
Abstract
Background
Inhaled nitric oxide (INO), a selective pulmonary vasodilator, has revolutionized the treatment of neonatal hypoxemic respiratory failure (NHRF). However, there is lack of sustained improvement in 30 to 46% of infants. Aerosolized prostaglandins I2 (PGI2) and E1 (PGE1) have been reported to be effective selective pulmonary vasodilators. The objective of this study was to evaluate the feasibility of a randomized controlled trial (RCT) of inhaled PGE1 (IPGE1) in NHRF.
Methods
Two pilot multicenter phase II RCTs are included in this report. In the first pilot, late preterm and term neonates with NHRF, who had an oxygenation index (OI) of ≥15 and <25 on two arterial blood gases and had not previously received INO, were randomly assigned to receive two doses of IPGE1 (300 and 150 ng/kg/min) or placebo. The primary outcome was the enrollment of 50 infants in six to nine months at 10 sites. The first pilot was halted after four months for failure to enroll a single infant. The most common cause for non-enrollment was prior initiation of INO. In a re-designed second pilot, co-administration of IPGE1 and INO was permitted. Infants with suboptimal response to INO received either aerosolized saline or IPGE1 at a low (150 ng/kg/min) or high dose (300 ng/kg/min) for a maximum duration of 72 hours. The primary outcome was the recruitment of an adequate number of patients (n = 50) in a nine-month-period, with fewer than 20% protocol violations.
Results
No infants were enrolled in the first pilot. Seven patients were enrolled in the second pilot; three in the control, two in the low-dose IPGE1, and two in the high-dose IPGE1 groups. The study was halted for recruitment futility after approximately six months as enrollment targets were not met. No serious adverse events, one minor protocol deviation and one pharmacy protocol violation were reported.
Conclusions
These two pilot RCTs failed to recruit adequate eligible newborns with NHRF. Complex management RCTs of novel therapies for persistent pulmonary hypertension of the newborn (PPHN) may require novel study designs and a longer period of time from study approval to commencement of enrollment.
Trial registration: ClinicalTrials.gov
Pilot one: NCT number: 00598429 registered on 10 January 2008. Last updated: 3 February 2011.
Pilot two: NCT number: 01467076 17 October 2011. Last updated: 13 February 2013.
Similar content being viewed by others


Background
Neonatal hypoxemic respiratory failure (NHRF) is usually associated with widespread vasoconstriction of the pulmonary microvasculature, giving rise to intra- and extra-pulmonary shunting and profound hypoxemia. Due to the delay in the physiologic fall in pulmonary vascular resistance, it is also known as persistent pulmonary hypertension of the newborn (PPHN). The goal of therapy is to decrease the regional pulmonary vascular resistance of ventilated lung areas, thus decreasing intrapulmonary shunting and selectively reducing the pulmonary-artery pressure without causing systemic vasodilation. Intravenously administered vasodilators lack pulmonary selectivity leading to systemic side effects. Inhaled nitric oxide (INO), a selective pulmonary vasodilator, has revolutionized the treatment of respiratory failure in the newborn. However, there is lack of sustained improvement in 30 to 46% of infants [1–4]. Moreover, INO requires specialized delivery systems, making the treatment expensive.
Aerosolized prostaglandins I2 (PGI2) and E1 (PGE1) have been reported to be effective selective pulmonary vasodilators in animals, adults, and preterm and term newborns [5–15]. Compared to PGI2, PGE1 has a shorter half-life, lower acid dissociation constant (pKa, 6.3 versus 10.5), bronchodilator action, and anti-proliferative and anti-inflammatory effects on the alveolar, interstitial, and vascular spaces of the lung [10, 16–20]. In addition, PGE1 is readily available in pharmacies of hospitals with neonatal services and has a proven safety record from its intravenous use in ductal-dependent cardiac anomalies. Prostaglandin nebulization can be performed without the sophisticated technical equipment needed for INO and hence is less expensive. Prostaglandins and nitric oxide relax the vascular smooth muscle through two different second-messenger systems; therefore, in combination, INO and inhaled PGE1 (IPGE1) may have a synergistic effect [21].
We reported the safety and feasibility of short-term administration of IPGE1 in an un-blinded phase I/II dose-escalation single center study [12] suggesting the need for placebo-controlled randomized studies to establish the efficacy and safety of this drug for NHRF. In that study, four doses ranging from 25 to 300 ng/kg/min were tested for a maximum duration of three hours. The purpose of the two studies included in this report was to determine the feasibility of conducting a future large randomized control trial (RCT) to evaluate the safety and efficacy of prolonged IPGE1 at high-volume level III to IV neonatal intensive care units (NICUs) within the National Institute of Child Health and Human Development (NICHD) Neonatal Research Network (NRN).
Methods
Pilot one: a randomized clinical trial of inhaled PGE1 in neonatal hypoxemic respiratory failure
Following the availability of INO for the treatment of NHRF, several interventional clinical trials for NHRF were halted prematurely for lack of enrollment [22–25]. Therefore, we sought to quantify the number of potentially eligible patients and perform a pilot trial before embarking upon a RCT of IPGE1. Late preterm and term infants with NHRF who had an oxygenation index (OI) (OI = mean airway pressure (MAP) × fractional inspired oxygen concentration (FiO2) × 100/arterial oxygen tension (PaO2)) ≥15 and <25 on two arterial blood gases (ABGs) and had not previously received INO were eligible for the study. Infants would be randomly assigned to receive two doses of IPGE1 (300 and 150 ng/kg/min) or placebo over a maximum duration of 72 hours. The primary outcome was the ability to enroll 50 infants in six to nine months at 10 sites, without excessive protocol violations or adverse effects. Secondary outcome variables included the definition of optimal dose and duration of treatment with the study aerosol, the need for INO or extra-corporeal membrane oxygenation (ECMO) mortality, and duration of mechanical ventilation, O2 administration, and INO administration.
The research study was approved by the institutional review boards (IRBs) of all participating sites (see Additional file 1). The first pilot RCT was halted after four months for failure to enroll any infants. The most common cause for inability to enroll infants was the rapidly changing condition of the infants, resulting in prior initiation of INO in transport or soon after admission. The next most frequent causes of inability to recruit infants were lack of an arterial line and hypothermia treatment for neonatal encephalopathy, both of which were exclusion criteria.
Pilot two: a randomized clinical trial of inhaled PGE1 in neonates with sub-optimal response to inhaled nitric oxide
After evaluating the causes for lack of enrollment in the first pilot, we determined that co-administration of IPGE1 and INO could potentially have a substantial positive impact on study feasibility, while allowing us to test for synergy between INO and IPGE1. In addition, we elected to include infants receiving therapeutic hypothermia, as this was now standard therapy in participating centers.
In order to design and meaningfully interpret the data from the second pilot RCT in a manner that would be most helpful for designing the main trial, we created a definition of the primary outcome and conducted sample size calculations for the main trial. Using the composite outcome of need for ECMO or death as the most concrete clinically meaningful outcome for the future main trial, a sample size of 149 patients per group would be required to detect a 16% absolute risk reduction in the composite outcome of ECMO or death from 50% in the control arm to 34% in the study arm, with α of 0.05, a power of 80%, and a two-tailed test. To get an estimate of the number of infants in the NRN sites that might be eligible for the main trial, we queried all NRN sites. Between 2009 and 2010, 775 infants received INO for NHRF in 17 sites in the NRN; 30 to 35% met the composite outcome of death or ECMO and 10 to 13% died without receiving ECMO. Based on these numbers, it would be feasible to perform the main trial of IPGE1 in NHRF over three years or less.
Study eligibility was modified in the second pilot to include infants who had a suboptimal response to INO defined as an OI of ≥15 on any two arterial blood gases, obtained 15 minutes to 12 hours apart, and during the first 72 hours of INO use. The research study was approved by the IRBs of all participating sites and informed parental consent was obtained for all enrolled infants once eligibility was confirmed and before institution of study intervention.
Subjects
Late preterm (34 0/7 to 36 6/7 weeks’ gestation) and term infants at less than or equal to seven days postnatal age, undergoing conventional ventilation (CNV) or high-frequency oscillatory ventilation (HFOV) for NHRF (including perinatal aspiration syndrome, suspected or proven pneumonia or sepsis, respiratory distress syndrome, idiopathic PPHN, or suspected pulmonary hypoplasia), and with a suboptimal response to INO were eligible. An indwelling arterial line was a requisite for study participation, as was informed parental consent. Exclusion criteria included the decision not to provide full treatment, a known structural congenital heart disease except patent ductus arteriosus and atrial or ventricular level shunts, congenital diaphragmatic hernia, and thrombocytopenia (platelet count <80,000/μl) unresponsive to platelet transfusion.
Study procedures
There were three arms to the study: low-dose IPGE1 (150 ng/kg/min), high-dose IPGE1 (300 ng/kg/min), and placebo (aerosolized saline). The two doses of IPGE1 were selected on the basis of the results of the pre-clinical toxicity study and the phase I/II open label study of IPGE1 in NHRF [12]. Eligible infants were centrally randomized by telephone using permuted block randomization. The IPGE1 was double-masked. Only the participating center’s pharmacist was aware of the infant’s assigned group.
PGE1 solution for aerosolization was prepared from synthetic PGE1 (Prostin VR (Alprostadil) 500 μg/ml in 1 ml dehydrated ethanol, Pfizer, New York, NY, USA) by dilution in sterile 0.9% saline (Sodium Chloride 0.9% Injection USP, Hospira Inc., Lake Forest, IL, USA). Fresh solutions were prepared every 24 hours. Aerosol administration began with a PGE1 dosage of either 300 or 150 ng/kg/min diluted in 4 ml preservative-free sterile normal saline/hr in study patients, and 4 ml sterile normal saline/hr in control patients. The study medication was delivered using a syringe pump into the nebulizer chamber of the MiniHeart low flow jet nebulizer (WestMed Inc., Tucson, Arizona, United States) (Figure 1). The nebulizer was placed approximately 50 cm from the endotracheal tube for CNV, and approximately 35 cm from the endotracheal tube in HFOV, based on bench studies to determine the emitted dose [26]. The nebulizer chamber was primed with 2 ml of study medication at study aerosol initiation. For weaning purposes, normal saline was administered using a second syringe pump with a Y-connection into the nebulizer chamber with increasing dose as the study drug aerosol dose was decreased, such that the total volume delivered was always 4 ml/hour. During aerosol administration, FiO2 delivered in the gas flow through the nebulizer was matched to that delivered in the ventilator circuit.

Diagram of nebulizer setup in neonatal ventilator circuit. Figure legend: ETT, endotracheal tube; INO, inhaled nitric oxide; CMV, conventional mechanical ventilation; HFV, high frequency ventilation; IPGE1, inhaled PGE1 (Pfizer, New York, NY, USA).
Conventional management including ventilation, hemodynamic support, and sedation were optimized prior to randomization as per usual care at site. Treatment with INO was recommended if an OI ≥25 was documented on two consecutive ABGs at least 15 minutes apart; however, depending on institutional practice, treatment with INO could be instituted at an OI <25 [4, 25]. It was recommended that an echocardiogram and cranial sonogram be obtained prior to, or soon after, the initiation of INO to rule out structural heart lesions, establish the presence of pulmonary hypertension, and evaluate for intracranial hemorrhage.
Response to study medication
Arterial blood gas analysis was performed 60 ± 15 minutes after study aerosol initiation. Response was defined on the basis of an increase in PaO2 (mmHg) above baseline after 60 ± 15 minutes exposure to the study aerosol as full (≥20), partial (10 to 19) or no (<10) increase in PaO2. All infants were continued on the assigned study aerosol regardless of initial response until they weaned off, met treatment failure criteria, or completed 72 hours of study aerosol administration. Arterial blood gases were drawn at four ± two hours and every 12 ± two hours thereafter. Additional blood gases were obtained as clinically indicated. After the initial 60 ± 15 minutes observation period, therapeutic decisions were left to the clinical team.
Primary outcome
The primary aim of the study was the ability to recruit an adequate number of patients (n = 50) in a nine-month-period without excessive (more than 20%) protocol violations.
Secondary outcomes
Secondary outcomes included improvement in PaO2 and OI at 60 ± 15 minutes and four ± two hours after study aerosol initiation; need for ECMO; death; combined outcome of need for ECMO or death in the first 120 days of life; need for supplemental O2 at 28 days of life; duration of INO, mechanical ventilation, supplemental oxygen, and hospitalization; and occurrence of intracerebral hemorrhage, intraventricular hemorrhage, periventricular hemorrhagic infarction, cystic leukomalacia, areas of low attenuation in the white matter (edema or ischemia), atrophy, and ventriculomegaly.
Treatment failure criteria
Treatment failure was defined as an acute deterioration on initiation of, or during administration of, the study aerosol with an absolute fall in pulse oximeter O2 saturation (SPO2) by more than 10% for over 10 minutes, or an absolute SPO2 of less than 85%. Mechanical causes, including pneumothorax or plugged or malpositioned endotracheal tube (ETT) needed to be excluded. It was recommended that ECMO be considered in infants with an OI >40, or an alveolar arterial PaO2 gradient >620 mmHg for eight hours, a PaO2 < 40 to 50 mmHg for two to four hours, or if institutional guidelines were met.
Weaning of study aerosol
A weaning algorithm was proposed for the study aerosol to avoid a potential drop in PaO2. For any given weaning time point, the dose was halved and weaning was attempted only if the PaO2 was >60 torr. The first weaning attempt was at 12 ± two hours. Further weaning attempts were mandated at 12 ± two hour intervals. Total duration of study aerosol administration could not exceed 72 hours. Weaning to the third dose had to be accomplished by 48 hours on study aerosol.
Weaning of ventilator during study
Once the study aerosol was initiated, attempts to wean INO dose and/or ventilator settings were protocol-driven and every 12 hours or more frequently as deemed appropriate by the clinical team.
Monitoring of study aerosol administration
Enrolled infants were monitored continuously throughout study aerosol administration for possible adverse effects of PGE1, including hyperthermia (>38°C), bradycardia (heart rate <70/min, arrhythmia, hypotension, seizures, bleeding tendency, pulmonary hemorrhage, diarrhea, and seizures. Study aerosol was weaned if hypotension or arrhythmia persisted despite maximal therapy. In addition, all newborns enrolled in the study were clinically evaluated for signs and symptoms of patent ductus arteriosus (PDA at well-defined time points (three ± one and seven ± one days after discontinuation of the study medication and within one week before discharge).
Protocol violations
Protocol violations included: wrong study aerosol administered; wean to dose three not completed by 48 +/- four hours; study aerosol given for more than 72 +/- four hours; and unmasking of the study aerosol.
Sample size
The pilot RCTs were primarily designed to evaluate the feasibility and safety of prolonged IPGE1 administration and determination of optimal dose in 50 patients recruited at high-volume sites within the NRN. Comparison of the combined IPGE1 groups (n = 100) with the control group (n = 50) would have a power of 76.9% to detect a difference of 16% absolute risk reduction in the composite outcome of ECMO or death, from 50% in the control arm to 34% in the study arm, with α of 0.25 and a two-tailed test. The higher type I error rate could erroneously lead to a conclusion of efficacy of IPGE1 but would not increase the risk of missing an efficacious treatment [27–29]. While statistically significant evidence of treatment efficacy was unlikely in these small pilot trials, we expected a relative risk of 67% (range: 50 to 80%) for the treatment compared to the control arm for the combined outcome of need for ECMO or death in 50 patients recruited over a time period of nine months to be able to justify embarking upon the full-scale RCT.
Data Safety Monitoring Committee
The NICHD NRN Data Safety Monitoring Committee (DSMC) monitored the progress of the trial for feasibility and safety. Feasibility was defined a priori as the ability to recruit at least four subjects per month over any rolling three-month-period once 50% of the participating sites had IRB approval; and six subjects per month over any rolling three-month-period once 75% of the participating sites had IRB approval. Given those accrual targets, if fewer than 20 subjects had been enrolled in the six-month-period after at least 75% of the sites had achieved IRB approval, the DSMC could recommend that the trial be stopped for futility.
Results
The second pilot was undertaken because no infants could be enrolled in the first pilot; factors that hindered feasibility of the first pilot guided the study design of the second pilot. Detailed results for the second pilot RCT are discussed in the following. A total of 10 sites participated in the second pilot RCT. There was a lag from 33 to up to 90 days between IRB approval and site readiness to enroll patients due to the complexity of education regarding drug preparation and administration (Figure 2). Enrollment was halted for lack of feasibility after approximately six months; at that time, only seven patients had been enrolled.During the study period, 46 infants were screened at eight sites; 14 met eligibility criteria and seven were randomized (50%) (Figure 3). The reasons for non-enrollment were: parent unavailable or refused consent (three), met ECMO criteria (three), or cardiorespiratory arrest (one).

Timeline of Institutional Review Board (IRB) approval and readiness to enroll patients at various sites.

Flow diagram of infants.
Of the seven infants randomized, three received aerosolized saline, and two each received high-dose and low-dose IPGE1. Aggregate data are presented for the control group and the two IPGE1 groups combined because of the small numbers of subjects in each group (Tables 1 and 2, Figures 4 and 5). The small numbers precluded formal statistical testing. Detailed individual patient data are presented in Table 3.

Boxplots of timeline of events prior to initiation of study aerosol in control and combined IPGE 1 groups (low-dose and high-dose IPGE 1 ). INO, inhaled nitric oxide; OI, oxygenation index. (a) Age at INO start (hours; (b) Age at randomization (hours); (c) Interval from 1st OI > 15 and start of study aerosol (hours); (d) Interval from randomization to start of study aerosol (hours).

Profile of temperature, heart rate, and mean blood pressure during study aerosol administration in control and combined IPGE 1 groups (low-dose and high-dose IPGE 1 ). Solid line with solid squares represents control subjects. Dotted line with open circles represents subjects receiving IPGE1 (combined data for low-dose and high-dose IPGE1). (a) Temperature; (b) Heart rate; (c) Mean blood pressure.
Delivery room characteristics of the enrolled infants in the control group and the two IPGE1 groups combined are presented in Table 1. Of note, all four infants assigned to low- or high-dose IPGE1 were intubated in the delivery room; three of these infants also received chest compressions. None of the infants in the control group were intubated or received chest compressions in the delivery room.
The underlying cause of NHRF was meconium aspiration syndrome in five infants and idiopathic in two infants; both infants with idiopathic NHRF were randomized to the control group (Table 2). Postnatal age at the start of INO ranged from two to 28 hours (Figure 4a). Duration of treatment with INO prior to start of study aerosol ranged from 3.5 to 23.5 hours, with the mean and median values being higher for infants in the combined IPGE1 groups. Infants were randomized to the study at an age range of 7.1 to 58.3 hours; all infants in the control group were >24 hours of age at time of randomization, whereas three of the four infants randomized to low- or high-dose IPGE1 were <16 hours of age (Figure 4b).
All infants had a trial of HFOV; five of the seven infants were receiving HFOV and two were receiving conventional mechanical ventilation at the time of randomization. All seven infants received fluid boluses prior to randomization, six infants received dopamine with or without dobutamine, and three infants received milrinone before randomization (Tables 2 and 3). Two infants in the control group received milrinone both before and during study aerosol administration; in one infant in the IPGE1 group, milrinone was discontinued 18 hours after study aerosol initiation. Only one infant received sildenafil before randomization; this infant belonged to the control group and received concurrent milrinone. All infants received sedation and/or analgesia, and five of the seven received neuromuscular blockade. Four of the seven infants received steroids before randomization.
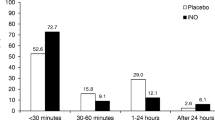
All infants met the criterion of suboptimal response to INO. Data on the first OI ≥15 were available in six of the seven enrolled infants; four of the six infants had OI ≥29 (highest OI: 99). OI on the first qualifying arterial blood gas ranged from 16 to 69, with five of the seven infants having OI ≥25. OI on the second qualifying arterial blood gas ranged from 20 to 44, with six of the seven infants having OI ≥25. All seven infants had at least one documented OI >25, with six infants having OI >35 prior to starting the study aerosol. The interval between the first and second qualifying arterial blood gas ranged from one to nine hours. The time from randomization to study aerosol initiation ranged from 3.0 to 20.0 hours, with the interval being less than six in all infants in the control group and more than six hours in all infants in the combined IPGE1 groups (Figure 4c). The time from randomization to study aerosol initiation ranged from 1.1 to 2.9 hours, with all infants in the control group receiving study aerosol within 1.5 hours of randomization and all infants in the IPGE1 group receiving it after 1.5 hours (Figure 4d).
An echocardiogram was performed in all seven enrolled infants before study aerosol initiation. PDA was visualized in five infants, with direction of shunting being right to left or bidirectional in three, left to right in one, and no shunt documented in one.
Only one infant met the criteria for a complete response based on the one-hour ABG and this infant belonged to the control group. On the ABG obtained four hours after the start of the study aerosol, there was improvement in oxygenation in all three groups compared to the ABG obtained after 60 minutes (mean change in PaO2 being 30, 85, and 43 mmHg in control, low-dose, and high-dose IPGE1 groups, respectively). The duration of treatment with study aerosol ranged from 10 to 72 hours (Table 3). All seven enrolled infants survived. Six infants received ECMO; five of these received ECMO within 24 hours of discontinuation of the study aerosol. During study aerosol administration, acute deterioration was noted in three infants, two in the low-dose and one in the high-dose IPGE1 group, at 10, 36, and 34 hours of age, respectively. The first wean of the study aerosol had been attempted in two of these infants and was successful in one infant.
All enrolled infants were closely monitored for predefined adverse events. The lowest mean blood pressure recorded during study aerosol administration was 37, 43, and 45 mmHg in the control, low-dose, and high-dose IPGE1 groups, respectively (Table 3, Figure 5). The highest heart rate recorded during study aerosol administration was 200, 131, and 142 bpm in the control, low-dose, and high-dose IPGE1 groups, respectively. The highest temperature recorded during study aerosol administration was 38.2, 33.6, and 33.7°C in the control, low-dose, and high-dose IPGE1 groups, respectively. Two adverse events were reported during the study and nether was attributed to the study aerosol. One infant in the control group developed transient, self-resolved mild fever within five minutes of starting the study aerosol, which was completely resolved within six hours after onset. One infant in the control group developed moderate hypotension 66 hours after initiation of the study aerosol. It was completely resolved within an hour of its onset. All enrolled infants were carefully clinically monitored for signs of PDA after study aerosol discontinuation. None of the infants had clinical signs of failure secondary to PDA prompting an echocardiogram. One infant (in the high-dose IPGE1 group) developed seizures later in the hospital course, but the infant was not receiving study aerosol at that time. No protocol violations were reported during the study. There was one minor protocol deviation (ABG obtained >60 ± 15 minutes after aerosol initiation) and one pharmacy protocol violation (infant incorrectly received 150 ng/kg/min instead of the assigned 300 ng/kg/min dose of PGE1; this was recognized on the 2nd day of study aerosol administration).
Discussion
We assessed the feasibility of two pilot RCTs to evaluate the use of two doses of IPGE1 in critically ill neonates with NHRF and were unable to recruit sufficient eligible subjects in the time period allowed. No infants were recruited in the first pilot. The second pilot was designed taking into account factors that hindered the feasibility of the first pilot. We demonstrated that continuous administration of aerosols was feasible in the second pilot. Of the 46 infants screened in this second pilot, only 14 met the eligibility criteria and seven were enrolled. Though the goal was to identify patients with OI ≥15, in fact all infants had much higher OIs at enrollment. All infants survived, and six of the seven received ECMO; five of these received ECMO within 24 hours of discontinuation of the study aerosol. There were no serious adverse events. Acute deterioration was documented in three infants in the IPGE1 groups 10 to 36 hours after start of study aerosol. There were systematic differences between the infants in the three groups; most notably, infants in both of the IPGE1 groups had lower Apgar scores, more need for delivery room resuscitation, an earlier age at presentation, and significantly lower temperatures throughout the study aerosol administration. The small sample size and clinical differences between the three groups precludes any inference regarding the efficacy of IPGE1 in PPHN.
The results of the two pilot RCTs reported in this study highlight the substantial investments of capital, human resources, technological expertise, and time in evaluating emerging new therapies [30]. Planning for these studies required performance of preclinical studies mandated by the United States Food and Drug Administration (FDA; addressing concerns about aerosol drug delivery with various modes of neonatal ventilation and/or INO; reaching consensus regarding patient management protocols, eligibility criteria, definition of meaningful outcome parameters, and use of nebulizer device in the study; development of study documents, training of site personnel, and obtaining IRB approvals.
The feasibility of conducting a large RCT was recognized to be an important limitation as several intervention clinical trials for NHRF were halted prematurely for lack of enrollment [22–25]; thus pilot studies were undertaken. Even though a survey of NRN sites between 2009 and 2010 suggested adequate numbers of patients eligible for this study, the pilot studies identified that the available patient population was very small. Foremost among factors contributing to lack of feasibility of this study was the decreased number of patients. This was attributed to improved perinatal care practices leading to decreased rates of both post-maturity and respiratory morbidity, as well as improved results in NHRF with the use of INO, surfactant, and HFOV [31, 32]. It is likely that with improved lung recruitment strategies, early use of surfactant and optimal delivery of INO, the current failure rate of INO may be considerably lower than that quoted in the literature [33]. Secondly, the consent process was challenging because of the short time window available, the high degree of stress parents’ experienced, frequent administration of medications that affect maternal cognition, and the lack of availability of parents when infants were transferred to tertiary care centers soon after birth for the management of PPHN.
The third factor that made these trials challenging was the novel nature of the study intervention. These pilot RCTs were the first trials of aerosolized selective pulmonary vasodilators in critically ill neonates with NHRF. The study protocol was complex and difficult to master for investigative teams. At the time of inception of this study, the MiniHeart low flow jet nebulizers were the only FDA approved nebulizers available for use in neonates; preliminary data were obtained using the MiniHeart nebulizer [26, 12, 34–38] and consequently the protocol allowed the use of MiniHeart nebulizers only in this study. Since then, ultrasonic and vibrating mesh nebulizers have been developed and are increasingly being used clinically [39, 40]. As there are advantages and disadvantages associated with each aerosol device, studies are needed to evaluate these devices for the delivery of IPGE1.
Another factor that contributed to the lack of feasibility of these trials was the long time lapse between IRB approval and initiation of enrollment. As defined by feasibility criteria in the second pilot, 75% of centers were IRB approved; however, readiness for study enrollment did not coincide with IRB approval. This may be due to the need for hospital approvals of research studies, as well as the need for training of site nurses, respiratory therapists, and pharmacists in implementing this novel intervention.
These studies highlight the challenges of performing clinical trials in critically ill neonates with hypoxemic respiratory failure. To date, only three interventions for NHRF (ECMO, INO, and HFOV) have been shown in large RCTs to significantly improve outcomes [4, 41, 42]. Objective evidence for other interventions for the treatment of term NHRF is lacking. Although INO has revolutionized outcomes, 40% of babies still do not respond, and thus further therapeutic improvements are critical. Other drugs, including sildenafil, inhaled iloprost, endothelin antagonists, and antioxidants, are actively being explored for the treatment of NHRF, but no large RCTs in critically ill neonates have been reported. A failure to recruit patients in this trial has important implications for other clinical trials in this patient population. Inability to design definitive clinical trials to evaluate newer therapies in NHRF will propagate the practice of ‘off-label’ use of drugs that have been approved for use in adults first [43]. From developmental and metabolic standpoints, the disease processes in children and their responses to therapies are very different from those in adults [44]. The search for robust evidence to guide the safe therapy of children and neonates with pulmonary hypertensive vascular disease is crucial.
Important lessons can be learned from clinical trials in adult and pediatric pulmonary hypertension where similar challenges with recruitment are encountered. The prevalence of pulmonary hypertension in adults, children, and neonates is insufficient to support formal examination of all potential drug targets [45]. Recognizing the need for innovative approaches [43, 46, 47], a workshop was established to consider alternative clinical trial designs in pulmonary hypertension, in order to raise the likelihood of new treatments reaching patients. Acknowledging that classical sample size calculations often lead to unrealistic expectations of the number of subjects to be recruited, Offringa and van der Lee described statistical and epidemiological aspects of small sample approaches and emphasized the importance of maximizing information obtained from the few subjects enrolled [48]. Development of national and international databases with common entry criteria may be an alternative approach. Clinical studies in pulmonary hypertension might be enhanced by a consortium approach that utilizes the expertise of academic medicine, the treatment initiatives of the pharmaceutical industry, and study design from funding agencies interested in biological mechanisms [49]. To address this challenge, the Pulmonary Hypertension Academic Research Consortium was created as a forum to openly discuss strategies for clinical trials in pulmonary hypertension that would benefit all of the stakeholders. The goals of the consortium were to establish consensus clinical endpoint definitions for future clinical trials, advance the conduct of clinical research in the field, identify modern strategies for clinical trials, and provide guidance to the pharmaceutical industry to allow them to better identify and develop treatments with the most promise. Similar approaches need to be explored in NHRF.
Conclusions
In conclusion, there are important lessons to be learned from the two pilot RCTs described in this report that will be instrumental in designing future studies in this critically ill population. The second pilot demonstrates that it is possible to administer aerosols continuously in the Neonatal Intensive Care Unit for extended periods of time. When defining feasibility criteria, it needs to be recognized that readiness to commence study enrollment does not coincide with IRB approval. This lag needs to be adjusted for in future clinical trials. In this small sample size, there were no serious adverse events reported with the two doses of IPGE1; future studies should explore the efficacy of the higher dose and consider dose escalation to identify the optimal therapeutic dose. The second pilot also demonstrates that though the goal was to identify patients with OI ≥15, in fact all the babies had much higher OIs at enrollment as has been shown in other RCTs in NHRF [4]. This suggests that future trials randomize infants at an even earlier stage, preferably at the time of initiation of INO. Another approach would be to randomize infants to placebo and treatments soon after PPHN is diagnosed, and then add specific therapy, such as INO. Future studies should allow the use of nebulizer devices approved by the FDA for use in neonates with provision for a subgroup analysis by the nebulizer device during the evaluation of data. Ventilator strategy, use of supportive therapies (such as surfactant and inotropes), and underlying cause of PPHN should be taken into account while designing future studies. Both of the pilot trials reinforce the challenges of performing RCTs in this critically ill population of patients and the need for pragmatic study designs reflective of the real-world situations with a realistic timeline.
Abbreviations
- ABG:
-
Arterial blood gas
- CNV:
-
Conventional ventilation
- DSMC:
-
Data Safety Monitoring Committee
- FiO2:
-
fractional inspired oxygen concentration
- HFOV:
-
High frequency oscillatory ventilation
- INO:
-
Inhaled nitric oxide
- IPGE1 :
-
Inhaled Prostaglandin E1
- IRB:
-
Institutional review board
- MAP:
-
mean airway pressure)
- NHRF:
-
Neonates with hypoxemic respiratory failure
- NICHD:
-
National Institute of Child Health and Human Development
- NICU:
-
Neonatal intensive care unit
- NRN:
-
Neonatal Research Network
- OI:
-
oxygenation index
- PaO2:
-
arterial oxygen tension
- PGE1 :
-
Prostaglandins E1
- PGI2 :
-
Prostaglandins I2
- PPHN:
-
Persistent pulmonary hypertension of the newborn
- RCT:
-
Randomized control trial
- SPO2 :
-
Pulse oximeter O2 saturation.
References
Roberts JD, Fineman JR, Morin FC, Shaul PW, Rimar S, Schreiber MD, Polin RA, Zwass MS, Zayek MM, Gross I, Heymann MA, Zapol WM: Inhaled nitric oxide and persistent pulmonary hypertension of the newborn: the inhaled nitric oxide study group. N Engl J Med. 1997, 336: 605-610. 10.1056/NEJM199702273360902.
Clark RH, Kueser TJ, Walker MW, Southgate WM, Huckaby JL, Perez JA, Roy BJ, Keszler M, Kinsella JP: Low-dose nitric oxide therapy for persistent pulmonary hypertension of the newborn: clinical inhaled nitric oxide research group. N Engl J Med. 2000, 342: 469-474. 10.1056/NEJM200002173420704.
Truog WE, Castor CA, Sheffield MJ: Neonatal nitric oxide use: predictors of response and financial implications. J Perinatol. 2003, 23: 128-132. 10.1038/sj.jp.7210864.
NINOS: Inhaled nitric oxide in full-term and nearly full-term infants with hypoxic respiratory failure: the neonatal inhaled nitric oxide study group. N Engl J Med. 1997, 336: 597-604.
Krieg P, Wahlers T, Giess W, Rohde R, Hartrumpf M, Bund M, Haverich A: Inhaled nitric oxide and inhaled prostaglandin E1: effect on left ventricular contractility when used for treatment of experimental pulmonary hypertension. Eur J Cardiothorac Surg. 1998, 14: 494-502. 10.1016/S1010-7940(98)00210-3.
Walmrath D, Schermuly R, Pilch J, Grimminger F, Seeger W: Effects of inhaled versus intravenous vasodilators in experimental pulmonary hypertension. Eur Respir J. 1997, 10: 1084-1092. 10.1183/09031936.97.10051084.
Kumar VH, Swartz DD, Rashid N, Lakshminrusimha S, Ma C, Ryan RM, Morin FC: Prostacyclin and milrinone by aerosolization improve pulmonary hemodynamics in newborn lambs with experimental pulmonary hypertension. J Appl Physiol. 2010, 109: 677-684. 10.1152/japplphysiol.01082.2009.
Putensen C, Hormann C, Kleinsasser A, Putensen-Himmer G: Cardiopulmonary effects of aerosolized prostaglandin E1 and nitric oxide inhalation in patients with acute respiratory distress syndrome. Am J Respir Crit Care Med. 1998, 157: 1743-1747. 10.1164/ajrccm.157.6.9609017.
Olschewski H, Simonneau G, Galie N, Higenbottam T, Naeije R, Rubin LJ, Nikkho S, Speich R, Hoeper MM, Behr J, Winkler J, Sitbon O, Popov W, Ghofrani HA, Manes A, Kiely DG, Ewert R, Meyer A, Corris PA, Delcroix M, Gomez-Sanchez M, Siedentop H, Seeger W: Inhaled iloprost for severe pulmonary hypertension. N Engl J Med. 2002, 347: 322-329. 10.1056/NEJMoa020204.
Meyer J, Theilmeier G, Van Aken H, Bone HG, Busse H, Waurick R, Hinder F, Booke M: Inhaled prostaglandin E1 for treatment of acute lung injury in severe multiple organ failure. Anesth Analg. 1998, 86: 753-758.
Ivy DD, Doran AK, Smith KJ, Mallory GB, Beghetti M, Barst RJ, Brady D, Law Y, Parker D, Claussen L, Abman SH: Short- and long-term effects of inhaled iloprost therapy in children with pulmonary arterial hypertension. J Am Coll Cardiol. 2008, 51: 161-169. 10.1016/j.jacc.2007.09.031.
Sood BG, Delaney-Black V, Aranda JV, Shankaran S: Aerosolized PGE1: a selective pulmonary vasodilator in neonatal hypoxemic respiratory failure results of a Phase I/II open label clinical trial. Pediatr Res. 2004, 56: 579-585. 10.1203/01.PDR.0000139927.86617.B6.
Hsiao R, Omar SA: Inhaled aerosolized Prostacyclin (IAP) fpr premature infants with PPHN. Pediatr Res. 2004, 55: 544A-
Kelly LK, Porta NF, Goodman DM, Carroll CL, Steinhorn RH: Inhaled prostacyclin for term infants with persistent pulmonary hypertension refractory to inhaled nitric oxide. J Pediatr. 2002, 141: 830-832. 10.1067/mpd.2002.129849.
De Luca D, Cogo P, Zecca E, Piastra M, Pietrini D, Tridente A, Conti G, Carnielli VP: Intrapulmonary drug administration in neonatal and paediatric critical care: a comprehensive review. Eur Respir J. 2011, 37: 678-689. 10.1183/09031936.00024910.
van Heerden PV, Caterina P, Filion P, Spagnolo DV, Gibbs NM: Pulmonary toxicity of inhaled aerosolized prostacyclin therapy–an observational study. Anaesth Intensive Care. 2000, 28: 161-166.
Wasserman MA, Griffin RL, Marsalisi FB: Inhibition of bronchoconstriction by aerosols of prostaglandins E1 and E2. J Pharmacol Exp Ther. 1980, 214: 68-73.
Borok Z, Gillissen A, Buhl R, Hoyt RF, Hubbard RC, Ozaki T, Rennard SI, Crystal RG: Augmentation of functional prostaglandin E levels on the respiratory epithelial surface by aerosol administration of prostaglandin E. Am Rev Respir Dis. 1991, 144: 1080-1084. 10.1164/ajrccm/144.5.1080.
Kato S, Sugimura H, Kishiro I, Machida M, Suzuki H, Kaneko N: Suppressive effect of pulmonary hypertension and leukocyte activation by inhaled prostaglandin E1 in rats with monocrotaline-induced pulmonary hypertension. Exp Lung Res. 2002, 28: 265-273. 10.1080/01902140252964357.
Cox JW, Andreadis NA, Bone RC, Maunder RJ, Pullen RH, Ursprung JJ, Vassar MJ: Pulmonary extraction and pharmacokinetics of prostaglandin E1 during continuous intravenous infusion in patients with adult respiratory distress syndrome. Am Rev Respir Dis. 1988, 137: 5-12. 10.1164/ajrccm/137.1.5.
Lowson SM: Inhaled alternatives to nitric oxide. Anesthesiology. 2002, 96: 1504-1513. 10.1097/00000542-200206000-00034.
Davidson D, Barefield ES, Kattwinkel J, Dudell G, Damask M, Straube R, Rhines J, Chang CT: Inhaled nitric oxide for the early treatment of persistent pulmonary hypertension of the term newborn: a randomized, double-masked, placebo-controlled, dose–response, multicenter study: the I-NO/PPHN Study Group. Pediatrics. 1998, 101: 325-334. 10.1542/peds.101.3.325.
Sadiq HF, Mantych G, Benawra RS, Devaskar UP, Hocker JR: Inhaled nitric oxide in the treatment of moderate persistent pulmonary hypertension of the newborn: a randomized controlled, multicenter trial. J Perinatol. 2003, 23: 98-103. 10.1038/sj.jp.7210878.
Mercier JC, Thébaud B, Onody P, Storme L, van Overmeire B, Bréart G: Early compared with delayed inhaled nitric oxide in moderately hypoxaemic neonates with respiratory failure: a randomised controlled trial: the Franco-Belgium Collaborative NO Trial Group. Lancet. 1999, 354: 1066-1071.
Konduri GG, Solimano A, Sokol GM, Singer J, Ehrenkranz RA, Singhal N, Wright LL, Van Meurs K, Stork E, Kirpalani H, Peliowski A: A randomized trial of early versus standard inhaled nitric oxide therapy in term and near-term newborn infants with hypoxic respiratory failure. Pediatrics. 2004, 113: 559-564. 10.1542/peds.113.3.559.
Sood BG, Peterson J, Malian M, Galli R, Geisor-Walter M, McKinnon J, Sharp J, Maddipati KR: Jet nebulization of prostaglandin E1 during neonatal mechanical ventilation: stability, emitted dose and aerosol particle size. Pharmacol Res. 2007, 56: 531-541. 10.1016/j.phrs.2007.09.017.
Piantadosi S: Clinical Trials: A Methodologic Perspective. Edited by: Balding DJ, Cressie NAC, Fisher NI, Johnstone IM, Kadane JB, Molenberghs G, Ryan LM, Scott DW, Smith AFM, Teugels JL. 2005, New Jersey: John Wiley & Sons, 251-308. Sample size and power, 2, Wiley Series in Probability and Statistics.
Stallard N, Whitehead J, Todd S, Whitehead A: Stopping rules for phase II studies. Br J Clin Pharmacol. 2001, 51: 523-529. 10.1046/j.0306-5251.2001.01381.x.
Schoenfeld D: Statistical considerations for pilot studies. Int J Radiat Oncol Biol Phys. 1980, 6: 371-374. 10.1016/0360-3016(80)90153-4.
Dickson M, Gagnon JP: The cost of new drug discovery and development. Discov Med. 2004, 4: 172-179.
Yoder BA, Gordon MC, Barth WH: Late-preterm birth: does the changing obstetric paradigm alter the epidemiology of respiratory complications?. Obstet Gynecol. 2008, 111: 814-822. 10.1097/AOG.0b013e31816499f4.
Qureshi FG, Jackson HT, Brown J, Petrosyan M, Rycus PT, Nadler EP, Oyetunji TA: The changing population of the United States and use of extracorporeal membrane oxygenation. J Surg Res. 2013, 184: 572-576. 10.1016/j.jss.2013.04.027.
Konduri GG, Sokol GM, Van Meurs KP, Singer J, Ambalavanan N, Lee T, Solimano A: Impact of early surfactant and inhaled nitric oxide therapies on outcomes in term/late preterm neonates with moderate hypoxic respiratory failure. J Perinatol. 2013, 33: 944-949. 10.1038/jp.2013.83.
Sood BG, Glibetic M, Aranda JV, Delaney-Black V, Chen X, Shankaran S: Systemic levels following PGE1 inhalation in neonatal hypoxemic respiratory failure. Acta Paediatr. 2006, 95: 1093-1098. 10.1080/08035250600580511.
Sood BG, Chen X, Dawe EJ, Malian M, Maddipati KR: Tissue distribution, metabolism and excretion of PGE1 following prolonged high-dose inhalation in neonatal pigs. Int J Pharmacol. 2010, 6: 224-230.
Sood BG, Shen Y, Latif Z, Chen X, Sharp J, Neelavalli J, Joshi A, Slovis TL, Haacke EM: Aerosol delivery in ventilated newborn pigs: an MRI evaluation. Pediatr Res. 2008, 64: 159-164. 10.1203/PDR.0b013e3181761841.
Sood BG, Shen Y, Latif Z, Galli R, Dawe EJ, Haacke EM: Effective aerosol delivery during high-frequency ventilation in neonatal pigs. Respirology. 2010, 15: 551-555. 10.1111/j.1440-1843.2010.01714.x.
Sood BG, Dawe EJ, Rao Maddipati K, Malian M, Chen X, Galli R, Rabah R: Toxicity of prolonged high dose inhaled PGE1 in ventilated neonatal pigs. Pulmonary Pharmacol Ther. 2008, 21: 565-572. 10.1016/j.pupt.2008.01.011.
Dolovich MB, Dhand R: Aerosol drug delivery: developments in device design and clinical use. Lancet. 2011, 377: 1032-1045. 10.1016/S0140-6736(10)60926-9.
Ari A, Fink JB, Dhand R: Inhalation therapy in patients receiving mechanical ventilation: an update. J Aerosol Med Pulm Drug Deliv. 2012, 25: 319-332. 10.1089/jamp.2011.0936.
Field D, Davis C, Elbourne D, Grant A, Johnson A, Macrae D: UK collaborative randomised trial of neonatal extracorporeal membrane oxygenation: UK Collaborative ECMO Trail Group. Lancet. 1996, 348: 75-82.
Kinsella JP, Truog WE, Walsh WF, Goldberg RN, Bancalari E, Mayock DE, Redding GJ, De Lemos RA, Sardesai S, McCurnin DC, Moreland SG, Cutter GR, Abman SH: Randomized, multicenter trial of inhaled nitric oxide and high-frequency oscillatory ventilation in severe, persistent pulmonary hypertension of the newborn. J Pediatr. 1997, 131: 55-62. 10.1016/S0022-3476(97)70124-0.
Adatia I, Haworth SG, Wegner M, Barst RJ, Ivy D, Stenmark KR, Karkowsky A, Rosenzweig E, Aguilar C: Clinical trials in neonates and children: report of the pulmonary hypertension academic research consortium pediatric advisory committee. Pulm Circ. 2013, 3: 252-266. 10.4103/2045-8932.109931.
Fike CD, Aschner JL: Spread the word, children are still not “small adults”. Pulm Circ. 2013, 3: 3-4. 10.4103/2045-8932.109909.
McLaughlin VV, Badesch DB, Delcroix M, Fleming TR, Gaine SP, Galie N, Gibbs JS, Kim NH, Oudiz RJ, Peacock A, Provencher S, Sitbon O, Tapson VF, Seeger W: End points and clinical trial design in pulmonary arterial hypertension. J Am Coll Cardiol. 2009, 54: S97-S107. 10.1016/j.jacc.2009.04.007.
Grieve AP, Chow SC, Curram J, Dawe S, Harnisch LO, Henig NR, Hung HM, Ivy DD, Kawut SM, Rahbar MH, Xiao S, Wilkins MR: Advancing clinical trial design in pulmonary hypertension. Pulm Circ. 2013, 3: 217-225. 10.4103/2045-8932.109933.
Wilkins MR: Developing treatments for pulmonary arterial hypertension. Pulm Circ. 2013, 3: 156-159. 10.4103/2045-8932.109961.
Offringa M, van der Lee H: Small sample approach, and statistical and epidemiological aspects. Handb Exp Pharmacol. 2011, 205: 181-202. 10.1007/978-3-642-20195-0_9.
Newman JH, Elliott GC, Haworth GS, Zampaglione E, Brar S, Gibbs SJ, Sandoval J: Clinical trials in pulmonary hypertension: time for a consortium. Pulm Circ. 2013, 3: 245-251. 10.4103/2045-8932.109922.
Acknowledgements
We thank the parents and families, neonatologists, nurses, respiratory therapists and pharmacists that enabled this study.
Sources of support
The National Institutes of Health, the Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD), the National Center for Research Resources, and the National Center for Advancing Translational Sciences provided grant support for the Neonatal Research Network’s IPGE1 Studies through cooperative agreements. While NICHD staff did have input into the study design, conduct, analysis, and manuscript drafting, the comments and views of the authors do not necessarily represent the views of the NICHD.
Data collected at participating sites of the NICHD Neonatal Research Network (NRN) were transmitted to RTI International, the data coordinating center (DCC) for the network, which stored, managed, and analyzed the data for this study. On behalf of the NRN, Drs Abhik Das (DCC Principal Investigator) and Shampa Saha (DCC Statistician) had full access to all of the data in the study, and with the NRN Center Principal Investigators, take responsibility for the integrity of the data and accuracy of the data analysis. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
We are indebted to our medical and nursing colleagues and the infants and their parents who agreed to take part in this study. The following investigators, in addition to those listed as authors, participated in this study:
NRN Steering Committee Chairs: Division of Neonatology, College of Physicians and Surgeons, Columbia University: Richard A Polin, MD (2011 to present).
Alpert Medical School of Brown University and Women and Infants Hospital of Rhode Island (U10 HD27904): Abbot R. Laptook, MD; Michael R Muller, PharmD; Angelita M Hensman, RNC-NIC BSN.
Duke University School of Medicine, University Hospital, University of North Carolina, Alamance Regional Medical Center, and Durham Regional Hospital (U10 HD40492, UL1 RR24128): Ronald N Goldberg, MD; Joanne Finkle, RN JD; Kimberley A Fisher, PhD FNP-BC IBCLC.
Eunice Kennedy Shriver National Institute of Child Health and Human Development: Stephanie Wilson Archer, MA.
Nationwide Children’s Hospital and the Ohio State University Medical Center (U10 HD68278): Gail E Besner, MD; Patricia Luzader, RN; Louis G Chicoine, MD.
RTI International (U10 HD36790): Jenna Gabrio BS CCRP; Jeanette O’Donnell Auman BS; Kristin M Zaterka-Baxter RN BSN CCRP.
Stanford University and Lucile Packard Children’s Hospital (U10 HD27880, M01 RR70, UL1 TR93): David K Stevenson, MD; M Bethany Ball, BS CCRC; Steven Chinn, PharmD; Melinda S Proud, RCP.
University of Alabama at Birmingham Health System and Children’s Hospital of Alabama (U10 HD34216, M01 RR32): Waldemar A Carlo, MD; Monica V Collins, RN BSN MaEd; Shirley S Cosby, RN BSN; Zahra Davis, PharmD; Rebecca J Quinn, PharmD; Brenda Reed Denson, PharmD.
University of California - Los Angeles, Mattel Children’s Hospital (U10 HD68270): Uday Devaskar, MD; Teresa Chanlaw, MPH; Rachel Geller, RN BSN.
University of Iowa Children’s Hospital and Mercy Medical Center (U10 HD53109, M01 RR59, UL1 TR442): Edward F Bell, MD; John A Widness, MD; Karen J Johnson, RN BSN; Donia B Campbell, RNC-NIC; Tarah T Colaizy, MD MPH; John M Dagle, MD PhD; Dan L Ellsbury, MD; Kristine M Johnson, BS Pharm RPh; Lynn Messina, RPh; Joanna L Nohr, PharmD RPh.
University of New Mexico Health Sciences Center (U10 HD53089, UL1 TR41): Kristi L Watterberg, MD; Conra Backstrom Lacy, RN; Nancy A Morgan, RPh MBA.
University of Rochester Medical Center, Golisano Children’s Hospital, the University of Buffalo Women’s, and Children’s Hospital of Buffalo (U10 HD68263, UL1 TR42): Carl T D’Angio, MD; Ronnie Guillet, MD PhD; Patricia Chess, MD; Stephen A Bean, RPh; Carol A Cole, RPh; Ann Marie Heine, PharmD; Rosemary Jensen; Michael G Sacilowski, BS; Karen Wynn, NNP RN; Patrick Conway, MS; Holly IM Wadkins; Ashley Williams, MSEd.
University of Texas Southwestern Medical Center at Dallas, Parkland Health and Hospital System, and Children’s Medical Center Dallas (U10 HD40689, M01 RR633): Luc P Brion, MD; Lijun Chen, RN, PhD; Andre Finley, RRT-NPS; Tram DoNguyen Meeks, PharmD; Ruth Merryman, PharmD RPh; Glenn Metoyer, RRT; Azadeh Mozaffari, PharmD RPh; Diana M Vasil, RNC-NIC; Brian Walsh, RRT-NPS.
Wayne State University, University of Michigan, Hutzel Women’s Hospital, and Children’s Hospital of Michigan (5K23HD041423, U10 HD21385): Robert J Galli, RRT; Maria Batts, RRT; Mirjana Lulic-Botica RPh.
Author information
Authors and Affiliations
Consortia
Corresponding author
Additional information
Competing interests
Nebulizers for the study were provided by WestMed Inc. (Tucson, Arizona, United States). Beena G Sood has received research support from Covidien™ for an unrelated research study. Martin Keszler, MD is a member of an Advisory Board and Speakers’ Bureau for Ikaria. Krisa Van Meurs, MD was the site Principal Investigator for a clinical trial supported by Ikaria and has served as a consultant for Ikaria.
Authors’ contributions
Below we detail the contributions made by each author for this manuscript. This manuscript represents a pilot study evaluate the feasibility of a randomized placebo-controlled trial of aerosolized selective pulmonary vasodilator, inhaled prostaglandin E1 (IPGE1), in neonates with hypoxemic respiratory failure in the NICHD Neonatal Research Network (NRN). The investigators had regular conference calls during protocol development and implementation and correspondence for manuscripts reviews and input. The following authors have made significant contributions as determined by the Uniform Requirements for Manuscripts Submitted to Biomedical Journals: BGS, MD MS, is the Co-Principal Investigator (PI) at Wayne State University and the chair of the IPGE1 Protocol Subcommittee. She developed the study, trained all NRN Coordinators and PIs in how to administer the drug, and drafted the manuscript. As the site investigator and co-PI, she oversaw study recruitment and implementation at the site, screened eight infants and enrolled one for this study. BGS drafted the manuscript and received input from the authors below as part of manuscript revision. MK, MD FAAP, is the NRN Co-PI at Brown University and the vice-chair of the IPGE1 Protocol Subcommittee. He oversaw recruitment and study implementation at Brown University, which screened 15 infants and enrolled one for this study. MK contributed to critical revision of the manuscript and approved the final manuscript for submission. MG, MD, is the NRN Co-PI at the University of California, Los Angeles, and a member of the IPGE1 Protocol Subcommittee. She oversaw recruitment and study implementation at UCLA, which screened 11 infants and enrolled four for this study. MG contributed to critical revision of the manuscript and approved the final manuscript for submission. JMK, MD, is the site investigator at the University of Iowa and a member of the IPGE1 Protocol Subcommittee. He oversaw recruitment and study implementation at the University of Iowa, which screened two infants and enrolled one for this study. JMK contributed to critical revision of the manuscript and approved the final manuscript for submission. RO, MD, is the NRN Co-PI at the University of New Mexico and a member of the IPGE1 Protocol Subcommittee. She oversaw recruitment and study implementation at the University of New Mexico, which screened four infants for this study. RO contributed to critical revision of the manuscript and approved the final manuscript for submission. NA, MD, is the NRN Co-PI at the University of Alabama at Birmingham and a member of the IPGE1 Protocol Subcommittee. He oversaw recruitment and study implementation at the University of Alabama, which screened three infants for this study. NA contributed to critical revision of the manuscript and approved the final manuscript for submission. CMC, MD, is the NRN Co-PI at Duke University and a member of the IPGE1 Protocol Subcommittee. CMC contributed to critical revision of the manuscript and approved the final manuscript for submission. MM, RPh, is a pharmacist at Wayne State University and a member of the IPGE1 Protocol Subcommittee. She provided pharmacological input into the study design, implementation, and data analysis. MM contributed to critical revision of the manuscript and approved the final manuscript for submission. PJS, MD, is the NRN PI at the University of Texas Southwestern and a member of the IPGE1 Protocol Subcommittee. He oversaw recruitment and study implementation at the University of Texas Southwestern, which screened one infant for this study. PJS contributed to critical revision of the manuscript and approved the final manuscript for submission. SL, MD, is the NRN site investigator at the University of Buffalo and a member of the IPGE1 Protocol Subcommittee. He oversaw recruitment and study implementation at the University of Buffalo, which screened two infants for this study. SL contributed to critical revision of the manuscript and approved the final manuscript for submission. LDN, MD, is the NRN PI at Nationwide Children’s Hospital and a member of the IPGE1 Protocol Subcommittee. LDN contributed to critical revision of the manuscript and approved the final manuscript for submission. KPVM, MD, is the NRN PI at Stanford University and a member of the IPGE1 Protocol Subcommittee. KPVM contributed to critical revision of the manuscript and approved the final manuscript for submission. RB, RN BSN, is the NRN coordinator at Wayne State University and a member of the IPGE1 Protocol Subcommittee. She assisted in training the NRN coordinators and PIs in study drug administration, and consulted with coordinators during recruitment and implementation. She assisted in implementation at Wayne State University, which screened eight infants and enrolled one for this study. RB contributed to critical revision of the manuscript and approved the final manuscript for submission. SS, PhD, served as the primary statistician for the study, providing statistical input for protocol development and completing the statistical analyses for the paper. She developed the tables for the paper and provided critical revision to the manuscript and approved the final version of the manuscript. AD, PhD, is the principal investigator for the NRN Data Coordinating Center and a member of the IPGE1 Protocol Subcommittee. AD oversaw all aspects of the statistical analysis, provided critical revisions to the manuscript and approved the final version of the manuscript. DW, PhD, is the Co-PI for the NRN Data Coordinating Center and a member of the IPGE1 Protocol Subcommittee. DW assisted in overseeing the statistical analysis, provided critical revisions to the manuscript and approved the final version of the manuscript. RDH, MD, served as the Program Scientist for the NICHD NRN and a member of the IPGE1 Protocol Subcommittee. RDH helped developed the protocol, oversaw study implementation, and assisted with data edits from the sites. She also provided critical revision to the manuscript and approved the final version of the manuscript. SS, MD, is the NRN PI at Wayne State University and a member of the IPGE1 Protocol Subcommittee. She oversaw recruitment and study implementation at Wayne State University, which screened eight infants and enrolled one for this study. SS contributed to critical revision of the manuscript and approved the final manuscript for submission. All authors read and approved the final manuscript.
Electronic supplementary material
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Sood, B.G., Keszler, M., Garg, M. et al. Inhaled PGE1 in neonates with hypoxemic respiratory failure: two pilot feasibility randomized clinical trials. Trials 15, 486 (2014). https://doi.org/10.1186/1745-6215-15-486
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1745-6215-15-486