation test, tests for surfactant deficiency, and laryngotracheobronchoscopy with bronchoalveolar lavage (BAL) were normal. Nasal brushing showed ciliary dyskinesia due to chronic inflammation. Periodic chest CT scans showed bronchial wall thickening, bronchiectasis, areas of consolidation with ground glass opacities and air trapping especially in the medium and upper right lobes. Due to recurrent pulmonary infections, PEP-mask (positive expiratory pressure) physiotherapy, and treatment with bronchodilators, inhaled corticosteroids, and anti-leukotriene agents were started. Physical examination during the first years of life showed broad and sparse eyebrows, convergent strabismus, broad nasal bridge and hypertelorism, nose with broad columella and nares, ears with prominent helix and antihelix and large lobes, long philtrum, thin upper lip, short neck, low set posterior hairline. Skeletal evaluation showed pectus excavatum, short fingers and toes, broad thumbs and big toes, short nails, joint hyperlaxity of lower limbs, and flat feet (Figure 1). Moreover, neurological examination showed mild psychomotor delay and especially speech delay, attention deficit, and hyperactivity. Since these features were associated with focal epilepsy, initially poorly responsive to conventional drugs, brain MRI was performed at the age of four years, showing mild symmetrical enlargement of supratentorial ventricular cavities and enlargement of fronto-insular periencephalic spaces (Figure 2). At the age of six years thoracic surgery for pectus excavatum was performed with ensuing improvement of respiratory dynamics and reduction in the frequency of respiratory infections.

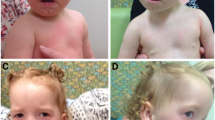
Patient at the age of 7 years. A) B) Note broad and sparse eyebrows, convergent strabismus, broad nasal bridge and hypertelorism,nose with broad columella and nares, ears with prominent helix and antihelix and large lobes, long philtrum, thin upper lip, short neck, low set posterior hairline; C) D) Pectus excavatum; E) F) Short fingers and nails, broad thumbs; G) H) Short toes and nails, broad big toes and flat feet.

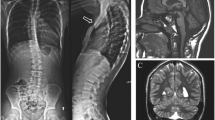
Chest CT scan before thoracic surgery. A) B) severe pectus excavatum with a Haller index of 4.07, bronchiectases and ground glass opacities C) area of consolidation and air trapping.
Results
Cytogenetic analysis, performed on GTG-banded metaphases from cultured lymphocytes of the patient and his parents, showed normal karyotypes. Considering the phenotypic abnormalities of the patient, array CGH analysis was performed, showing a 5.345 Mb de novo interstitial deletion at 14q24.1q24.3 bands. The deletion spanned from probe A_16_P40228390 (69,500,246 bps) to probe A_16_P20096718 (74,845,355 bps) flanked by probe A_16_P02937773 (69,482,103 bps) and probe A_16_P20096719 (74,852,834 bps) (Figure 3). The complete region covered by these deletions harbours more than 50 RefSeq genes with both known and unknown functions, but we focused our attention on the genes C14orf169 (NO66) (nucleolar protein 66; MIM 611919), NUMB (numb Drosophila homolog; MIM 603728), and PSEN1 (presenilin 1; MIM 104311) which could be candidate genes for the patient’s phenotype in addition to those discussed by Oehl-Jaschkowitz et al., 2014[1].

Results of array-CGH analysis. Array CGH revealed a de novo interstitial deletion of 5.345 Mb at q24.1q24.3 band of a chromosome 14: arr[hg19]14q24.1q24.3(69,500,246-74,845,355)x1. A) Array-CGH profile of patient's chromosome 14 showing the 14q deletion, B) enlargement of the deleted region 14q.
Discussion
Interstitial deletions of chromosome 14 involving the 14q24.2 band are very rare, and most deletions reported in the literature involve the proximal or distal end of chromosome 14. To our knowledge, Oehl-Jaschkowitz et al.[1] reported the first three patients, but only patient 1 had a deletion overlapping that of our case, while patients 2 (DECIPHER 251926) and 3 (DECIPHER 250043) showed a smaller deletion (Figure 4). Clinical features of the four patients are reported in Table 1. The two patients with overlapping deletion (our patient and patient 1) presented both atrial septal defect and wheezing or asthma, short fingers, hypertelorism, thin upper lip, language delay, and intellectual disability. Similarly to the other two patients, patient 2 and 3 showed different cardiac defects, hypertelorism, language delay, and mild intellectual disability. Another two subjects, with partial deletions, have been reported in DECIPHER database (http://decipher.sanger.ac.uk). The patient DECIPHER 249722 showed a 2.07 Mb deletion at band 14q24.3 (Figure 4) and presented preauricular skin tag, behavioural/psychiatric abnormalities, intellectual disability, and proportionate short stature. Another patient (DECIPHER 282030) presented a 185.2 Kb partial deletion at band 14q24.2 including only two OMIM genes: PSEN1 and NUMB (Figure 4). The only reported clinical features were autism and delayed speech.

Overview of the 14q24.1q24.3 region and its gene contents, according to the UCSC Genome Browser (GRCh37/hg19 assembly). The circles indicate the genes, which could be responsible for the phenotypic features of the 14q24.1q24.3 deletion patients. Bars indicate the deleted region of patients.
Given the large number of deleted genes comprised in the deletion, the comparison of our patient with a second case could contribute to the identification of candidate genes responsible for the phenotypic features of patients with 14q24.1q24.3 deletion.
The clinical features shared by the three patients reported by Oehl-Jaschkowitz et al.[1] were congenital heart defects, brachydactyly, mild intellectual disability, and facial dysmorphic signs. These authors suggested a possible causative role of SMOC1 and DCAF5 genes in the phenotypic features of their patients.
It is interesting to note that both patients 2 and 3, carrying a smaller deletion, had more severe heart defects and patient 2 had anomalies of hands and feet. This could be due to the haploinsufficiency of more genes or to their different expressivity. However, we can hypothesize that other genes or genetic factors could modify the phenotype in each particular case.
In addition to the above-mentioned genes, we considered C14orf169 (NO66), NUMB, and PSEN1 as possible causative genes for the phenotype of our patient and for patient 1 reported by Oehl-Jaschkowitz et al.[1].
The gene product of NO66 (C14orf169) is a jumonji C-containing protein identified as Osterix-interacting polypeptides expressed in bone that exhibits an in vitro demethylase activity with dual specificity for lysines 4 and 36 of histone H3. According to Sinha et al.[2], interactions of NO66 demethylase with Osterix should be considered physiologically significant in regulating osteoblast differentiation through modulation of Osterix activity. The authors concluded that NO66 helps gene repression through histone demethylation and/or formation of a repressor complex, which results in multilayered control of chromatin architecture of specific osteoblast genes[3].
Another interesting gene is NUMB, which is a cell fate determinant and plays a key role in asymmetric cell division (ACD) versus symmetric cell division (SCD)[4–6].
In ACD, a dividing mother cell segregates cell fate determinants asymmetrically into only one of the two daughter cells, and this process is indeed crucial for balancing self-renewal, cell differentiation, and correct spatial and temporal specification of cell lineages during development[7–9].
In the lung, ACD plays an essential role in mediating the balance between lung epithelial stem/progenitor cell maintenance and differentiation of cell populations at distal epithelial tips during lung development[10, 11].
Lethal defects of gas diffusion capacity such as the common congenital forms of lung hypoplasia and bronchopulmonary dysplasia as well as the limited capacity of the lung to recover from these defects could be explained by a significant deficiency of stem/progenitor cells[11, 12]. Therefore, proper balance between self-renewal and differentiation of lung-specific progenitors, which is mediated by ACD, is essential for normal morphogenesis and regeneration of the lung.
Moreover, loss of epithelial cell polarity is also involved in lung epithelial cancers and chronic obstructive pulmonary disease, which are likewise related to disruption of lung epithelial differentiation and cellular function[13].
As regards neurogenesis, it has been reported that Numb and Numblike (Numbl) are functionally related proteins that critically regulate progenitor differentiation and neuroepithelial integrity during embryonic neurogenesis[14–17]. They function during neural precursor ACD to antagonize Notch function in one of the daughter cells[18]. In mouse, the loss of Numb and Numbl causes premature progenitor cell depletion and, consequently, a highly specific malformation of the neocortex and hippocampus[14].
Recently, Zhao et al.[19] studied NUMB functions in cardiac progenitor cell differentiation and cardiac morphogenesis. Heart development is a spatiotemporal multistep morphogenetic process that depends on the addition of progenitor cells from four different sources, including cells from the first heart field and the second heart field (FHF and SHF), cells derived from cardiac neural crest cells, and cells derived from the pro-epicardial organ[20–24].
Perturbations in different cardiac cell populations determine a spectrum of congenital heart defects. The posterior SHF contributes to create chambre septation. Abnormal differentiation and development of the cells of this area were found associated with atrial septal defect and atrioventricular septal defect[25, 26]. In fact, as demonstrated in knockout mice, the deletion of NUMB and NUMBL in SHF-derived cells resulted in atrioventricular septation defects, which indicates their role in cardiac morphogenesis[19].
Finally, another deleted gene in our patient, PSEN1 (Presenilin 1), is the catalytic component of the γ-secretase complex, a membrane-embedded aspartyl protease that plays a central role in biology and in the pathogenesis of Alzheimer's disease.
Mutations in the PSEN1 gene are the most common cause of autosomal dominant Alzheimer's disease (AD), with around 180 mutations described to date. PSEN1 AD has a broad clinical phenotype, encompassing not only dementia but also a variety of other neurological features that may include epileptic seizures[27]. Recently, in a transgenic mouse model, it has been shown that altered expression of Numb isoforms in vulnerable neurons occurs during AD pathogenesis, which suggests a role for Numb in the disease process[28].
The other genes in this interval with known disease associations are DNAL1, COQ6, ALDH6A1, CHX10, and ABCD4. Their mutations cause autosomal recessive syndromes and no abnormalities have been reported in heterozygous carriers.
Therefore, in our opinion NUMB and PSEN1 could be suggestive of cardiac, neurological, and respiratory phenotypes. Moreover, NO66 deletion could play a role in the skeletal anomalies of our patient.
Conclusions
In conclusion, we identified a new very rare case of a 14q24.1q24.3 deletion in a boy affected by cardiac, neurological, bronchopulmonary, and skeletal anomalies. This region encompasses about 50 RefSeq genes. We suggest that NUMB, PSEN1, and NO66 genes, in addition to those reported by Oehl-Jaschkowitz et al.[1], may play a role in the phenotypic features of our patient. Furthermore, the patient 1[1] and our patient are the first human cases of a deletion of the NUMB gene, which are consistent with its importance for the cardiac, neurologic and lung normal development. On the other hand, we cannot exclude some influence of the many other genes included in the deleted region.
Methods
Standard GTG banding was performed at a resolution of 400-550 bands on metaphase chromosomes from peripheral blood lymphocytes of the patient and his parents. Molecular karyotyping was performed on the proband and his parents using Human Genome CGH Microarray Kit G3 180 (Agilent Technologies, Palo Alto, USA) with ~13 Kb overall median probe spacing. Labelling and hybridization were performed following the protocols provided by the manufacturers. A graphical overview was obtained using the Agilent Genomic Workbench Lite Edition Software 6.5.0.18.
Consent
Written informed consent was obtained from the patient’s parents for publication of this paper and any accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal.
References
Oehl-Jaschkowitz B, Vanakker OM, De Paepe A, Menten B, Martin T, Weber G, Christmann A, Krier R, Scheid S, McNerlan SE, McKee S, Tzschach A: Deletions in 14q24.1q24.3 are associated with congenital heart defects, brachydactyly, and mild intellectual disability. Am J Med Genet A 2014, 164: 620–626. 10.1002/ajmg.a.36321
Sinha KM, Yasuda H, Coombes MM, Dent SY, de Crombrugghe B: Regulation of the osteoblast-specific transcription factor Osterix by NO66, a Jumonji family histone demethylase. EMBO J 2010, 29: 68–79. 10.1038/emboj.2009.332
Sinha KM, Yasuda H, Zhou X, Decrombrugghe B: Osterix and NO66 histone demethylase control the chromatin of osterix target genes during osteoblast differentiation. J Bone Miner Res 2014, 29: 855–865. 10.1002/jbmr.2103
Knoblich JA: Asymmetric cell division during animal development. Nat Rev Mol Cell Biol 2001, 2: 11–20. 10.1038/35048085
Knoblich JA: Asymmetric cell division: recent developments and their implications for tumour biology. Nat Rev Mol Cell Biol 2010, 11: 849–860. 10.1038/nrm3010
El-Hashash AH, Warburton D: Numb expression and asymmetric versus symmetric cell division in distal embryonic lung epithelium. J Histochem Cytochem 2012, 60: 675–682. 10.1369/0022155412451582
Knoblich JA: Mechanisms of asymmetric stem cell division. Cell 2008, 132: 583–597. 10.1016/j.cell.2008.02.007
Zhong W, Chia W: Neurogenesis and asymmetric cell division. Curr Opin Neurobiol 2008, 18: 4–11. 10.1016/j.conb.2008.05.002
Siller KH, Doe CQ: Spindle orientation during asymmetric cell division. Nat Cell Biol 2009, 11: 365–374. 10.1038/ncb0409-365
Warburton D, Schwarz M, Tefft D, Flores-Delgado G, Anderson KD, Cardoso WV: The molecular basis of lung morphogenesis. Mech Dev 2000, 92: 55–81. 10.1016/S0925-4773(99)00325-1
Warburton D, Perin L, Defilippo R, Bellusci S, Shi W, Driscoll B: Stem/progenitor cells in lung development, injury repair, and regeneration. Proc Am Thorac Soc 2008, 5: 703–706. 10.1513/pats.200801-012AW
Shi W, Xu J, Warburton D: Development, repair and fibrosis: what is common and why it matters. Respirology 2009, 14: 656–665. 10.1111/j.1440-1843.2009.01565.x
Xu J, Tian J, Grumelli S, Haley K, Shapiro SD: Stage-specific effects of cAMP signaling during distal lung epithelial development. J Biol Chem 2006, 281: 38894–38904. 10.1074/jbc.M609339200
Petersen PH, Zou K, Hwang JK, Jan YN, Zhong W: Progenitor cell maintenance requires numb and numblike during mouse neurogenesis. Nature 2002, 419: 929–934. 10.1038/nature01124
Shen Q, Zhong W, Jan YN, Temple S: Asymmetric Numb distribution is critical for asymmetric cell division of mouse cerebral cortical stem cells and neuroblasts. Development 2002, 129: 4843–4853.
Li HS, Wang D, Shen Q, Schonemann MD, Gorski JA, Jones KR, Temple S, Jan LY, Jan YN: Inactivation of Numb and Numblike in embryonic dorsal forebrain impairs neurogenesis and disrupts cortical morphogenesis. Neuron 2003, 40: 1105–1118. 10.1016/S0896-6273(03)00755-4
Petersen PH, Zou K, Krauss S, Zhong W: Continuing role for mouse Numb and Numbl in maintaining progenitor cells during cortical neurogenesis. Nat Neurosci 2004, 7: 803–811. 10.1038/nn1289
Roegiers F, Jan YN: Asymmetric cell division. Curr Opin Cell Biol 2004, 16: 195–205. 10.1016/j.ceb.2004.02.010
Zhao C, Guo H, Li J, Myint T, Pittman W, Yang L, Zhong W, Schwartz RJ, Schwarz JJ, Singer HA, Tallquist MD, Wu M: Numb family proteins are essential for cardiac morphogenesis and progenitor differentiation. Development 2014, 141: 281–295. 10.1242/dev.093690
Kelly RG, Brown NA, Buckingham ME: The arterial pole of the mouse heart forms from Fgf10-expressing cells in pharyngeal mesoderm. Dev Cell 2001, 1: 435–440. 10.1016/S1534-5807(01)00040-5
Mjaatvedt CH, Nakaoka T, Moreno-Rodriguez R, Norris RA, Kern MJ, Eisenberg CA, Turner D, Markwald RR: The outflow tract of the heart is recruited from a novel heart-forming field. Dev Biol 2001, 238: 97–109. 10.1006/dbio.2001.0409
Waldo KL, Kumiski DH, Wallis KT, Stadt HA, Hutson MR, Platt DH, Kirby ML: Conotruncal myocardium arises from a secondary heart field. Development 2001, 128: 3179–3188.
Verzi MP, McCulley DJ, De Val S, Dodou E, Black BL: The right ventricle, outflow tract, and ventricular septum comprise a restricted expression domain within the secondary/anterior heart field. Dev Biol 2005, 287: 134–145. 10.1016/j.ydbio.2005.08.041
Vincent SD, Buckingham ME: How to make a heart: the origin and regulation of cardiac progenitor cells. Curr Top Dev Biol 2010, 90: 1–41.
Snarr BS, O'Neal JL, Chintalapudi MR, Wirrig EE, Phelps AL, Kubalak SW, Wessels A: Isl1 expression at the venous pole identifies a novel role for the second heart field in cardiac development. Circ Res 2007, 101: 971–974. 10.1161/CIRCRESAHA.107.162206
Briggs LE, Kakarla J, Wessels A: The pathogenesis of atrial and atrioventricular septal defects with special emphasis on the role of the dorsal mesenchymal protrusion. Differentiation 2012, 84: 117–130. 10.1016/j.diff.2012.05.006
Larner AJ: Presenilin-1 mutation Alzheimer's disease: a genetic epilepsy syndrome? Epilepsy Behav 2011, 21: 20–22. 10.1016/j.yebeh.2011.03.022
Chigurupati S, Madan M, Okun E, Wei Z, Pattisapu JV, Mughal MR, Mattson MP, Chan SL: Evidence for altered Numb isoform levels in Alzheimer's disease patients and a triple transgenic mouse model. J Alzheimers Dis 2011, 24: 349–361.
Acknowledgments
We thank the patient’s parents for their kind participation and support. We are grateful to Marco Bertorello and Corrado Torello for their technical assistance. This work was supported by “Cinque per mille dell’IRPEF- Finanziamento della ricerca sanitaria” and “Finanziamento Ricerca Corrente, Ministero Salute (contributo per la ricerca intramurale).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
All authors have made substantial contributions to conception and design, acquisition of data, analysis and interpretation of data. All authors have been involved in drafting the manuscript and revising it critically for important intellectual content. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Tassano, E., Accogli, A., Panigada, S. et al. Phenotypic and genetic characterization of a patient with a de novo interstitial 14q24.1q24.3 deletion. Mol Cytogenet 7, 49 (2014). https://doi.org/10.1186/1755-8166-7-49
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1755-8166-7-49