
Abstract
Pseudomyxoma peritonei (PMP, ORPHA26790) is a clinical syndrome characterized by progressive dissemination of mucinous tumors and mucinous ascites in the abdomen and pelvis. PMP is a rare disease with an estimated incidence of 1–2 out of a million. Clinically, PMP usually presents with a variety of unspecific signs and symptoms, including abdominal pain and distention, ascites or even bowel obstruction. It is also diagnosed incidentally at surgical or non-surgical investigations of the abdominopelvic viscera. PMP is a neoplastic disease originating from a primary mucinous tumor of the appendix with a distinctive pattern of the peritoneal spread. Computed tomography and histopathology are the most reliable diagnostic modalities. The differential diagnosis of the disease includes secondary peritoneal carcinomatoses and some rare peritoneal conditions. Optimal elimination of mucin and the mucin-secreting tumor comprises the current standard of care for PMP offered in specialized centers as visceral resections and peritonectomy combined with intraperitoneal chemotherapy. This multidisciplinary approach has reportedly provided a median survival rate of 16.3 years, a median progression-free survival rate of 8.2 years and 10- and 15-year survival rates of 63% and 59%, respectively. Despite its indolent, bland nature as a neoplasm, PMP is a debilitating condition that severely impacts quality of life. It tends to be diagnosed at advanced stages and frequently recurs after treatment. Being ignored in research, however, PMP remains a challenging, enigmatic entity. Clinicopathological features of the PMP syndrome and its morbid complications closely correspond with the multifocal distribution of the secreted mucin collections and mucin-secreting implants. Novel strategies are thus required to facilitate macroscopic, as well as microscopic, elimination of mucin and its source as the key components of the disease. In this regard, MUC2, MUC5AC and MUC5B have been found as the secreted mucins of relevance in PMP. Development of mucin-targeted therapies could be a promising avenue for future research which is addressed in this article.
Similar content being viewed by others


Introduction
With an estimated incidence of 1–2 out of a million [1], pseudomyxoma peritonei (PMP, ORPHA26790)-also known as adenomucinosis or gelatinous ascites- is listed as a rare disease by the NIH Office of Rare Diseases Research (ORDR) and National Organization for Rare Disorders (NORD). As an indolent neoplasm with unspecific manifestations, PMP tends to be misdiagnosed, or discovered at advanced stages. Moreover, it is a challenging entity with debilitating, even fatal complications. Despite a multidisciplinary approach composed of an extensive surgical procedure and chemotherapy, PMP frequently recurs and increasingly jeopardizes quality of life. Being ignored in research, however, PMP remains poorly understood and enigmatic. This is despite the fact that “rare diseases are rare, but rare disease patients are numerous”[2] and those with progressive, life-threatening courses deserve to be further explored. To enhance outcomes of the conventional therapy, novel approaches based on in-depth understanding of the pathological processes and biological events in the pathogenesis of the disease are warranted. Since PMP and mucin are inextricably linked, any therapeutic intervention needs to properly target the mucin ectopy. In this article, the current knowledge on the crucial role of mucin in the pathogenesis of the disease is reviewed and a number of potential therapeutic strategies for mucin elimination in PMP are addressed.
Definition
PMP is characterized by dissemination of mucinous tumor implants on peritoneal surfaces and progressive accumulation of mucinous ascites throughout the peritoneal cavity resulting in the so-called “jelly belly”.
Etiology
Since initial descriptions of PMP as a syndrome in association with an ovarian tumor [3] or an appendiceal mucocele [4], a pre-existing intraperitoneal mucinous neoplasm has been implicated as the primary cause of PMP. As follows, emerging evidence supports the appendiceal rather than ovarian origin of the disease.
Classification
PMP has been broadly applied to a heterogeneous group of pathological conditions with a similar clinical presentation, with the site of the primary tumor, neoplastic phenotype of the peritoneal tumor cells and classification of the disease being a matter of controversy. However, attempts have been made to better define and classify the condition based on the clinicopathological characteristics. Ronnett et al. [5] suggested a classification of multifocal peritoneal mucinous tumors into three groups. These include disseminated peritoneal adenomucinosis (DPAM), peritoneal mucinous carcinomatosis (PMCA) and a third hybrid group called peritoneal mucinous carcinomatosis with intermediate or discordant features (PMCA I/D), also known as intermediate features group (IFG). According to this clinicopathological classification, DPAM includes histologically benign peritoneal lesions associated with ruptured appendiceal mucinous adenomas as well as those with similar pathology but lacking a demonstrable appendiceal adenoma. Subsequently, Sugarbaker defined PMP as a grade I mucinous adenocarcinoma that arises from an appendiceal adenoma [6]. Later, they stipulated that the term PMP should be exclusively used to describe the clinical syndrome of mucinous ascites accompanied by a characteristic distribution of peritoneal mucinous tumors with the pathologic features of DPAM [7]. Further studies strongly supported the notion that PMP is a generally low-grade, indolent neoplasm of appendiceal origin with rare distant metastases and unlikely involvement of solid organs [1, 8, 9]. Since neither adenoma nor adenocarcinoma precisely represents the neoplastic nature of PMP, “mucinous neoplasm of low malignant potential” and “low-grade appendiceal mucinous neoplasm” have been proposed in the literature as the alternative terms for pathological description of the PMP tumor [10].
Pathogenesis
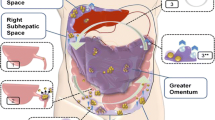
The pathological process starts with neoplastic transformation of the appendiceal goblet cells and subsequent formation of a primary mucinous tumor. While proliferating, tumor cells maintain their constitutive level of mucin expression. As a result, the overall secretion of mucin dramatically rises [8]. This is followed by intraluminal accumulation of mucin and eventual development of an appendiceal mucocele. A small perforation or rupture of the mucocele is the key event towards the development of PMP through which tumor cells gain access into the peritoneal cavity. Lacking cell surface adhesion molecules, the exfoliated tumor cells passively circulate with the peritoneal fluid and redistribute throughout the peritoneal cavity. As a result, tumor implants and mucin collections form at the peritoneal fluid reabsorption sites as well as within the dependent portions of the peritoneal cavity to create PMP’s characteristic pattern of the peritoneal dissemination (Figure 1) [11]. Accumulating mucin increases intraabdominal pressure and compresses visceral organs. Furthermore, extensive involvement of the peritoneal surface promotes variable inflammatory and fibrotic responses in the peritoneal environment and hence the development of bowel obstruction as a fatal complication of the disease [12, 13]. The detailed role of mucin in the pathogenesis of PMP will be discussed later.

Schematic representation of the events resulting in the development of PMP. The pathologic process starts with a neoplastic transformation of the appendiceal goblet cells and development of a primary mucinous tumor (1). Overproduction of mucin and obstruction of the appendiceal lumen lead to the development, and subsequent rupture, of a mucocele (2). Shredded tumor cells gain access to the peritoneal cavity and circulate with the peritoneal fluid (3). Accordingly, tumor cells redistribute and accumulate within the dependent portions of the peritoneal cavity (3*, downward arrows) as well as at the peritoneal fluid reabsorption sites (3**, upward arrows).
Clinical presentation
The disease is usually diagnosed after the age of 40 [14], with an average age at diagnosis of 53 [15]. PMP clinically presents with a variety of unspecific and sometimes uncommon signs and symptoms. PMP’s clinical manifestations can be roughly classified based on the disease progression (Table 1) [16]. In advanced disease, increased abdominal girth and complaints of abdominal pain related to intestinal obstruction are the most presenting symptom seen in 30-50% of the PMP patients as a result of disseminated mucinous tumor and ascites classically presenting at laparotomy with jelly belly. In less advanced disease, local symptoms are seen in 50-80% of PMP patients without jelly belly ascites and might correspond to the site of the primary tumor, such as appendicitis-like symptoms in 25% of cases, or the location of the peritoneal implants, including lower abdominal pain, pelvic pressure and gynecological complaints in females due to the ovarian deposits of the mucinous tumor in 20-30% of the patients [17, 18]. Finally, PMP is coincidentally found in up to 20% of patients undergoing such procedures as laparotomy, laparoscopy or imaging for other medical conditions, e.g. hernia [17–27], bladder tumor [28], jelly like material in urine [29], total uterovaginal prolapse [30], recurrent rectal cancer [31], pregnancy [32] and cesarean section [33]. PMP cases presenting with an ulcerated skin fistula on the right flank [34] or a subcutaneous nontender umbilical nodule [35] have also been reported.
Diagnosis
Imaging
- Ultrasound and parallel fine needle biopsy. Ultrasound is accessible and inexpensive. However, conclusions cannot be drawn from ultrasound alone since the mucinous ascites resembles free intraperitoneal fluid [36]. Although cytological investigation of the mucin collections seems to be a useful accompanying procedure, sampling errors, dry taps and false negative results due to low amount of mucin and/or low cellular density are considered as the disadvantages of parallel fine needle biopsy [16].
- Computed tomography. Computed tomography (CT) remains the most widely used imaging modality in PMP. Higher densities of mucinous ascites compared to the nonmucinous collections [40], characteristic pattern of the mucinous accumulation [41] and the extent of the disease for preoperative planning and prognostic purposes [36] can be evaluated by CT.
- Other imaging methods. Magnetic resonance imaging (MRI) has been described to show the location of mucocele and its morphologic criteria identically to CT. T1- and T2-weighted MRI are more sensitive in distinguishing between mucin and fluid ascites [42–44]. Although positron emission tomography (PET) has been suggested for predicting the peritoneal dissemination [45] and preoperative evaluation of pathological grade and potential for complete cytoreduction [46], its value in PMP remains controversial [47, 48].
Circulating tumor markers
Although relatively non-specific [14], the following tumor markers have been reported to be of value in PMP:
- Carcinoembryonic antigen (CEA). This tumor marker has been shown to serve as a valuable diagnostic [49] and prognostic tool [49, 50] in the management of PMP.
- Carbohydrate antigen 19.9 (CA19.9). Practical value of CA19.9 in diagnostic [49] and prognostic [49, 51–54] evaluation of PMP has been reported.
- Carbohydrate antigen 125 (CA125) also known as MUC16. Although suggested as a marker with diagnostic sensitivity for PMP [52], CA125 is not widely used as a tumor marker for PMP. Instead, as a gynecological tumor marker, it is recommended for exclusion of an ovarian neoplasm [16].
These tumor markers are also used as a baseline value for postoperative follow-up. Moreover, they have been reported as predictors of the completeness of cytoreductive surgery (CRS) [50, 52, 53, 55], a significant prognostic factor for PMP.
Histopathological analysis
Apart from the characteristic feature of PMP as acellular to paucicellular mucin pools with variable amounts of neoplastic mucinous epithelium, immunohistochemical markers can help to identify the organ of origin. These include positive cytokeratin 20 (CK20), CEA, caudal-type homeobox protein 2 (CDX-2), and MUC2 as well as negative cytokeratin 7 (CK7) and CA125 [16]. Of particular interest is the secreted mucin MUC2 that is extensively positive in the patients. Although MUC2 has been suggested as a biological marker of PMP [56–60], its significance as a prognostic factor is a matter of controversy [61].
Differential diagnosis
The main entity to be considered in the differential diagnosis of PMP is peritoneal mucinous carcinomatosis arising from a primary mucinous carcinoma. Other differential diagnoses reported in the literature include endometriosis with myxoid change [62], melioidosis (a lethal infectious disease caused by Burkholderia pseudomallei) [63] and those with abdominal CT resemblance, e.g. extensive abdominal plexiform neurofibromatosis [64].
Treatment
Traditional treatment consists of repetitive surgical debulking. Due to the presence of tumor deposits after the first debulking surgery, this approach could results in short term palliation with imminent recurrence or progression, hence redo procedures and a shorter 5- to 10-year overall survival (OS) rate of approximately 50% [65–67]. A more aggressive approach by Sugarbaker [68, 69] utilizes peritonectomy and visceral resections, called cytoreductive surgery (CRS), in combination with hyperthermic intraperitoneal chemotherapy (HIPEC) that is featured by direct targeting of the microscopic disease, locoregional drug availability, minimal systemic exposure and improved drug penetration through hyperthermia. This strategy comprises the current standard of care for PMP with known benefits well-documented by us [15, 53, 54, 70–81] and others [82–92]. Other proposed modalities include laparoscopy for less advanced disease [93, 94], whole abdominopelvic radiotherapy (WAPRT) as a palliative treatment [95] and use of mucolytic [96–100], antibacterial [58, 101, 102] and anti-inflammatory [103] agents with potential values as complementary/adjuvant therapies.
Prognosis
Although PMP as a neoplastic disease runs a chronic, indolent course with late invasion and only rare metastasis outside the peritoneum, it is a morbid, recurrent condition with life-threatening complications. Biological features of the tumor [61, 104] and access to the current standard of care at specialized oncology centers with a peritoneal surface malignancy program [105–110] comprise the most important prognostic determinants of PMP. Through a retrospective, multi-institutional study on 2298 patients treated at 16 specialized centers affiliated with the Peritoneal Surface Oncology Group International [15], Chua et al. reported a median survival rate of 196 months (16.3 years) and a median progression-free survival rate of 98 months (8.2 years) as well as 10- and 15-year survival rates of 63% and 59%, respectively.
Mucin: from intestinal physiology to PMP
Mucin family
Mucins are a diverse family of high molecular weight, heavily glycosylated proteins (Table 2). Also known as MUC glycoproteins, mucins are differentially expressed by specialized epithelial cells of mucosal surfaces throughout the body in a relatively organ- and cell type-specific manner [111]. Mucins are categorized into membrane-associated and secreted types, with the latter being divided to gel-forming and non-gel-forming subtypes [112, 113]. Membrane-associated mucins communicate information about extracellular conditions, mediate intracellular signal transduction, and contribute to morphological and behavioral characteristics of the epithelial cells [114, 115].
Secreted mucins provide a physical barrier for epithelial cells lining the respiratory and gastrointestinal tracts and form the ductal surfaces of such organs as liver, breast, pancreas and kidney [116]. Moreover, they are part of a defensive system at the mucosal surfaces, including intestinal mucosa.
Mucin in intestinal physiology
While facilitating the transit of intestinal contents [117], secreted mucins participate in the front line of the enteric host defense generated by the alliance of the epithelial cells, immune cells and resident microbiota [118]. This interactive ecosystem is essential for the maintenance of intestinal homeostasis and the normal function and activity of digestive system [119]. The gastrointestinal epithelium and the overlying mucus layer also function as a barrier against intestinal luminal hazards [120]. Colonic mucus is composed of two layers. The outer, loose layer is the habitat of the microbial flora. The inner, dense layer is bacteria-free and firmly attached to the epithelium. This organization keeps the flora well separated from the mucosal surface. The gel-forming mucin MUC2, which is specifically secreted in the small intestine and colon, comprises the substantial component of this double-layered mucus compartment (Figure 2). MUC2 of the inner layer is uncleaved. To form the outer layer, however, MUC2 undergoes proteolytic cleavage to allow expansion of the polymeric structure [121].

MUC2 in colonic mucosa. A Synthesis, secretion and organization of colonic mucosa. The first stage in the biosynthesis of MUC2 is the formation of MUC2 monomer as an N-glycosylated apoprotein in the endoplasmic reticulum. Subsequently, MUC2 dimers are formed when intermolecular disulfide bonds bridge between the C-terminal cysteine knot domains. During transit through the Golgi apparatus, MUC2 dimers become heavily O-glycosylated. Complete glycosylation of the dimers occurs in Golgi where trimerization through disulfide bonds at the N-terminus forms protease-resistant trimers. The fully glycosylated and processed MUC2 is densely packed and stored in secretory granules/vesicles and released through constitutive or stimulated secretory mechanisms. Once released, MUC2 is organized into the firmly adherent inner layer. At a certain distance from the epithelium, this layer is converted into the loose outer layer through proteolytic cleavage and expansion. Mucus also contains immunoglobulins and other proteins. B MUC2 structure. The protein core consists of five different regions. Segment (a) and (b) are two central repetitive regions rich in potential O-glycosylation sites, to which branched carbohydrate chains of 4–12 sugars are O-glycosidically linked to form a closely packed sheath around the central protein core. Segment (a), also known as VNTR domain, is a large domain that contains 50–100 “variable number of tandem repeats (VNTRs)” of 23 amino acids, in particular, threonine. Segment (b), also called PTS domain, is a 347 amino acid domain, containing irregular repeats rich in proline, threonine and serine (PTS). These two segments are linked together by segment (c) which is a 148 amino acid, cysteine containing region. Segments (d) and (e) are extensive peptide chains rich in cysteine located at the C and N terminal ends, respectively, containing D domains with sequence homology to von Willebrand factor. These regions are the presumed sites for end to end polymerization of mucin subunits.
Mucin in PMP
Under normal conditions, metabolic turnover of intestinal mucin is maintained by the constitutive expression against enzymatic degradation, and, elimination. In PMP, however, mucin is ectopically secreted and increasingly deposited in the peritoneal cavity where it is unable to degrade or drain away. Accumulating mucin causes a major part of the morbidity in PMP. The typical syndrome develops after secreted mucin forms voluminous gels over months and years. Mucin also plays a key role in the biology of the PMP tumor. Most of the tumor cells are surrounded by a mucin coat that allows them to freely move, disseminate and “redistribute” within the peritoneal cavity to create the distinctive feature of PMP. This coating also seems to act as a protective shield against immune recognition and chemotherapy. MUC2, MUC5AC and MUC5B are the gel-forming mucins reportedly found in the PMP secretions. The intestinal mucin MUC2 is known as the PMP-specific mucin. According to O’Connell et al. [8, 56], primary ovarian mucinous tumors essentially express MUC5AC whereas solitary appendiceal mucinous tumors and different categories of PMP express MUC2 along with MUC5AC. This finding also supports the notion that PMP is a neoplasm of appendiceal origin. In their studies, O’Connell et al. also showed that MUC2 is behind the high degree of gelation formed in PMP. Since MUC2 is more extensively glycosylated, it is more voluminous than MUC5AC on an equimolar basis, hence formation of abundant mucinous collections where the average mucin:cell ratio is higher than 10:1. Taken together, the investigators concluded that PMP is a disease of the MUC2-secreting goblet cells and that MUC2 could serve as a molecular marker for PMP [8, 56]. Since the expression level of MUC2 in PMP is seemingly independent of the degree of the malignant transformation, prognostic significance of MUC2 is controversial [61].
In two case studies, Mall et al. reported the presence of MUC5B, in addition to MUC2 and MUC5AC, in the PMP material [122, 123]. Based on the investigations by Sheehan et al. implicating a low-charge glycoform of MUC5B in the production of a tenacious respiratory mucus plug [124, 125], Mall et al. speculated that it may be MUC5B that is responsible for the semisolid material found in some PMP patients. Given the high protein content of the PMP secretions, they also raised the possibility that interactions between mucin and non-mucin proteins could contribute to the viscous nature of the PMP exudates [122, 123]. Table 3 summarizes a number of studies in which disease-specific expression pattern of MUC2 and other mucins in PMP has been explored.
Mucin elimination in the management of PMP
Mucin comprises the cornerstone of the PMP pathogenesis. However, optimal removal of mucin and mucinous implants via conventional therapies remains challenging, with the residual disease accounting for the recurrence of the condition. In order to enhance the current standard of care, novel strategies are required to more effectively eliminate mucin and its source. As such, some efforts have been made to disrupt PMP production of MUC2. In this regard, targeting biological mechanisms regulating the mucin synthesis at transcriptional and post-transcriptional levels could be of potential value. Since MAPK pathway has been implicated in the pathological induction of MUC2 by Pseudomonas aeruginosa infection [131] and mucoepidermoid carcinogenesis [132], targeted inhibition of MAPK might be of therapeutic benefit in PMP. On the basis of the cross-talk between MAPK and other inflammation-associated signaling pathways [133], multi-targeted agents may be more effective in PMP where the disease develops within an inflammatory milieu [134].
In addition, PMP inflammatory microenvironment with a unique profile of cytokines [13] contributes to the mucin overproduction. Cytokines reportedly upregulate the expression of MUC2 and enhance the mucin secretion [135–137]. Glucocorticoids, on the other hand, have been shown to downregulate MUC2 via direct inhibition of glucocorticoid response elements (GREs) and indirect transrepression of inflammation-associated transcription factors [138, 139]. Choudry et al. recently demonstrated dexamethasone- and celecoxib-induced inhibition of the mucin production in a mucin-secreting cancer cell line with goblet cell phenotype as well as in a murine model of PMP [103]. Methylation of the MUC2 promoter has also been suggested as a potential tool for manipulating the expression of MUC2. Okudaira et al. showed that colorectal cancer (CRC) cell lines of mucinous type exhibit low-level methylation at the MUC2 promoter as compared to non-mucinous cell lines. They concluded that low methylation status of the MUC2 gene plays a predominant role in the high level MUC2 expression in mucinous CRC [140]. Through inhibiting the MUC2 promoter methylation, the Sp-family of transcription factors augments MUC2 expression. Thus, mithramycin, a Sp1 binding site inhibitor, effectively blocks the MUC2 expression in colorectal cancer [132, 141].
Breakdown of mucin as well as enhancement of cytoreduction through locoregional therapies outlines our intended approach to this challenge. In this regard, we aim to develop a novel treatment for facilitated, enhanced removal of the mucin-tumor burden at both macroscopic and microscopic levels. For this purpose, a panel of mucin-secreting cancer cells of gastrointestinal and peritoneal origin and animal models of PMP [142] and peritoneal carcinomatosis [143] are employed in our preclinical investigations. Among a number of mucolytic agents studied [99, 100], bromelain and N-acetyl cysteine (NAC), two generally-safe mucolytics of plant sources [144, 145], have shown promise. Our preliminary results indicate bromelain- [146–148] and NAC-induced inhibition of the growth and proliferation of the mucin-producing cancer cells in vitro, with the cytotoxicity being augmented when the two are used in combination (unpublished data). We have also observed the ability of these mucolytics in dissolving both the mucin samples from PMP patients ex vivo and the mucin implants in a xenograft model of PMP in vivo with no treatment-related toxicity in rats [149]. We are currently performing final preclinical testing of our mucolytic compound before proceeding to the clinical phase.
Conclusion
Despite its morbid, debilitating nature with severe impact on quality of life, PMP remains orphan and enigmatic. Essential for physiological function of the gastrointestinal tract, mucin is the major contributor to the pathophysiology of PMP. Peritoneal implantation of the tumor cells originating from an appendiceal mucinous tumor results in the progressive accumulation of ectopic mucin. Given the clinicopathological profile of the disease compatible with an indolent, generally low grade malignancy, multifocal collections of voluminous mucin and the ensuing complications are the main determinants of the disease prognosis. As the predominant, gel-forming mucin secreted in PMP, MUC2 is responsible for the high degree of gelation and the characteristic feature of the clinical syndrome. Despite the current standard of care as extensive surgical resection combined with chemotherapy, PMP frequently recurs; with treatmen options being limited at recurrence. On this basis, in-depth investigations are warranted to illuminate unknown aspects of the disease and to seek novel therapeutic approaches for an enhanced treatment. Owing to the substantial role of mucin in the pathogenesis of PMP, development of strategies for targeting mucin and its biology seems to be of particular significance that needs to be further explored in future studies.
Abbreviations
- CA125:
-
Carbohydrate antigen 125
- CA19.9:
-
Carbohydrate antigen 19.9
- CDX-2:
-
Caudal-type homeobox protein 2
- CEA:
-
Carcinoembryonic antigen
- CK7:
-
Cytokeratin 7
- CK20:
-
Cytokeratin 20
- CRC:
-
Colorectal cancer
- CRS:
-
Cytoreductive surgery
- CT:
-
Computed tomography
- DPAM:
-
Disseminated peritoneal adenomucinosis
- GREs:
-
Glucocorticoid response elements
- HIPEC:
-
Hyperthermic intraperitoneal chemotherapy
- IFG:
-
Intermediate features group
- MRI:
-
Magnetic resonance imaging
- NAC:
-
N-acetyl cysteine
- NORD:
-
National organization for rare disorders
- ORDR:
-
NIH office of rare diseases research
- PET:
-
Positron emission tomography
- PMCA:
-
Peritoneal mucinous carcinomatosis
- PMCA I/D:
-
Peritoneal mucinous carcinomatosis with intermediate or discordant features
- PMP:
-
Pseudomyxoma peritonei
- WAPRT:
-
Whole abdominopelvic radiotherapy.
References
Smeenk RM, van Velthuysen ML, Verwaal VJ, Zoetmulder FA: Appendiceal neoplasms and pseudomyxoma peritonei: a population based study. Eur J Surg Oncol. 2008, 34: 196-201. 10.1016/j.ejso.2007.04.002.
The portal for rare diseases and orphan drugs. [http://www.orpha.net/consor/cgi-bin/index.php?lng=EN]
Werth R: Klinische und anatomische untersuchungen zur lehre von den bauchgeschwülsten und der laparatomie. Archiv Fur Gynakologie. 1884, 24: 100-118. 10.1007/BF01837425.
Fraenkel E: Ueber das sogennante pseudomyxoma perotonei. Munch Med Wochenschr. 1901, 48: 965-971.
Ronnett BM, Zahn CM, Kurman RJ, Kass ME, Sugarbaker PH, Shmookler BM: Disseminated peritoneal adenomucinosis and peritoneal mucinous carcinomatosis. A clinicopathologic analysis of 109 cases with emphasis on distinguishing pathologic features, site of origin, prognosis, and relationship to “pseudomyxoma peritonei”. Am J Surg Pathol. 1995, 19: 1390-1408. 10.1097/00000478-199512000-00006.
Sugarbaker PH: Pseudomyxoma peritonei. Cancer Treat Res. 1996, 81: 105-119. 10.1007/978-1-4613-1245-1_10.
Ronnett BM, Yan H, Kurman RJ, Shmookler BM, Wu L, Sugarbaker PH: Patients with pseudomyxoma peritonei associated with disseminated peritoneal adenomucinosis have a significantly more favorable prognosis than patients with peritoneal mucinous carcinomatosis. Cancer. 2001, 92: 85-91. 10.1002/1097-0142(20010701)92:1<85::AID-CNCR1295>3.0.CO;2-R.
O’Connell JT, Tomlinson JS, Roberts AA, McGonigle KF, Barsky SH: Pseudomyxoma peritonei is a disease of MUC2-expressing goblet cells. Am J Pathol. 2002, 161: 551-564. 10.1016/S0002-9440(10)64211-3.
Misdraji J: Appendiceal mucinous neoplasms: controversial issues. Arch Pathol Lab Med. 2010, 134: 864-870.
Buell-Gutbrod R, Gwin K: Pathologic diagnosis, origin, and natural history of pseudomyxoma peritonei. Am Soc Clin Oncol Educ Book. 2013, 33: 221-225.
Sugarbaker PH: Pseudomyxoma peritonei. A cancer whose biology is characterized by a redistribution phenomenon. Ann Surg. 1994, 219: 109-111. 10.1097/00000658-199402000-00001.
Sugarbaker PH: New standard of care for appendiceal epithelial neoplasms and pseudomyxoma peritonei syndrome?. Lancet Oncol. 2006, 7: 69-76. 10.1016/S1470-2045(05)70539-8.
Lohani K, Shetty S, Sharma P, Govindarajan V, Thomas P, Loggie B: Pseudomyxoma Peritonei: Inflammatory Responses in the Peritoneal Microenvironment. Ann Surg Oncol. 2014, 21: 1441-1447. 10.1245/s10434-013-3261-6.
Pseudomyxoma peritonei. [http://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=EN&Expert=26790]
Chua TC, Moran BJ, Sugarbaker PH, Levine EA, Glehen O, Gilly FN, Baratti D, Deraco M, Elias D, Sardi A, Liauw W, Yan TD, Barrios P, Gómez Portilla A, de Hingh IH, Ceelen WP, Pelz JO, Piso P, González-Moreno S, Van Der Speeten K, Morris DL: Early- and long-term outcome data of patients with pseudomyxoma peritonei from appendiceal origin treated by a strategy of cytoreductive surgery and hyperthermic intraperitoneal chemotherapy. J Clin Oncol. 2012, 30: 2449-2456. 10.1200/JCO.2011.39.7166.
Smeenk RM, Bruin SC, van Velthuysen ML, Verwaal VJ: Pseudomyxoma peritonei. Curr Probl Surg. 2008, 45: 527-575. 10.1067/j.cpsurg.2008.04.003.
Sugarbaker PH, Ronnett BM, Archer A, Averbach AM, Bland R, Chang D, Dalton RR, Ettinghausen SE, Jacquet P, Jelinek J, Koslowe P, Kurman RJ, Shmookler B, Stephens AD, Steves MA, Stuart OA, White S, Zahn CM, Zoetmulder FA: Pseudomyxoma peritonei syndrome. Adv Surg. 1996, 30: 233-280.
Esquivel J, Sugarbaker PH: Clinical presentation of the Pseudomyxoma peritonei syndrome. Br J Surg. 2000, 87: 1414-1418. 10.1046/j.1365-2168.2000.01553.x.
Ben-Hur H, Schachter M, Mashiah A, Lifschitz-Mercer B, Pfeffermann R: Recurrent mucinous adenocarcinoma of the ovary presenting as an inguino-labial hernia. Eur J Gynaecol Oncol. 1996, 17: 299-302.
Young RH, Rosenberg AE, Clement PB: Mucin deposits within inguinal hernia sacs: a presenting finding of low-grade mucinous cystic tumors of the appendix. A report of two cases and a review of the literature. Mod Pathol. 1997, 10: 1228-1232.
Edwards DP, Scott HJ: The use of laparoscopy in a case of appendiceal cystadenoma presenting as pseudomyxoma peritonei in an inguinal hernial sac. J R Coll Surg Edinb. 1998, 43: 112-113.
Esquivel J, Sugarbaker PH: Pseudomyxoma peritonei in a hernia sac: analysis of 20 patients in whom mucoid fluid was found during a hernia repair. Eur J Surg Oncol. 2001, 27: 54-58. 10.1053/ejso.2000.1031.
Rezkalla MA, Peterson KG, Ryan JJ: Pseudomyxoma peritonei: a case of mucinous adenocarcinoma of the appendix presenting as inguinal hernia. S D Med. 2006, 59: 54-55. 57
Shinohara T, Misawa K, Sano H, Okawa Y, Takada A: Pseudomyxoma peritonei due to mucinous cystadenocarcinoma in situ of the urachus presenting as an inguinal hernia. Int J Clin Oncol. 2006, 11: 416-419. 10.1007/s10147-006-0594-1.
Campbell P, Dawson S, Wali J, Kenny B, Whiteside MC: Pseudomyxoma peritonei presenting as inguinal hernia. Ulster Med J. 2009, 78: 189-190.
Ghidirim G, Mishin I, Zastavnitsky G: Pseudomyxoma peritonei presenting with inguinal hernia. Chirurgia (Bucur). 2011, 106: 527-529.
Morris-Stiff G, Falk GA, Joyce D, Chalikonda S: Unusual contents of a scrotal swelling. BMJ case reports. 2011, 2011: doi:10.1136/bcr.03.2011.4023
Skaane P, Isachsen MM, Hoiseth A: Computed tomography of mucin-producing adenocarcinoma of the appendix presenting as a bladder tumor. Comput Assist Tomogr. 1985, 9: 566-567. 10.1097/00004728-198505000-00031.
Gandhi VV, Nagral S: Pseudomyxoma peritonea: uncommon presentation. Indian J Surg. 2012, 74: 172-173. 10.1007/s12262-012-0408-9.
Snyder TE, Vandivort MR: Mucinous cystadenocarcinoma of the appendix with pseudomyxoma peritonei presenting as total uterine prolapse. A case report. J Reprod Med. 1992, 37: 103-106.
Newman CM, Moran BJ: Pseudomyxoma peritonei presenting as recurrent rectal cancer: a preventable condition?. Tech Coloproctol. 2011, 15: 89-90. 10.1007/s10151-010-0654-4.
Koyama S, Tomimatsu T, Sawada K, Kanagawa T, Tsutsui T, Kimura T: Pseudomyxoma peritonei originating from colorectal cancer during pregnancy. J Obstet Gynaecol Res. 2011, 37: 254-258. 10.1111/j.1447-0756.2010.01379.x.
Abdu B, Hobgood D, Stallings S, Depasquale S: Incidental finding of pseudomyxoma peritonei at primary cesarean section. Am J Perinatol. 2009, 26: 633-635. 10.1055/s-0029-1220791.
Cakmak A, Karakayali F, Bayar S, Unal E, Akyol C, Kocaoglu H: Pseudomyxoma retroperitonei presenting with a skin fistula. Turk J Gastroenterol. 2009, 20: 79-80.
Srinivasaiah N, Retnasingam G, Kasarneni R, Slater B: Pseudomyxoma peritonei: a rare presentation as an umbilical nodule. Ir J Med Sci. 2009, 178: 219-221. 10.1007/s11845-008-0118-2.
Smeenk RM, Verwaal VJ, Zoetmulder FA: Pseudomyxoma peritonei. Cancer Treat Rev. 2007, 33: 138-145. 10.1016/j.ctrv.2006.11.001.
Kalu E, Croucher C: Appendiceal mucocele: a rare differential diagnosis of a cystic right adnexal mass. Arch Gynecol Obstet. 2005, 271: 86-88. 10.1007/s00404-004-0663-5.
Gortchev G, Tomov S, Dimitrov D, Nanev V, Betova T: Appendiceal mucocele presenting as a right adnexal mass: a case report. Obstet Gynecol Int. 2010, 2010: doi:10.1155/2010/281053
Khan S, Patel AG, Jurkovic D: Incidental ultrasound diagnosis of pseudomyxoma peritonei in an asymptomatic woman. Ultrasound Obstet Gynecol. 2002, 19: 410-412. 10.1046/j.1469-0705.2002.00611.x.
Kreel L, Bydder GM: Computed tomography of fluid collections within the abdomen. J Comput Tomogr. 1980, 4: 105-115. 10.1016/S0149-936X(80)80004-X.
Sulkin TV, O’Neill H, Amin AI, Moran B: CT in pseudomyxoma peritonei: a review of 17 cases. Clin Radiol. 2002, 57: 608-613. 10.1053/crad.2002.0942.
Buy JN, Malbec L, Ghossain MA, Guinet C, Ecoiffier J: Magnetic resonance imaging of pseudomyxoma peritonei. Eur J Radiol. 1989, 9: 115-118.
Matsuoka Y, Masumoto T, Suzuki K, Terada K, Ushimi T, Yokoyama Y, Abe K, Kamata N, Yasuno M, Hishima T: Pseudomyxoma retroperitonei. Eur Radiol. 1999, 9: 457-459. 10.1007/s003300050692.
Fairise A, Barbary C, Derelle A, Tissier S, Granger P, Marchal F, Laurent V: Regent D: [Mucocele of the appendix and pseudomyxoma peritonei]. J Radiol. 2008, 89: 751-762. 10.1016/S0221-0363(08)73781-8.
Yang QM, Bando E, Kawamura T, Tsukiyama G, Nemoto M, Yonemura Y, Furukawa H: The diagnostic value of PET-CT for peritoneal dissemination of abdominal malignancies. Gan To Kagaku Ryoho. 2006, 33: 1817-1821.
Passot G, Glehen O, Pellet O, Isaac S, Tychyj C, Mohamed F, Giammarile F, Gilly FN, Cotte E: Pseudomyxoma peritonei: role of 18 F-FDG PET in preoperative evaluation of pathological grade and potential for complete cytoreduction. Eur J Surg Oncol. 2010, 36: 315-323. 10.1016/j.ejso.2009.09.001.
Stewart JH, Shen P, Levine EA: Intraperitoneal hyperthermic chemotherapy for peritoneal surface malignancy: current status and future directions. Ann Surg Oncol. 2005, 12: 765-777. 10.1245/ASO.2005.12.001.
Rohani P, Scotti SD, Shen P, Stewart JH, Russell GB, Cromer M, Levine EA: Use of FDG-PET imaging for patients with disseminated cancer of the appendix. Am Surg. 2010, 76: 1338-1344.
Carmignani CP, Hampton R, Sugarbaker CE, Chang D, Sugarbaker PH: Utility of CEA and CA 19–9 tumor markers in diagnosis and prognostic assessment of mucinous epithelial cancers of the appendix. J Surg Oncol. 2004, 87: 162-166. 10.1002/jso.20107.
Alexander-Sefre F, Chandrakumaran K, Banerjee S, Sexton R, Thomas JM, Moran B: Elevated tumour markers prior to complete tumour removal in patients with pseudomyxoma peritonei predict early recurrence. Colorectal Dis. 2005, 7: 382-386. 10.1111/j.1463-1318.2005.00773.x.
van Ruth S, Hart AA, Bonfrer JM, Verwaal VJ, Zoetmulder FA: Prognostic value of baseline and serial carcinoembryonic antigen and carbohydrate antigen 19.9 measurements in patients with pseudomyxoma peritonei treated with cytoreduction and hyperthermic intraperitoneal chemotherapy. Ann Surg Oncol. 2002, 9: 961-967. 10.1245/ASO.2002.04.020.
Baratti D, Kusamura S, Martinetti A, Seregni E, Laterza B, Oliva DG, Deraco M: Prognostic value of circulating tumor markers in patients with pseudomyxoma peritonei treated with cytoreductive surgery and hyperthermic intraperitoneal chemotherapy. Ann Surg Oncol. 2007, 14: 2300-2308. 10.1245/s10434-007-9393-9.
Chua TC, Chong CH, Liauw W, Zhao J, Morris DL: Inflammatory markers in blood and serum tumor markers predict survival in patients with epithelial appendiceal neoplasms undergoing surgical cytoreduction and intraperitoneal chemotherapy. Ann Surg. 2012, 256: 342-349. 10.1097/SLA.0b013e3182602ad2.
Koh JL, Liauw W, Chua T, Morris DL: Carbohydrate antigen 19–9 (CA 19–9) is an independent prognostic indicator in pseudomyxoma peritonei post cytoreductive surgery and perioperative intraperitoneal chemotherapy. J Gastrointest Oncol. 2013, 4: 173-181.
Kusamura S, Hutanu I, Baratti D, Deraco M: Circulating tumor markers: predictors of incomplete cytoreduction and powerful determinants of outcome in pseudomyxoma peritonei. J Surg Oncol. 2013, 108: 1-8. 10.1002/jso.23329.
O’Connell JT, Hacker CM, Barsky SH: MUC2 is a molecular marker for pseudomyxoma peritonei. Mod Pathol. 2002, 15: 958-972. 10.1097/01.MP.0000026617.52466.9F.
Bibi R, Pranesh N, Saunders MP, Wilson MS, O’Dwyer ST, Stern PL, Renehan AG: A specific cadherin phenotype may characterise the disseminating yet non-metastatic behaviour of pseudomyxoma peritonei. Br J Cancer. 2006, 95: 1258-1264. 10.1038/sj.bjc.6603398.
Semino-Mora C, Liu H, McAvoy T, Nieroda C, Studeman K, Sardi A, Dubois A: Pseudomyxoma peritonei: is disease progression related to microbial agents? A study of bacteria, MUC2 AND MUC5AC expression in disseminated peritoneal adenomucinosis and peritoneal mucinous carcinomatosis. Ann Surg Oncol. 2008, 15: 1414-1423. 10.1245/s10434-007-9778-9.
Flatmark K, Davidson B, Kristian A, Stavnes HT, Forsund M, Reed W: Exploring the peritoneal surface malignancy phenotype–a pilot immunohistochemical study of human pseudomyxoma peritonei and derived animal models. Hum Pathol. 2010, 41: 1109-1119. 10.1016/j.humpath.2009.12.013.
Guo AT, Song X, Wei LX, Zhao P: Histological origin of pseudomyxoma peritonei in Chinese women: clinicopathology and immunohistochemistry. World J Gastroenterol. 2011, 17: 3531-3537. 10.3748/wjg.v17.i30.3531.
Baratti D, Kusamura S, Nonaka D, Cabras AD, Laterza B, Deraco M: Pseudomyxoma peritonei: biological features are the dominant prognostic determinants after complete cytoreduction and hyperthermic intraperitoneal chemotherapy. Ann Surg. 2009, 249: 243-249. 10.1097/SLA.0b013e31818eec64.
Clement PB, Granai CO, Young RH, Scully RE: Endometriosis with myxoid change. A case simulating pseudomyxoma peritonei. Am J Surg Pathol. 1994, 18: 849-853. 10.1097/00000478-199408000-00013.
Sugi Subramaniam RV, Karthikeyan VS, Sistla SC, Ali SM, Sistla S, Ram D, Sudhagar R: Melioidosis presenting as pseudomyxoma peritonei: yet another pretense of the great mimicker: an unreported entity. Surg Infect (Larchmt). 2013, 14: 415-417. 10.1089/sur.2012.140.
Mirich DR, Gray RR, Grosman H: Abdominal plexiform neurofibromatosis simulating pseudomyxoma peritonei on computed tomography. J Comput Assist Tomogr. 1989, 13: 709-711. 10.1097/00004728-198907000-00034.
Gough DB, Donohue JH, Schutt AJ, Gonchoroff N, Goellner JR, Wilson TO, Naessens JM, O’Brien PC, van Heerden JA: Pseudomyxoma peritonei. Long-term patient survival with an aggressive regional approach. Ann Surg. 1994, 219: 112-119. 10.1097/00000658-199402000-00002.
Miner TJ, Shia J, Jaques DP, Klimstra DS, Brennan MF, Coit DG: Long-term survival following treatment of pseudomyxoma peritonei: an analysis of surgical therapy. Ann Surg. 2005, 241: 300-308. 10.1097/01.sla.0000152015.76731.1f.
Jarvinen P, Jarvinen HJ, Lepisto A: Survival of patients with pseudomyxoma peritonei treated by serial debulking. Colorectal Dis. 2010, 12: 868-872. 10.1111/j.1463-1318.2009.01947.x.
Sugarbaker PH: Cytoreductive surgery and intraperitoneal chemotherapy with peritoneal spread of cystadenocarcinoma. Eur J Surg Suppl = Acta chirurgica Supplement. 1991, 561: 75-82.
Sugarbaker PH: Cytoreductive surgery and peri-operative intraperitoneal chemotherapy as a curative approach to pseudomyxoma peritonei syndrome. Eur J Surg Oncol. 2001, 27: 239-243. 10.1053/ejso.2000.1038.
Hadi R, Saunders V, Utkina O, Clingan P, Kam P, Links M, Morris DL: Review of patients with peritoneal malignancy treated with peritonectomy and heated intraperitoneal chemotherapy. ANZ J Surg. 2006, 76: 156-161. 10.1111/j.1445-2197.2006.03579.x.
Yan TD, Links M, Xu ZY, Kam PC, Glenn D, Morris DL: Cytoreductive surgery and perioperative intraperitoneal chemotherapy for pseudomyxoma peritonei from appendiceal mucinous neoplasms. Br J Surg. 2006, 93: 1270-1276. 10.1002/bjs.5427.
Chua TC, Yan TD, Smigielski ME, Zhu KJ, Ng KM, Zhao J, Morris DL: Long-term survival in patients with pseudomyxoma peritonei treated with cytoreductive surgery and perioperative intraperitoneal chemotherapy: 10 years of experience from a single institution. Ann Surg Oncol. 2009, 16: 1903-1911. 10.1245/s10434-009-0341-8.
Chua TC, Liauw W, Morris DL: The St George Hospital peritoneal surface malignancy program–where are we now?. ANZ J Surg. 2009, 79: 416-418.
Saxena A, Yan TD, Chua TC, Morris DL: Critical assessment of risk factors for complications after cytoreductive surgery and perioperative intraperitoneal chemotherapy for pseudomyxoma peritonei. Ann Surg Oncol. 2010, 17: 1291-1301. 10.1245/s10434-009-0875-9.
Chua TC, Baker B, Yan TD, Zhao J, Morris DL: Palliative effects of an incomplete cytoreduction combined with perioperative intraperitoneal chemotherapy. Am J Clin Oncol. 2010, 33: 568-571. 10.1097/COC.0b013e3181b9cf47.
Chua TC, Al-Zahrani A, Saxena A, Liauw W, Zhao J, Morris DL: Secondary cytoreduction and perioperative intraperitoneal chemotherapy after initial debulking of pseudomyxoma peritonei: a study of timing and the impact of malignant dedifferentiation. J Am Coll Surg. 2010, 211: 526-535. 10.1016/j.jamcollsurg.2010.06.011.
Chua TC, Martin S, Saxena A, Liauw W, Yan TD, Zhao J, Lok I, Morris DL: Evaluation of the cost-effectiveness of cytoreductive surgery and hyperthermic intraperitoneal chemotherapy (peritonectomy) at the St George Hospital peritoneal surface malignancy program. Ann Surg. 2010, 251: 323-329. 10.1097/SLA.0b013e3181c9b53c.
Chua TC, Al-Zahrani A, Saxena A, Glenn D, Liauw W, Zhao J, Morris DL: Determining the association between preoperative computed tomography findings and postoperative outcomes after cytoreductive surgery and perioperative intraperitoneal chemotherapy for pseudomyxoma peritonei. Ann Surg Oncol. 2011, 18: 1582-1589. 10.1245/s10434-010-1492-3.
Chua TC, Liauw W, Zhao J, Morris DL: Upfront compared to delayed cytoreductive surgery and perioperative intraperitoneal chemotherapy for pseudomyxoma peritonei is associated with considerably lower perioperative morbidity and recurrence rate. Ann Surg. 2011, 253: 769-773. 10.1097/SLA.0b013e31820b4dba.
Chua TC, Liauw W, Morris DL: Early recurrence of pseudomyxoma peritonei following treatment failure of cytoreductive surgery and perioperative intraperitoneal chemotherapy is indicative of a poor survival outcome. Int J Colorectal Dis. 2012, 27: 381-389. 10.1007/s00384-011-1303-8.
Kirby R, Liauw W, Zhao J, Morris D: Quality of life study following cytoreductive surgery and intraperitoneal chemotherapy for pseudomyxoma peritonei including redo procedures. Int J Surg Oncol. 2013, 2013: 461041.
Sugarbaker PH, Chang D: Results of treatment of 385 patients with peritoneal surface spread of appendiceal malignancy. Ann Surg Oncol. 1999, 6: 727-731. 10.1007/s10434-999-0727-7.
Deraco M, Baratti D, Inglese MG, Allaria B, Andreola S, Gavazzi C, Kusamura S: Peritonectomy and intraperitoneal hyperthermic perfusion (IPHP): a strategy that has confirmed its efficacy in patients with pseudomyxoma peritonei. Ann Surg Oncol. 2004, 11: 393-398. 10.1245/ASO.2004.07.002.
Loungnarath R, Causeret S, Bossard N, Faheez M, Sayag-Beaujard AC, Brigand C, Gilly F, Glehen O: Cytoreductive surgery with intraperitoneal chemohyperthermia for the treatment of pseudomyxoma peritonei: a prospective study. Dis Colon Rectum. 2005, 48: 1372-1379. 10.1007/s10350-005-0045-5.
Murphy EM, Sexton R, Moran BJ: Early results of surgery in 123 patients with pseudomyxoma peritonei from a perforated appendiceal neoplasm. Dis Colon Rectum. 2007, 50: 37-42. 10.1007/s10350-006-0741-9.
Smeenk RM, Verwaal VJ, Antonini N, Zoetmulder FA: Survival analysis of pseudomyxoma peritonei patients treated by cytoreductive surgery and hyperthermic intraperitoneal chemotherapy. Ann Surg. 2007, 245: 104-109. 10.1097/01.sla.0000231705.40081.1a.
Moran B, Baratti D, Yan TD, Kusamura S, Deraco M: Consensus statement on the loco-regional treatment of appendiceal mucinous neoplasms with peritoneal dissemination (pseudomyxoma peritonei). J Surg Oncol. 2008, 98: 277-282. 10.1002/jso.21054.
Elias D, Gilly F, Quenet F, Bereder JM, Sideris L, Mansvelt B, Lorimier G, Glehen O: Pseudomyxoma peritonei: a French multicentric study of 301 patients treated with cytoreductive surgery and intraperitoneal chemotherapy. Eur J Surg Oncol. 2010, 36: 456-462. 10.1016/j.ejso.2010.01.006.
Andreasson H, Graf W, Nygren P, Glimelius B, Mahteme H: Outcome differences between debulking surgery and cytoreductive surgery in patients with Pseudomyxoma peritonei. Eur J Surg Oncol. 2012, 38: 962-968. 10.1016/j.ejso.2012.07.009.
Dayal S, Taflampas P, Riss S, Chandrakumaran K, Cecil TD, Mohamed F, Moran BJ: Complete cytoreduction for pseudomyxoma peritonei is optimal but maximal tumor debulking may be beneficial in patients in whom complete tumor removal cannot be achieved. Dis Colon Rectum. 2013, 56: 1366-1372. 10.1097/DCR.0b013e3182a62b0d.
Kuijpers AM, Mirck B, Aalbers AG, Nienhuijs SW, de Hingh IH, Wiezer MJ, van Ramshorst B, van Ginkel RJ, Havenga K, Bremers AJ, de Wilt JH, Te Velde EA, Verwaal VJ: Cytoreduction and HIPEC in the Netherlands: nationwide long-term outcome following the Dutch protocol. Ann Surg Oncol. 2013, 20: 4224-4230. 10.1245/s10434-013-3145-9.
McBride K, McFadden D, Osler T: Improved survival of patients with pseudomyxoma peritonei receiving intraperitoneal chemotherapy with cytoreductive surgery: a systematic review and meta-analysis. J Surg Res. 2013, 183: 246-252. 10.1016/j.jss.2012.12.040.
Raj J, Urban LM, ReMine SG, Raj PK: Laparoscopic management of pseudomyxoma peritonei secondary to adenocarcinoma of the appendix. J Laparoendosc Adv Surg Tech A. 1999, 9: 299-303. 10.1089/lap.1999.9.299.
Fish R, Selvasekar C, Crichton P, Wilson M, Fulford P, Renehan A, O’Dwyer S: Risk-reducing laparoscopic cytoreductive surgery and hyperthermic intraperitoneal chemotherapy for low-grade appendiceal mucinous neoplasm: early outcomes and technique. Surg Endosc. 2014, 28: 341-345. 10.1007/s00464-013-3189-8.
Berkovic P, van de Voorde L, De Meerleer G, Delrue L, Speleers B, Van Belle S, Vandecasteele K: Whole abdominopelvic radiotherapy in the palliative treatment of pseudomyxoma peritonei. Strahlenther Onkolo: Organ der Deutschen Rontgengesellschaft [et al.]. 2014, 190: 223-228. 10.1007/s00066-013-0470-7.
Green N, Gancedo H, Smith R, Bernett G: Pseudomyxoma peritonei-nonoperative management and biochemical findings. A case report. Cancer. 1975, 36: 1834-1837. 10.1002/1097-0142(197511)36:5<1834::AID-CNCR2820360539>3.0.CO;2-6.
Piver MS, Lele SB, Patsner B: Pseudomyxoma peritonei: possible prevention of mucinous ascites by peritoneal lavage. Obstet Gynecol. 1984, 64: 95S-96S. 10.1097/00006250-198409001-00025.
Machado MA, Rodrigues JG, Laurino RM, Garrido Junior A, Pinotti HW: [Conservative treatment of pseudomyxoma peritonei]. Rev Hosp Clin Fac Med Sao Paulo. 1993, 48: 301-304.
Pillai K, Akhter J, Chua TC, Morris DL: Mucolysis by ascorbic acid and hydrogen peroxide on compact mucin secreted in pseudomyxoma peritonei. J Surg Res. 2012, 174: e69-73. 10.1016/j.jss.2011.10.038.
Pillai K, Akhter J, Chua TC, Morris DL: Potential mucolytic agents for mucinous ascites from pseudomyxoma peritonei. Invest New Drugs. 2012, 30: 2080-2086. 10.1007/s10637-012-9797-7.
Gilbreath JJ, Semino-Mora C, Friedline CJ, Liu H, Bodi KL, McAvoy TJ, Francis J, Nieroda C, Sardi A, Dubois A, Lazinski DW, Camilli A, Testerman TL, Merrell DS: A core microbiome associated with the peritoneal tumors of pseudomyxoma peritonei. Orphanet J Rare Dis. 2013, 8: 105-10.1186/1750-1172-8-105.
Semino-Mora C, Testerman TL, Liu H, Whitmire JM, Studeman K, Jia Y, McAvoy TJ, Francis J, Nieroda C, Sardi A, Merrell DS, Dubois A: Antibiotic treatment decreases microbial burden associated with pseudomyxoma peritonei and affects beta-catenin distribution. Clin Cancer Res. 2013, 19: 3966-3976. 10.1158/1078-0432.CCR-13-0616.
Choudry HA, Mavanur A, O’Malley ME, Zeh HJ, Guo Z, Bartlett DL: Chronic anti-inflammatory drug therapy inhibits gel-forming mucin production in a murine xenograft model of human pseudomyxoma peritonei. Ann Surg Oncol. 2012, 19: 1402-1409. 10.1245/s10434-012-2242-5.
Shetty S, Natarajan B, Thomas P, Govindarajan V, Sharma P, Loggie B: Proposed classification of pseudomyxoma peritonei: influence of signet ring cells on survival. Am Surg. 2013, 79: 1171-1176.
Solkar MH, Akhtar NM, Khan Z, Parker MC: Pseudomyxoma extraperitonei occurring 35 years after appendicectomy: a case report and review of literature. World J Surg Oncol. 2004, 2:19. 10.1186/1477-7819-2-19.
Thompson MA, Ashton RW, Pitot HC: Mucinous appendiceal adenocarcinoma presenting 5 years after appendectomy. Ann Intern Med. 2004, 140: W33-10.7326/0003-4819-140-1-200401060-00009.
Vana J, Adamicova K, Johanes R, Zacharova O, Ammerova E, Solek R: [Appendiceal mucocele causing pseudomyxoma peritonei]. Zentralblatt fur Chirurgie. 2005, 130: 177-180. 10.1055/s-2005-836390.
Ruiz-Tovar J, Teruel DG, Castineiras VM, Dehesa AS, Quindos PL, Molina EM: Mucocele of the appendix. World J Surg. 2007, 31: 542-548. 10.1007/s00268-006-0454-1.
Taii A, Sakagami J, Shinoda M, Taniguchi H, Tosa M, Baba T, Motoyoshi T, Ito R, Mitsufuji S, Kataoka K, Okanoue T: Pseudomyxoma peritonei occurring after an uneventful 23 years interval from appendectomy. Intern Med. 2007, 46: 1109-1112. 10.2169/internalmedicine.46.0079.
Spiliotis J, Efstathiou E, Halkia E, Vaxevanidou A, Datsis A, Sugarbaker P: The influence of tumor cell entrapment phenomenon on the natural history of Pseudomyxoma peritonei syndrome. Hepatogastroenterology. 2012, 59: 705-708.
Gum JR: Mucin genes and the proteins they encode: structure, diversity, and regulation. Am J Respir Cell Mol Biol. 1992, 7: 557-564. 10.1165/ajrcmb/7.6.557.
Rose MC, Voynow JA: Respiratory tract mucin genes and mucin glycoproteins in health and disease. Physiol Rev. 2006, 86: 245-278. 10.1152/physrev.00010.2005.
Williams OW, Sharafkhaneh A, Kim V, Dickey BF, Evans CM: Airway mucus: from production to secretion. Am J Respir Cell Mol Biol. 2006, 34: 527-536. 10.1165/rcmb.2005-0436SF.
Hollingsworth MA, Swanson BJ: Mucins in cancer: protection and control of the cell surface. Nat Rev Cancer. 2004, 4: 45-60. 10.1038/nrc1251.
Rachagani S, Torres MP, Moniaux N, Batra SK: Current status of mucins in the diagnosis and therapy of cancer. Biofactors. 2009, 35: 509-527. 10.1002/biof.64.
Kufe DW: Mucins in cancer: function, prognosis and therapy. Nat Rev Cancer. 2009, 9: 874-885. 10.1038/nrc2761.
Cone RA: Barrier properties of mucus. Adv Drug Deliv Rev. 2009, 61: 75-85. 10.1016/j.addr.2008.09.008.
Lievin-Le Moal V, Servin AL: The front line of enteric host defense against unwelcome intrusion of harmful microorganisms: mucins, antimicrobial peptides, and microbiota. Clin Microbiol Rev. 2006, 19: 315-337. 10.1128/CMR.19.2.315-337.2006.
McCracken VJ, Lorenz RG: The gastrointestinal ecosystem: a precarious alliance among epithelium, immunity and microbiota. Cell Microbiol. 2001, 3: 1-11. 10.1046/j.1462-5822.2001.00090.x.
Dharmani P, Srivastava V, Kissoon-Singh V, Chadee K: Role of intestinal mucins in innate host defense mechanisms against pathogens. J Innate Immun. 2009, 1: 123-135. 10.1159/000163037.
Johansson ME, Larsson JM, Hansson GC: The two mucus layers of colon are organized by the MUC2 mucin, whereas the outer layer is a legislator of host-microbial interactions. Proc Natl Acad Sci U S A. 2011, 108 (Suppl 1): 4659-4665.
Mall AS, Chirwa N, Govender D, Lotz Z, Tyler M, Rodrigues J, Kahn D, Goldberg P: MUC2, MUC5AC and MUC5B in the mucus of a patient with pseudomyxoma peritonei: biochemical and immunohistochemical study. Pathol Int. 2007, 57: 537-547. 10.1111/j.1440-1827.2007.02137.x.
Mall AS, Lotz Z, Tyler M, Goldberg P, Rodrigues J, Kahn D, Chirwa N, Govender D: Immunohistochemical and biochemical characterization of mucin in pseudomyxoma peritonei: a case study. Case Rep Gastroenterol. 2011, 5: 5-16. 10.1159/000323137.
Sheehan JK, Richardson PS, Fung DC, Howard M, Thornton DJ: Analysis of respiratory mucus glycoproteins in asthma: a detailed study from a patient who died in status asthmaticus. Am J Respir Cell Mol Biol. 1995, 13: 748-756. 10.1165/ajrcmb.13.6.7576713.
Sheehan JK, Howard M, Richardson PS, Longwill T, Thornton DJ: Physical characterization of a low-charge glycoform of the MUC5B mucin comprising the gel-phase of an asthmatic respiratory mucous plug. Biochem J. 1999, 338 (Pt 2): 507-513.
Mohamed F, Gething S, Haiba M, Brun EA, Sugarbaker PH: Clinically aggressive pseudomyxoma peritonei: a variant of a histologically indolent process. J Surg Oncol. 2004, 86: 10-15. 10.1002/jso.20038.
Kinkor Z, Michal M: [Syndrome of pseudomyxoma peritonei–description of three cases and survey of the problem]. Ceska Gynekol. 2005, 70: 67-72.
Nonaka D, Kusamura S, Baratti D, Casali P, Younan R, Deraco M: CDX-2 expression in pseudomyxoma peritonei: a clinicopathological study of 42 cases. Histopathology. 2006, 49: 381-387. 10.1111/j.1365-2559.2006.02512.x.
Ferreira CR, Carvalho JP, Soares FA, Siqueira SA, Carvalho FM: Mucinous ovarian tumors associated with pseudomyxoma peritonei of adenomucinosis type: immunohistochemical evidence that they are secondary tumors. Int J Gynecol Cancer. 2008, 18: 59-65.
Chang MS, Byeon SJ, Yoon SO, Kim BH, Lee HS, Kang GH, Kim WH, Park KJ: Leptin, MUC2 and mTOR in appendiceal mucinous neoplasms. Pathobiology. 2012, 79: 45-53. 10.1159/000332739.
Li JD, Feng W, Gallup M, Kim JH, Gum J, Kim Y, Basbaum C: Activation of NF-kappaB via a Src-dependent Ras-MAPK-pp90rsk pathway is required for Pseudomonas aeruginosa-induced mucin overproduction in epithelial cells. Proc Natl Acad Sci U S A. 1998, 95: 5718-5723. 10.1073/pnas.95.10.5718.
Perrais M, Pigny P, Copin MC, Aubert JP, Van Seuningen I: Induction of MUC2 and MUC5AC mucins by factors of the epidermal growth factor (EGF) family is mediated by EGF receptor/Ras/Raf/extracellular signal-regulated kinase cascade and Sp1. J Biol Chem. 2002, 277: 32258-32267. 10.1074/jbc.M204862200.
Orlowski RZ, Baldwin AS: NF-kappaB as a therapeutic target in cancer. Trends Mol Med. 2002, 8: 385-389. 10.1016/S1471-4914(02)02375-4.
Choudry HA, O’Malley ME, Guo ZS, Zeh HJ, Bartlett DL: Mucin as a therapeutic target in pseudomyxoma peritonei. J Surg Oncol. 2012, 106: 911-917. 10.1002/jso.23146.
Enss ML, Cornberg M, Wagner S, Gebert A, Henrichs M, Eisenblatter R, Beil W, Kownatzki R, Hedrich HJ: Proinflammatory cytokines trigger MUC gene expression and mucin release in the intestinal cancer cell line LS180. Inflamm Res. 2000, 49: 162-169. 10.1007/s000110050576.
Kim YD, Kwon EJ, Kwon TK, Baek SH, Song SY, Suh JS: Regulation of IL-1beta-mediated MUC2 gene in NCI-H292 human airway epithelial cells. Biochem Biophys Res Commun. 2000, 274: 112-116. 10.1006/bbrc.2000.3107.
Iwashita J, Sato Y, Sugaya H, Takahashi N, Sasaki H, Abe T: mRNA of MUC2 is stimulated by IL-4, IL-13 or TNF-alpha through a mitogen-activated protein kinase pathway in human colon cancer cells. Immunol Cell Biol. 2003, 81: 275-282. 10.1046/j.1440-1711.2003.t01-1-01163.x.
Kai H, Yoshitake K, Hisatsune A, Kido T, Isohama Y, Takahama K, Miyata T: Dexamethasone suppresses mucus production and MUC-2 and MUC-5 AC gene expression by NCI-H292 cells. Am J Physiol. 1996, 271: L484-L488.
Schoneveld OJ, Gaemers IC, Lamers WH: Mechanisms of glucocorticoid signalling. Biochim Biophys Acta. 2004, 1680: 114-128. 10.1016/j.bbaexp.2004.09.004.
Okudaira K, Kakar S, Cun L, Choi E, Wu Decamillis R, Miura S, Sleisenger MH, Kim YS, Deng G: MUC2 gene promoter methylation in mucinous and non-mucinous colorectal cancer tissues. Int J Oncol. 2010, 36: 765-775.
Aslam F, Palumbo L, Augenlicht LH, Velcich A: The Sp family of transcription factors in the regulation of the human and mouse MUC2 gene promoters. Cancer Res. 2001, 61: 570-576.
Chua TC, Akther J, Yao P, Morris DL: In vivo model of pseudomyxoma peritonei for novel candidate drug discovery. Anticancer Res. 2009, 29: 4051-4055.
Akhter J, Yao P, Johnson LA, Riordan SM, Morris DL: A new peritoneal carcinomatosis model in cyclosporine immunosuppressed rats. Anticancer Res. 2008, 28: 105-108.
Bromelain. Monograph. Altern Med Rev. 2010, 15: 361-368.
N-acetylcysteine. Altern Med Rev. 2000, 5: 467-471.
Amini A, Ehteda A, Masoumi Moghaddam S, Akhter J, Pillai K, Morris DL: Cytotoxic effects of bromelain in human gastrointestinal carcinoma cell lines (MKN45, KATO-III, HT29-5F12, and HT29-5M21). Onco Targets Ther. 2013, 2013: 403-409.
Pillai K, Akhter J, Chua TC, Morris DL: Anticancer property of bromelain with therapeutic potential in malignant peritoneal mesothelioma. Cancer Invest. 2013, 31: 241-250. 10.3109/07357907.2013.784777.
Pillai K, Ehteda A, Akhter J, Chua TC, Morris DL: Anticancer effect of bromelain with and without cisplatin or 5-FU on malignant peritoneal mesothelioma cells. Anti-cancer drugs. 2014, 25: 150-160. 10.1097/CAD.0000000000000039.
Pillai K, Akhter J, Chua TC, Morris DL: A formulation for in situ lysis of mucin secreted in pseudomyxoma peritonei. Int J Cancer. 2014, 134: 478-486. 10.1002/ijc.28380.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
AA designed and performed the systematic literature search supervised by DLM. AA drafted and edited the manuscript. SMM and AE reviewed the initial draft. SMM designed and prepared the figures. DLM critically assessed the initial draft. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Amini, A., Masoumi-Moghaddam, S., Ehteda, A. et al. Secreted mucins in pseudomyxoma peritonei: pathophysiological significance and potential therapeutic prospects. Orphanet J Rare Dis 9, 71 (2014). https://doi.org/10.1186/1750-1172-9-71
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1750-1172-9-71