
Abstract
Background
The pathophysiologic mechanisms classically involved in sickle-cell nephropathy include endothelial dysfunction and vascular occlusion. Arguments demonstrating that ischemia-reperfusion injury-related kidney damage might coincide with vaso-occlusive crisis (VOC) are lacking.
Methods
In this prospective study, we sought to determine whether tubular cells and glomerular permeability might be altered during VOC. Urine neutrophil gelatinase-associated lipocalin (NGAL) levels and albumin-excretion rates (AER) of 25 patients were evaluated prospectively during 25 VOC episodes and compared to their steady state (ST) values.
Results
During VOC, white blood-cell counts (WBC) and C-reactive protein (CRP) were significantly higher than at ST but creatinine levels were comparable. Urine NGAL levels were significantly increased during VOC vs ST (P = 0.007) and remained significant when normalized to urine creatinine (P = 0.004), while AER did not change significantly. The higher urine NGAL concentration was not associated with subsequent (24-48 hour) acute kidney injury. Univariate analysis identified no significant correlations between urine NGAL levels and laboratory parameters during VOC.
Conclusions
These results demonstrated that subclinical ischemia-reperfusion tubular injury is common during VOC and highlight the importance of hydroelectrolyte monitoring and correction during VOC.
Similar content being viewed by others

Background
Sickle-cell nephropathy (SCN), a major mortality risk factor in sickle-cell disease (SCD) patients, is characterized by early increased glomerular filtration rates (GFR), frequently associated with proteinuria, that promote progressive renal function decline[1–3]. Although the SCN pathophysiologic process remains hypothetical, chronic hemolysis-related endothelial dysfunction and the relative renal hypoxia triggered by repeated vaso-occlusive crises (VOC) are currently considered critical key factors[2, 4, 5]. Experimental data from murine SCD models demonstrated that ischemia-reperfusion (I-R) injury can induce direct kidney damage, characterized by marked renal vascular congestion and tubular injury, associated with renal hemodynamic changes[6, 7]. In SCD patients, VOC initiation, progression and resolution may be considered typical features of I-R injury[8, 9], but definitive proof of I-R–induced tubular injury in SCD patients during VOC is lacking. Renal tubular cells, a predominant site of cell metabolic activity and oxygen consumption, are highly susceptible to oxygen deprivation during I-R injury[10]. However, clinical demonstration that the kidneys are a major I-R site during SCD VOC remains unsubstantiated. We compared prospective urine neutrophil gelatinase-associated lipocalin (NGAL), a tubular injury biomarker[11], and albumin-excretion rate (AER) levels during VOC to steady state (ST) values to determine whether renal tubular cells and glomerular capillary permeability could be directly affected during VOC.
Methods
This prospective study, approved by our local Ethics Committee, was conducted in our Adult Sickle-Cell Referral Center in accordance with the Declaration of Helsinki, Good Clinical Practice guidelines, and local laws and regulations. Patients were enrolled after giving their written informed consent.
Homozygous SCD patients, at least 18 years old with severe VOC requiring hospitalization for pain (regardless of location) not controlled by grade-II analgesics, were eligible for inclusion once. VOC was defined as pain or tenderness requiring opioids and not attributable to other causes[12]. Exclusion criteria included: VOC with parenteral hydration lasting >24 hours; blood transfusion during the previous month; acute chest syndrome or severe complication requiring blood transfusion at inclusion; pregnancy and/or psychiatric disorder; preexisting renal insufficiency, defined as GFR ≤60 mL/min/1.73 m2 according to the modification of diet in renal disease (MDRD) formula, and/or Hydroxyurea use when the VOC occurred or at ST.
Standardized clinical practices included bed rest, fluid replacement with 5% glucose (2 L with NaCl 4 g/L), oral alkaline water (500 mL/day), folinic acid (5 mg/day), analgesia with morphine and intravenous paracetamol (1 g every 6 hours), and preventive incitative spirometry. Urine NGAL levels and biological parameters were determined simultaneously on samples collected on D1, D2 or D4 of hospitalization for each of the 25 patients’ single VOC and at an ST consultation. ST was defined as a consultation ≥1 months after a VOC and ≥3 months after blood transfusion. The Acute Kidney-Injury Network (AKIN) criteria stratify AKI into 3 stages reflecting severity of the percentage serum-creatinine change [measured using the Jaffe kinetic method (Gen.2, Roche Diagnostics, France)][13]. All room-temperature urine samples were centrifuged (2000 g × 5 minutes) within 2 hours of arriving at the lab and frozen at -80°C until assayed. An enzyme-linked immunosorbent assay kit (BioPorto Diagnostics A/S, Denmark) determined NGAL levels according to the manufacturer’s instructions. AER of <3, 3-30 or >30 mg/mmol creatinine defined normal, micro- or macroalbuminuria, respectively.
Statistical analyses
Results are expressed as means ± standard deviation or numbers (%). Between-group quantitative parameters were compared with paired Student t-tests. Simple linear regressions were computed for each biologic parameter to search for potential relationships with NGAL. Spearman’s correlation coefficient established correlations; P < 0.05 defined significance.
Results and discussion
Twenty-five SS SCD patients (12 men, 13 women; mean age: 34.6 ± 6 years), each hospitalized for a VOC, were included. The mean VOC-onset-to-hospitalization interval was 30.4 ± 23.1 hours. The mean pain visual analog scale at inclusion was 7.2 ± 1.3. None had received nonsteroidal anti-inflammatory drugs (NSAID) to treat VOC. Mean systolic and diastolic blood pressures during VOC were, respectively, 115 ± 13 and 71 ± 9 mm Hg. During VOC, white blood-cell counts (WBC) and C-reactive protein (CRP) were significantly higher than at ST but creatinine levels were comparable (Table 1). During hospitalization, no infection was documented and no patient had AKI satisfying AKIN criteria. The latter observation confirms our reported finding that AKI is rare during VOC[14]. Notably, AKI absence did not exclude possible VOC association with subclinical renal I-R injury.
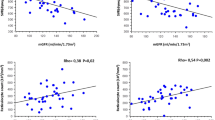
We next analyzed AER and urine NGAL level changes between VOC and ST. AER, an early marker of renal damage, reflects glomerular endothelial dysfunction and impaired glomerular barrier permeability[15]. It was reportedly elevated in 68% of adult SCD patients[1]. At ST, 15 (60%), 7 (28%) or 3 (12%) patients, respectively, had normal, micro- or macroalbuminuria. VOC and ST AER were comparable (Table 1), suggesting that, during VOC, I-R injury does not directly target glomerular permeability. NGAL, an extracellular, 25-kDa protein, is expressed at low levels in several human tissues, eg kidney[16], and is induced in tubular cells early after renal ischemic injury[17]. Urine or serum NGAL levels are considered relevant biomarkers of AKI[16, 18]. Patients with preexisting renal insufficiency were excluded from our analysis because recent findings suggested that urine and serum NGAL concentrations increased in parallel with renal disease severity[19]. Moreover, NSAID, known to cause tubular injury, were not used because our previous prospective, controlled trial results showed limited ketoprofen impact on VOC requiring hospitalization[12]. Sundaram et al recently demonstrated that ST urine NGAL levels were subnormal in 116 SCD patients and independent of the degree of albuminuria[20]. Herein, urine NGAL levels increased significantly during VOC (P = 0.007) and remained significant when normalized to urine creatinine (P = 0.004) (Figure 1). NGAL concentrations on hospital D1, D2 or D4 (n = 9, 11 and 5, respectively) were comparable (P = 0.9). Median urine NGAL values were significantly associated with AKI stage in a cohort of patients without AKI or with stage-3 AKIN admitted to the emergency department (respectively, 23 vs 153 ng/mL)[21]. Our patients’ mean NGAL-level increase in urine samples collected a mean of 30.4 ± 23.1 hours after VOC onset was relatively weak (76.7 ng/mL), probably reflecting subclinical renal injury, because none had AKI meeting AKIN criteria[22].

Urine NGAL levels (A) and urine NGAL/creatinine ratios (B) in 25 SCD patients at vaso-occlusive crises (VOC) and steady state (ST).
Based on the 50 samples from 25 patients, no correlation was found between urine NGAL levels and other laboratory parameters considered (WBC counts, hemoglobin, CRP, lactate dehydrogenase, ASAT, bilirubinemia and creatinine levels). These findings suggest that acute tubular injury during VOC is not directly related to hemolysis, anemia and inflammatory syndrome that may confound NGAL level interpretation in these patients[16]. Furthermore, they highlight the importance of correcting hydroelectrolyte disturbances, particularly acidosis, which raises the hemoglobin desaturation rate and favors HbS polymerization. Indeed, our therapeutic protocol for VOC includes bicarbonate administration. The first objective of our study was not to determine whether a potential increase of urine NGAL levels might be predictive of subsequent acute kidney injury (AKI) in these patients, as we previously demonstrated that AKI during VOC is a rare finding[14]. Our study’s primary aim was to determine whether increased urine NGAL levels might represent a pertinent biomarker attesting to subclinical tubular impairment in these patients. In agreement with that hypothesis, as observed herein, Haase et al recently identified, among 2322 critically ill patients, 19.2% failing to meet current creatinine-based consensus criteria for AKI but probably having acute tubular injury, based on increased NGAL levels[22]. We postulate that repeated tubular injury in the context of multiple VOC might promote hypoxic stress, thereby inducing a chronic inflammatory state in renal parenchyma, leading to progressive tubulointerstitial and glomerular damage[4, 23].
Conclusions
This study is the first to indicate that I-R tubular damage might occur simultaneously with VOC, as revealed by increased urine NGAL levels. The demonstration that tubular cells are indeed a major site of tissue-oxygen deprivation during SCD VOC is important for VOC management and emphasizes the need to improve intravenous hydration and patients’ tissue oxygenation during VOC. Further investigations are needed to evaluate a potential direct relationship between elevated NGAL levels during VOC and the risk of subsequent chronic kidney disease.
Author’s contributions
Contributions: PB and VA designed the study, analyzed and interpreted data, wrote the manuscript; SM, GV and SL conducted laboratory tests and reviewed data; AH, MK, PG, YL and BR, contributed patients and collected data; PL, PG, BG and FG reviewed the data and the paper; all authors approved the final manuscript.
References
Guasch A, Navarrete J, Nass K, Zayas CF: Glomerular involvement in adults with sickle cell hemoglobinopathies: prevalence and clinical correlates of progressive renal failure. J Am Soc Nephrol. 2006, 17 (8): 2228-2235. 10.1681/ASN.2002010084.
Haymann JP, Stankovic K, Levy P, Avellino V, Tharaux PL, Letavernier E, Grateau G, Baud L, Girot R, Lionnet F: Glomerular hyperfiltration in adult sickle cell anemia: a frequent hemolysis associated feature. Clin J Am Soc Nephrol. 2010, 5 (5): 756-761. 10.2215/CJN.08511109.
Powars DR, Chan LS, Hiti A, Ramicone E, Johnson C: Outcome of sickle cell anemia: a 4-decade observational study of 1056 patients. Medicine (Baltimore). 2005, 84 (6): 363-376. 10.1097/01.md.0000189089.45003.52.
Nath KA, Katusic ZS: Vasculature and kidney complications in sickle cell disease. J Am Soc Nephrol. 2012, 23 (5): 781-784. 10.1681/ASN.2011101019.
Day TG, Drasar ER, Fulford T, Sharpe CC, Thein SL: Association between hemolysis and albuminuria in adults with sickle cell anemia. Haematologica. 2012, 97 (2): 201-205. 10.3324/haematol.2011.050336.
Nath KA, Grande JP, Croatt AJ, Frank E, Caplice NM, Hebbel RP, Katusic ZS: Transgenic sickle mice are markedly sensitive to renal ischemia-reperfusion injury. Am J Pathol. 2005, 166 (4): 963-972. 10.1016/S0002-9440(10)62318-8.
Sabaa N, de Franceschi L, Bonnin P, Castier Y, Malpeli G, Debbabi H, Galaup A, Maier-Redelsperger M, Vandermeersch S, Scarpa A, Janin A, Levy B, Girot R, Beuzard Y, Leboeuf C, Henri A, Germain S, Dussaule JC, Tharaux PL: Endothelin receptor antagonism prevents hypoxia-induced mortality and morbidity in a mouse model of sickle-cell disease. J Clin Invest. 2008, 118 (5): 1924-1933. 10.1172/JCI33308.
Kaul DK, Hebbel RP: Hypoxia/reoxygenation causes inflammatory response in transgenic sickle mice but not in normal mice. J Clin Invest. 2000, 106 (3): 411-420. 10.1172/JCI9225.
Eltzschig HK, Eckle T: Ischemia and reperfusion – from mechanism to translation. Nat Med. 2011, 17 (11): 1391-1401. 10.1038/nm.2507.
Bonventre JV, Yang L: Cellular pathophysiology of ischemic acute kidney injury. J Clin Invest. 2011, 121 (11): 4210-4221. 10.1172/JCI45161.
Paragas N, Qiu A, Zhang Q, Samstein B, Deng SX, Schmidt-Ott KM, Viltard M, Yu W, Forster CS, Gong G, Liu Y, Kulkarni R, Mori K, Kalandadze A, Ratner AJ, Devarajan P, Landry DW, D'Agati V, Lin CS, Barasch J: The Ngal reporter mouse detects the response of the kidney to injury in real time. Nat Med. 2011, 17 (2): 216-222. 10.1038/nm.2290.
Bartolucci P, El Murr T, Roudot-Thoraval F, Habibi A, Santin A, Renaud B, Noël V, Michel M, Bachir D, Galactéros F, Godeau B: A randomized, controlled clinical trial of ketoprofen for sickle-cell disease vaso-occlusive crises in adults. Blood. 2009, 114 (18): 3742-3747. 10.1182/blood-2009-06-227330.
Mehta RL, Kellum JA, Shah SV, Molitoris BA, Ronco C, Warnock DG, Levin A: Acute kidney injury network: report of an initiative to improve outcomes in acute kidney injury. Crit Care. 2007, 11 (2): R31-10.1186/cc5713.
Audard V, Homs S, Habibi A, Galacteros F, Bartolucci P, Godeau B, Renaud B, Levy Y, Grimbert P, Lang P, Brun-Buisson C, Brochard L, Schortgen F, Maitre B, Mekontso DA: Acute kidney injury in sickle patients with painful crisis or acute chest syndrome and its relation to pulmonary hypertension. Nephrol Dial Transplant. 2010, 25 (8): 2524-2529. 10.1093/ndt/gfq083.
Sarnak MJ, Astor BC: Implications of proteinuria: CKD progression and cardiovascular outcomes. Adv Chronic Kidney Dis. 2011, 18 (4): 258-266. 10.1053/j.ackd.2011.04.002.
Devarajan P: Review: Neutrophil gelatinase-associated lipocalin: a troponin-like biomarker for human acute kidney injury. Nephrology (Carlton). 2010, 15 (4): 419-428. 10.1111/j.1440-1797.2010.01317.x.
Mishra J, Ma Q, Prada A, Mitsnefes M, Zahedi K, Yang J, Barasch J, Devarajan P: Identification of neutrophil gelatinase-associated lipocalin as a novel early urinary biomarker for ischemic renal injury. J Am Soc Nephrol. 2003, 14 (10): 2534-2543. 10.1097/01.ASN.0000088027.54400.C6.
Siew ED, Ware LB, Ikizler TA: Biological markers of acute kidney injury. J Am Soc Nephrol. 2011, 22 (5): 810-820. 10.1681/ASN.2010080796.
Bolignano D, Lacquaniti A, Coppolino G, Donato V, Campo S, Fazio MR, Nicocia G, Buemi M: Neutrophil gelatinase-associated lipocalin (NGAL) and progression of chronic kidney disease. Clin J Am Soc Nephrol. 2009, 4 (2): 337-344. 10.2215/CJN.03530708.
Sundaram N, Bennett M, Wilhelm J, Kim MO, Atweh G, Devarajan P, Malik P: Biomarkers for early detection of sickle nephropathy. Am J Hematol. 2011, 86 (7): 559-566. 10.1002/ajh.22045.
Schinstock CA, Semret MH, Wagner SJ, Borland TM, Bryant SC, Kashani KB, Larson TS, Lieske JC: Urinalysis is more specific and urinary neutrophil gelatinase-associated lipocalin is more sensitive for early detection of acute kidney injury. Nephrol Dial Transplant. 2013, 28 (5): 1175-1185. 10.1093/ndt/gfs127.
Haase M, Devarajan P, Haase-Fielitz A, Bellomo R, Cruz DN, Wagener G, Krawczeski CD, Koyner JL, Murray P, Zappitelli M, Goldstein SL, Makris K, Ronco C, Martensson J, Martling CR, Venge P, Siew E, Ware LB, Ikizler TA, Mertens PR: The outcome of neutrophil gelatinase-associated lipocalin-positive subclinical acute kidney injury: a multicenter pooled analysis of prospective studies. J Am Coll Cardiol. 2011, 57 (17): 1752-1761. 10.1016/j.jacc.2010.11.051.
Maigne G, Ferlicot S, Galactéros F, Belenfant X, Ulinski T, Niaudet P, Ronco P, Godeau B, Durrbach A, Sahali S, Lang P, Lambotte O, Audard V: Glomerular lesions in patients with sickle cell disease. Medicine (Baltimore). 2010, 89 (1): 18-27. 10.1097/MD.0b013e3181ca59b6.
Acknowledgments
We thank Jugurtha Berkenou and Christine Fauroux for data management; and Janet Jacobson for editorial assistance.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Competing interests
The authors declare no competing interests.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Audard, V., Moutereau, S., Vandemelebrouck, G. et al. First evidence of subclinical renal tubular injury during sickle-cell crisis. Orphanet J Rare Dis 9, 67 (2014). https://doi.org/10.1186/1750-1172-9-67
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1750-1172-9-67