
Abstract
Background
Bacteria affect oral health, but few studies have systematically examined the role of bacterial communities in oral diseases. We examined this relationship in a large population-based Chinese cancer screening cohort.
Methods
Human Oral Microbe Identification Microarrays were used to test for the presence of 272 human oral bacterial species (97 genera) in upper digestive tract (UDT) samples collected from 659 participants. Oral health was assessed using US NHANES (National Health and Nutrition Examination Survey) protocols. We assessed both dental health (total teeth missing; tooth decay; and the decayed, missing, and filled teeth (DMFT) score) and periodontal health (bleeding on probing (BoP) extent score, loss of attachment extent score, and a periodontitis summary estimate).
Results
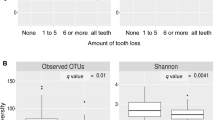
Microbial richness, estimated by number of genera per sample, was positively correlated with BoP score (P = 0.015), but negatively correlated with tooth decay and DMFT score (P = 0.008 and 0.022 respectively). Regarding β-diversity, as estimated by the UniFrac distance matrix for pairwise differences among samples, at least one of the first three principal components of the UniFrac distance matrix was correlated with the number of missing teeth, tooth decay, DMFT, BoP, or periodontitis. Of the examined genera, Parvimonas was positively associated with BoP and periodontitis. Veillonellacease [G-1] was associated with a high DMFT score, and Filifactor and Peptostreptococcus were associated with a low DMFT score.
Conclusions
Our results suggest distinct relationships between UDT microbiota and dental and periodontal health. Poor dental health was associated with a less microbial diversity, whereas poor periodontal health was associated with more diversity and the presence of potentially pathogenic species.
Similar content being viewed by others


Background
The human mouth harbors one of the most diverse microbial communities (microbiotas) in the human body with over 700 bacterial species identified [1, 2]. The oral microbiota is essential for the development of the mucosal immune system, the maintenance of a normal physical environment, and for the digestion of food [3]. Loss or imbalance of normal microbiota may lead to an overgrowth of opportunistic bacterial pathogens, leading to disease [4].
The oral microbiota is intimately related to oral health [5]. Dental caries occur when organic acids from bacterial fermentation of carbohydrates demineralize the teeth [6]. Periodontitis, the inflammatory response in the gingival tissue and surrounding connecting tissue, can result in the loss of collagen attachment between the tooth and the bone, leading to bone destruction and tooth loss [7]. The majority of oral microbiology studies have focused on individual candidate pathogens that could be cultured [8]. However, oral diseases may be related to the diversity and function of the entire oral microbial community, not just single species [5]. With the introduction of culture independent technology, researchers can now investigate the role of the entire oral microbial community [8–12]. However, the majority of such studies [8–12] have been small and have often focused on single oral diseases and people with good access to dental care.
Culture-independent approaches, like 16S rRNA gene-based methods can provide a comprehensive view of the human microbial communities, including currently un-culturable microorganisms. In this study, we used a 16S rRNA gene-based microarray method (the Human Oral Microbe Identification Microarray) to examine the relationship between the upper digestive tract (UDT) microbiota and oral health (including both dental and periodontal health) in 659 healthy Chinese adults from a cancer screening study in Linxian, China, a rural cohort with poor access to dental care and a high rate of esophageal cancer.
Methods
The Study Cohort
Study participants were enrolled from three villages in Linzhou (formally Linxian), Henan Province, People’s Republic of China. The description of the parent study (the Cytology Sampling Study 2, CSS2) was published previously [13]. Participants were apparently healthy volunteers, aged 40–65 years [13]. We successfully obtained UDT microbiota data in upper digestive tract samples from 659 out of the original 720 study participants. The missing of UDT microbiota data were at random due to failure in DNA extraction or microarray. This study was conducted under the auspices of the Institutional Review Boards of the Cancer Institute, Chinese Academy of Medical Sciences and the US National Cancer Institute, and all subjects provided written informed consent.
Sample collection, DNA extraction
Upper digestive tract (UDT) cell samples for DNA extraction were collected by one of two esophageal balloon cytology retrieval devices. One consisted of an inflatable rubber balloon covered with cotton mesh attached to a 0.2 cm diameter single lumen rubber tube (manufactured in China, and called the Chinese balloon type (CHB)). The second device was the Cytomesh Esophageal Cytology Device (WCH) (Wilson-Cook Medical, Inc, Winston-Salem, NC, USA). Allocation of the sampling devices was random. Each volunteer was given 2 ml of 2% lidocaine slurry by mouth, after which the balloon was inserted into the stomach, inflated, drawn through the esophagus, deflated at the upper esophageal sphincter, and withdrawan from the mouth. The head of the balloon was cut off and deposited in 40 ml of sterile saline in a 50 ml centrifuge tube and transferred to the lab on ice for processing. This centrifuge tube was then vortexed for 30 seconds, the balloon head was removed, and the remaining fluid was centrifuged at 1500 RPM for 5 minutes. After discarding the supernatant, the pellet was re-suspended in 1 ml saline, frozen in liquid nitrogen, and stored at -80 ° C until DNA extraction.
DNA was extracted by the Gentra Puregene Cell Kit (Qiagen, Valencia, CA). The quality and quantity of the DNA was checked by the 260:280 ratio, nanodrop, and picogreen. The presence of human and bacterial DNA was verified by TaqMan with species-specific primers.
DNA preparation for array hybridization and HOMIM array
The Human Oral Microbe Identification Microarray (HOMIM) analysis was conducted in the Paster laboratory as previously described [14]. The array uses 16S rRNA-based oligonucleotide probes printed on glass slides. The extracted DNA was used as the template in PCR reactions with universal forward and reverse primers and labeled in a second nested PCR. Bacterial 16S rRNA gene amplicons from the UDT samples were hybridized onto 16S rRNA gene microarrays [14]. After hybridization, the washed slides were scanned using GenePix Pro software. A normalized median intensity score was generated by subtracting the median feature intensity from the background intensity for each individual feature. The relative intensity of each probed species/strain was then estimated using feature-specific criteria.
HOMIM array was developed to profile human oral microbiota. Thus, we are studying oral bacterial species in the UDT samples although the UDT samples include cells from the stomach, esophagus and mouth.
Estimation of oral health parameters
The National Health and Nutrition Examination Survey (NHANES) oral health examination protocols were used to obtained information on oral health [15]. Dental health was assessed by the number of missing teeth (total permanent teeth missing, including third molars), the amount of untreated tooth decay (the total surfaces of all teeth with coronal decay), and the DMFT score (decayed, missing, and filled teeth, excluding third molars). Periodontal health information was assessed by bleeding on probing (BoP, the percent of probed sites with bleeding), attachment loss (AL, the percent of probed sites with loss of attachment > = 3 mm), and a periodontitis summary estimate (presence/absence of periodontal disease, present if one or more probed periodontal site had an attachment loss of 3 mm or greater and a pocket depth of 4 mm or greater). Assessments of untreated tooth decay, DMFT, bleeding on probing and attachment loss were restricted to participants with at least one tooth (dentate). Four examiners independently conducted the clinical visits and the details of examiner calibration were described previously [15].
Statistical analysis
As some of the probes on the arrays hybridized to more than one species, microbial richness was estimated as the number of genera in each sample. β-diversity, which estimates pairwise difference in microbial diversity between samples, was measured by constructing an unweighted UniFrac distance matrix via FastUniFrac (http://unifrac.colorado.edu) [16]. The phylogeny used for UniFrac estimation was constructed from Human Oral Microbiome Database [17]. The unweighted UniFrac matrix was analyzed by principle coordinate (PC) analysis. The first three PCs, which explain respectively 22%, 14%, 9% of the variance, were used for further analyses.
Associations between number of genera per sample, the first three PCs for β-diversity and oral health variables were evaluated by adjusted linear regression models and Spearman tests in R [18]. Both linear regression models and Spearman tests were adjusted for age (years), sex (female or male), history of smoking (yes or no), history of antibiotic use in the last three months (yes or no), and sample device (CHB or WCB). Adjusted Spearman tests used the residuals of each health variable and residuals of each microbial variable after linearly fitting them against the above adjustment variables. We used nonparametric Spearman tests due to the non-normal distribution of each oral health variable. A statistical significance level of 0.05 was applied. However we did not show Spearman tests results in the tables because they produced similar results. We conducted several sensitivity analyses including stratifying the analysis by sampling device and by removing patients that reported taking antibiotics in the last 3 months prior to enrollment.
To examine the relationship between oral health and the presence/absence of a specific genus, we used logistic regression models. Bonferroni correction was used to account for multiple testing.
Results
Characteristics of the study cohort
Table 1 describes the study cohort. Among the study participants, the average age was 55 years, 42% were male, 25.5% were never-smokers, and 11.5% had used antibiotics in the last three months. With regard to oral health, although most had no tooth decay (median = 0), 17.5% of participants had lost all of their teeth, and 43% of participants had periodontitis.
Associations between oral health variables and microbial richness
In both adjusted linear regression models and Spearman correlation tests, microbial richness, measured as number of genera per sample, was associated inversely with several measures of poor dental health (teeth missing, tooth decay, and DMFT score), but was associated positively with poor periodontal health (BoP, AL, and periodontitis). Many of these associations were statistically significant (Table 2). For example, increased tooth decay and DMFT score were significantly associated with fewer genera per sample (Plinear = 0.01, 0.02, respectively), and increased BoP was significantly associated with more genera per sample (Plinear = 0.02, PSpearman = 0.02).
We found no significant difference in microbial richness between toothless subjects and those with one or more teeth remaining (Table 2).
Significant association between Oral health variables and microbial β-diversity
We assessed associations between β-diversity and oral health using the first three principal components (PCs) of the unweighted UniFrac distance matrix (Table 3). Of the six tested oral health measures, five of them were associated with at least one of these three PCs. Tooth decay was associated with PC1 (Plinear = 0.01, Pspearman = 0.05). Teeth missing and BoP were associated with PC2 (teeth missing, Plinear = 0.01, PSpearman = 0.01; BoP, Plinear = 0.01, Pspearman = 0.01). DMFT score and periodontitis were significantly associated with PC2 and PC3. PC 2 and PC3 were also associated with whether or not participants had any teeth.
In addition, we used the Unweighted Pair Group Method with Arithmetic Mean (UPGMA) to create clusters based on the UniFrac distance matrix (Additional file 1). We focused on clusters B and C, as cluster A included only 11 participants. However, such clusters were not associated with any of our examined oral health measures (Additional file 2).
Association between oral health and presence/absence of specific genera
We also examined associations between the oral health variables and the presence/absence of specific genera (Additional file 3). Table 4 showed the associations that remained significant after Bonferroni correction. Subjects with Parvinomas had a significantly higher risk for periodontitis (p = 0.000219). Subjects with Parvimonas or Porphyromonas had a significantly higher extent of BoP (P = 0.000006 and 0.0002, respectively). Subjects with Filfactor or Peptostreptococcus had a significantly lower DMFT score (P = 0.0000006 and 0.000005, respectively) while subjects with Veillonellaceae [G-1] had a significantly higher DMFT score (P = 0.00009).
Discussion
In this study, we examined several aspects of UDT microbiota, including microbial richness, β-diversity, and specific bacterial genera. Each aspect was associated with one or more measures of poor dental or periodontal health, although these associations differed. Poor dental health was associated inversely with microbial richness, and certain bacterial species were notably absent among those with high DMFT scores. In contrast, poor periodontal health was associated positively with microbial richness and the presence of species that have previously been implicated in this disease.
An integral part of the tooth decay process is demineralization of the tooth structure, which can lead to the development of cavities. Untreated tooth decay can result in substantial loss of tooth structure, infection, and pain. We found significant inverse associations between microbial richness, tooth decay, and DMFT score. Our findings are concordant with the ecological plaque hypothesis [19], which suggests that caries stem from a shift in the balance of UDT microbiota in response to environmental pressures, such as frequent carbohydrate intake or acidification of the oral environment, and that these changes lead to an overall reduction of microbial richness.
Alternatively, two other hypotheses have been described: the specific plaque hypothesis and the nonspecific plaque hypothesis [20, 21]. The specific plaque hypothesis proposes that only a few specific species, such as Streptococcus mutans and Streptococcus sobrinus, are actively involved in the disease [20]. On the other hand, the nonspecific plaque hypothesis suggests that caries arise from the combined actions of the many different bacterial species residing in the plaque [21]. Carries, according to this hypothesis, may be caused by increasing presence of acid-producing oral micro-organisms and/or a decrease in alkali-producing bacteria. Consistent with the nonspecific hypothesis, recent studies have suggested the involvement of many different genera, including Lactobacillus, Actinomyces, Bifidobacterium, Veillonella, Corynebacterium or Leptotrichia[22, 23], in this process. Some of these bacteria, particularly Veillonella, have been shown to be predominant at all stages of caries progression [22] and under high-glucose conditions, and have been implicated in acid production [24]. In our study, we found that subjects with a high DMFT score were more likely to have Veillonellaceae [G-1], but less likely to have Filifactor and Peptostreptococcus. As Veillonellaceae [G-1] grows well in an acid environment, and Peptostreptococcus does better in a basic environment [25], our results are consistent with the non-specific plaque hypothesis.
BoP is an indicator of periodontal tissue inflammatory response to bacterial pathogens and may indicate active periodontitis, whereas attachment loss reflects the loss of supporting tissue as part of the periodontal disease process. We found a significant positive association between BoP and microbial richness and a consistent, though non-significant, association between attachment loss and periodontitis. Our results are consistent with previous findings [9, 26]. The association of increased microbial richness and increased BoP suggests that pathogenic periodontal species may colonize the mouth during periods of periodontal inflammation. Changes in the microbial community during periodic inflammatory events within the periodontium may be a progressive process, with the introduction of taxa associated with inflammation without the replacement of original resident species.
Previous studies suggest that several key species are associated with periodontitis [27–31]. These species include Porphyromonas gingivalis, Treponema denticola, Prevotella intermedia, Aggregatibacter actinomycetemcomitans, and Fusobacterium nucleatum. Culture-independent approaches, such as quantitative ribosomal 16S cloning and sequencing techniques have implicated other species including Parvimonas micra, Filifactor alocis, uncultivated clones from the Deferribacteres and Bacteroidetes phyla, Megasphaera clone BB166, and clone I025 from the TM7 phylum [32, 33]. Our results provide further support for the role of Porphyromonas and Parvimonas in periodontitis as previous studies suggested [34–38]. However, it remains unclear whether these bacterial species are responsible for initiation of the disease or whether periodontitis creates conditions that select for these bacteria. Studies with prospective designs and experiments on animal models are required.
Another important finding was the observed significant association between β-diversity and most of our examined health measures, highlighting the important association between the UDT microbiota and oral health. Previous studies of the gut microbiota have shown that diet, race, and geography contribute to the difference among gut microbial communities [39–41]. Our findings suggest that oral health makes a similarly important contribution to the UDT microbiota.
Our study has several strengths and limitations. The strengths include the relatively large sample size and the availability of detailed oral health and demographic information on participants. It is one of only a few studies that have focused on a population with poor access to dental care and have had potential confounders measured. The limitations include the cross-sectional design of the parent study, and the use of the HOMIM array with samples from a device that collected cellular and luminal material from the stomach, esophagus, and oral cavity. The HOMIM arrays are specifically designed to measure oral bacteria, and therefore did not allow us to study bacterial species not on the array. Therefore, the results may reflect only the oral bacteria and these bacteria when present in the esophagus and stomach.
Conclusions
Our study suggests that tooth decay and DMFT score were associated with a microbial community shift towards decreased microbial richness, which is likely due to an increase of aciduric and decrease of alkali-producing bacteria species. Participants with periodontitis had a distinct microbial community, characterized by increased microbial richness and the presence of additional, potentially pathogenic, bacterial genera such as Porphyromonas and Parvimonas. However, due to the cross-sectional design, we could not determine whether these changes in the UDT microbiota were the cause or the result of oral disease. Experimental studies and prospective cohorts with repeated assessment are needed to define the temporal relationship between microbial communities and oral health.
Abbreviations
- UDT:
-
Upper digestive tract
- NHANES:
-
National Health and Nutrition Examination Survey
- HOMIM:
-
Human Oral Microbe Identification Microarray
- DMFT:
-
Decayed, missing, and filled teeth score based on teeth excluding third molars
- BoP:
-
Bleeding of probe extent score, percent of probed sites with bleeding
References
Human Microbiome Project Consortion: Structure, function and diversity of the healthy human microbiome. Nature. 2012, 486 (7402): 207-214. 10.1038/nature11234.
Lamont R, Jenkinson H: Oral Microbiology at a Glance. 2010, Hoboken, New Jersey: Wiley-Blackwell
Cunningham-Rundles S, Ahrn S, Abuav-Nussbaum R, Dnistrian A: Development of immunocompetence: role of micronutrients and microorganisms. Nutr Rev. 2002, 60 (5 Pt 2): S68-72.
Pei Z, Bini EJ, Yang L, Zhou M, Francois F, Blaser MJ: Bacterial biota in the human distal esophagus. Proc Natl Acad Sci U S A. 2004, 101 (12): 4250-4255. 10.1073/pnas.0306398101.
Filoche S, Wong L, Sissons CH: Oral Biofilms: Emerging Concepts in Microbial Ecology. J Dent Res. 2010, 89 (1): 8-18. 10.1177/0022034509351812.
Gross EL, Leys EJ, Gasparovich SR, Firestone ND, Schwartzbaum JA, Janies DA, Asnani K, Griffen AL: Bacterial 16S Sequence Analysis of Severe Caries in Young Permanent Teeth. J Clin Microbiol. 2010, 48 (11): 4121-4128. 10.1128/JCM.01232-10.
Loesche W: Dental caries and periodontitis: Contrasting two infections that have medical implications. Infect Dis Clin N Am. 2007, 21 (2): 471-502. 10.1016/j.idc.2007.03.006.
Wade WG: The oral microbiome in health and disease. Pharmacol Res. 2012, 69 (1): 137-143.
Griffen AL, Beall CJ, Campbell JH, Firestone ND, Kumar PS, Yang ZK, Podar M, Leys EJ: Distinct and complex bacterial profiles in human periodontitis and health revealed by 16S pyrosequencing. ISME J. 2012, 6 (6): 1176-1185. 10.1038/ismej.2011.191.
Belda-Ferre P, Alcaraz LD, Cabrera-Rubio R, Romero H, Simon-Soro A, Pignatelli M, Mira A: The oral metagenome in health and disease. ISME J. 2012, 6 (1): 46-56. 10.1038/ismej.2011.85.
Abusleme L, Dupuy AK, Dutzan N, Silva N, Burleson JA, Strausbaugh LD, Gamonal J, Diaz PI: The subgingival microbiome in health and periodontitis and its relationship with community biomass and inflammation. ISME J. 2013, 7 (5): 1016-1025. 10.1038/ismej.2012.174.
Yang F, Zeng X, Ning K, Liu KL, Lo CC, Wang W, Chen J, Wang D, Huang R, Chang X, Chain PS, Xie G, Ling J, Xu J: Saliva microbiomes distinguish caries-active from healthy human populations. ISME J. 2012, 6 (1): 1-10. 10.1038/ismej.2011.71.
Wei WQ, Abnet CC, Lu N, Roth MJ, Wang GQ, Dye BA, Dong ZW, Taylor PR, Albert P, Qiao YL, Dawsey SM: Risk factors for oesophageal squamous dysplasia in adult inhabitants of a high risk region of China. Gut. 2005, 54 (6): 759-763. 10.1136/gut.2004.062331.
Colombo AP, Boches SK, Cotton SL, Goodson JM, Kent R, Haffajee AD, Socransky SS, Hasturk H, Van Dyke TE, Dewhirst F, Paster BJ: Comparisons of subgingival microbial profiles of refractory periodontitis, severe periodontitis, and periodontal health using the human oral microbe identification microarray. J Periodontol. 2009, 80 (9): 1421-1432. 10.1902/jop.2009.090185.
Dye BA, Wang R, Lashley R, Wei W, Abnet CC, Wang G, Dawsey SM, Cong W, Roth MJ, Li X, Qiao Y: Using NHANES oral health examination protocols as part of an esophageal cancer screening study conducted in a high-risk region of China. BMC Oral Health. 2007, 7: 10-10.1186/1472-6831-7-10.
Lozupone C, Lladser ME, Knights D, Stombaugh J, Knight R: UniFrac: an effective distance metric for microbial community comparison. ISME J. 2011, 5 (2): 169-172. 10.1038/ismej.2010.133.
Dewhirst FE, Chen T, Izard J, Paster BJ, Tanner AC, Yu WH, Lakshmanan A, Wade WG: The human oral microbiome. J Bacteriol. 2010, 192 (19): 5002-5017. 10.1128/JB.00542-10.
Team RDC: R: A language and environment for statistical computing. 2011, Vienna, Austria: R Foundation for Statistical Computing
Kanasi E, Dewhirst FE, Chalmers NI, Kent R, Moore A, Hughes CV, Pradhan N, Loo CY, Tanner AC: Clonal analysis of the microbiota of severe early childhood caries. Caries Res. 2010, 44 (5): 485-497. 10.1159/000320158.
Loesche WJ: The specific plaque hypothesis and the antimicrobial treatment of periodontal disease. Dent Update. 1992, 19 (2): 68-70-2, 74
Theilade E: The non-specific theory in microbial etiology of inflammatory periodontal diseases. J Clin Periodontol. 1986, 13 (10): 905-911. 10.1111/j.1600-051X.1986.tb01425.x.
Aas JA, Griffen AL, Dardis SR, Lee AM, Olsen I, Dewhirst FE, Leys EJ, Paster BJ: Bacteria of dental caries in primary and permanent teeth in children and young adults. J Clin Microbiol. 2008, 46 (4): 1407-1417. 10.1128/JCM.01410-07.
Becker MR, Paster BJ, Leys EJ, Moeschberger ML, Kenyon SG, Galvin JL, Boches SK, Dewhirst FE, Griffen AL: Molecular analysis of bacterial species associated with childhood caries. J Clin Microbiol. 2002, 40 (3): 1001-1009. 10.1128/JCM.40.3.1001-1009.2002.
Bradshaw DJ, Marsh PD: Analysis of pH-driven disruption of oral microbial communities in vitro. Caries Res. 1998, 32 (6): 456-462. 10.1159/000016487.
Liu H, Liu X, Zhang J, Chen J: Acetate accumulation and shift of bacterial community during anaerobic sewage sludge fermentation by pH adjustment. Wei Sheng Wu Xue Bao. 2009, 49 (12): 1643-1649.
Socransky SS, Haffajee AD, Smith C, Dibart S: Relation of Counts of Microbial Species to Clinical Status at the Sampled Site. J Clin Periodontol. 1991, 18 (10): 766-775. 10.1111/j.1600-051X.1991.tb00070.x.
Socransky SS, Haffajee AD, Cugini MA, Smith C, Kent RL: Microbial complexes in subgingival plaque. J Clin Periodontol. 1998, 25 (2): 134-144. 10.1111/j.1600-051X.1998.tb02419.x.
Ledder RG, Gilbert P, Huws SA, Aarons L, Ashley MP, Hull PS, McBain AJ: Molecular analysis of the subgingival microbiota in health and disease. Appl Environ Microbiol. 2007, 73 (2): 516-523. 10.1128/AEM.01419-06.
Paster BJ, Boches SK, Galvin JL, Ericson RE, Lau CN, Levanos VA, Sahasrabudhe A, Dewhirst FE: Bacterial diversity in human subgingival plaque. J Bacteriol. 2001, 183 (12): 3770-3783. 10.1128/JB.183.12.3770-3783.2001.
Paster BJ, Olsen I, Aas JA, Dewhirst FE: The breadth of bacterial diversity in the human periodontal pocket and other oral sites. Periodontol 2000. 2006, 42: 80-87. 10.1111/j.1600-0757.2006.00174.x.
Socransky SS, Haffajee AD: Periodontal microbial ecology. Periodontol 2000. 2005, 38: 135-187. 10.1111/j.1600-0757.2005.00107.x.
Brinig MM, Lepp PW, Ouverney CC, Armitage GC, Relman DA: Prevalence of bacteria of division TM7 in human subgingival plaque and their association with disease. Appl Environ Microbiol. 2003, 69 (3): 1687-1694. 10.1128/AEM.69.3.1687-1694.2003.
Kumar PS, Griffen AL, Barton JA, Paster BJ, Moeschberger ML, Leys EJ: New bacterial species associated with chronic periodontitis. J Dent Res. 2003, 82 (5): 338-344. 10.1177/154405910308200503.
Alpagot T, Wolff LF, Smith QT, Tran SD: Risk indicators for periodontal disease in a racially diverse urban population. J Clin Periodontol. 1996, 23 (11): 982-988. 10.1111/j.1600-051X.1996.tb00524.x.
Beck JD: Methods of assessing risk for periodontitis and developing multifactorial models. J Periodontol. 1994, 65 (5 Suppl): 468-478.
Grossi SG, Zambon JJ, Ho AW, Koch G, Dunford RG, Machtei EE, Norderyd OM, Genco RJ: Assessment of risk for periodontal disease. I. Risk indicators for attachment loss. J Periodontol. 1994, 65 (3): 260-267. 10.1902/jop.1994.65.3.260.
Haffajee AD, Socransky SS, Smith C, Dibart S: Relation of baseline microbial parameters to future periodontal attachment loss. J Clin Periodontol. 1991, 18 (10): 744-750. 10.1111/j.1600-051X.1991.tb00066.x.
Moore WE, Moore LH, Ranney RR, Smibert RM, Burmeister JA, Schenkein HA: The microflora of periodontal sites showing active destructive progression. J Clin Periodontol. 1991, 18 (10): 729-739. 10.1111/j.1600-051X.1991.tb00064.x.
Yatsunenko T, Rey FE, Manary MJ, Trehan I, Dominguez-Bello MG, Contreras M, Magris M, Hidalgo G, Baldassano RN, Anokhin AP, Heath AC, Warner B, Reeder J, Kuczynski J, Caporaso JG, Lozupone CA, Lauber C, Clemente JC, Knights D, Knight R, Gordon JI: Human gut microbiome viewed across age and geography. Nature. 2012, 486 (7402): 222-227.
Smith MI, Yatsunenko T, Manary MJ, Trehan I, Mkakosya R, Cheng J, Kau AL, Rich SS, Concannon P, Mychaleckyj JC, Liu J, Houpt E, Li JV, Holmes E, Nicholson J, Knights D, Ursell LK, Knight R, Gordon JI: Gut microbiomes of Malawian twin pairs discordant for kwashiorkor. Science (New York, NY). 2013, 339 (6119): 548-554. 10.1126/science.1229000.
Muegge BD, Kuczynski J, Knights D, Clemente JC, Gonzalez A, Fontana L, Henrissat B, Knight R, Gordon JI: Diet Drives Convergence in Gut Microbiome Functions Across Mammalian Phylogeny and Within Humans. Science (New York, NY). 2011, 332 (6032): 970-974. 10.1126/science.1198719.
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1471-2458/14/1110/prepub
Acknowledgements
This study was supported by the Intramural Research Program of the NIH, National Cancer Institute.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
All authors declare that they have no competing interests.
Authors’ contributions
GY and CCA conceived of the analysis; VKC and BJP performed the microarray assays; GY, MHG, JS and CCA analyzed the data; BAD, GQW, WQW, JHF, YLQ, and SMD participated in the acquisition of the samples; GY drafted the manuscript; BAD, MHG, JS, VKC, BJP, WQW, YLQ, SMD, NDF and CCA gave critical feedback on the manuscript. All authors read and approved the final manuscript.
Electronic supplementary material
12889_2014_7206_MOESM1_ESM.pdf
Additional file 1: Subjects grouped into clusters (designated as A, B, C) by the UniFrac distance matrix according to the UPGMA method.(PDF 32 KB)
12889_2014_7206_MOESM2_ESM.doc
Additional file 2: Comparisons of oral health variables between subjects in clusters B and C from the UPGMA analysis of the UniFrac distance matrix (clusters shown in Additional file 1 ).(DOC 33 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Yu, G., Dye, B.A., Gail, M.H. et al. The association between the upper digestive tract microbiota by HOMIM and oral health in a population-based study in Linxian, China. BMC Public Health 14, 1110 (2014). https://doi.org/10.1186/1471-2458-14-1110
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2458-14-1110