
Abstract
Background
The advance of high-throughput sequencing has made it possible to obtain new transcriptomes and study splicing mechanisms in non-model organisms. In these studies, there is often a need to investigate the transcriptomes of two related organisms at the same time in order to find the similarities and differences between them. The traditional approach to address this problem is to perform de novo transcriptome assemblies to obtain predicted transcripts for these organisms independently and then employ similarity comparison algorithms to study them.
Results
Instead of obtaining predicted transcripts for these organisms separately from the intermediate de Bruijn graph structures employed by de novo transcriptome assembly algorithms, we develop an algorithm to allow direct comparisons between paths in two de Bruijn graphs by first enumerating short paths in both graphs, and iteratively extending paths in one graph that have high similarity to paths in the other graph to obtain longer corresponding paths between the two graphs. These paths represent predicted transcripts that are present in both organisms.
We show that our algorithm recovers significantly more shared transcripts than traditional approaches by applying it to simultaneously recover transcripts in mouse against rat and in mouse against human from publicly available RNA-Seq libraries. Our strategy utilizes sequence similarity information within the paths that is often more reliable than coverage information.
Conclusions
Our approach generalizes the pairwise sequence alignment problem to allow the input to be non-linear structures, and provides a heuristic to reliably recover similar paths from the two structures. Our algorithm allows detailed investigation of the similarities and differences in alternative splicing between the two organisms at both the sequence and structure levels, even in the absence of reference transcriptomes or a closely related model organism.
Similar content being viewed by others


Background
The advance of next generation sequencing technologies has made it possible to perform detailed studies of splicing mechanisms among eukaryotic organisms. A popular strategy is to first sequence their transcriptomes, then map the reads to reference databases. In non-model organisms, such reference databases are often unavailable, and de novo transcriptome assembly algorithms are employed to obtain predicted transcripts [1–4]. This is often achieved by first constructing a de Bruijn graph structure that contains all branching possibilities [5, 6], then obtaining predicted transcripts based on coverage information along the paths.
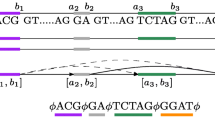
In many of these studies, there is often a need to investigate the transcriptomes of two related organisms at the same time in order to study their similarities and differences. In these cases, RNA-Seq libraries are obtained from both organisms under different experimental conditions and the goal is to compare their transcriptome assemblies. The traditional approach to address this problem is to perform transcriptome assemblies to obtain predicted transcripts for the two organisms separately (see Figure 1). Similarity comparison algorithms such as BLAST [7] are then employed to extract corresponding transcripts that are shared in the two organisms.

Difference between traditional strategy and our strategy.
Since predicted transcripts are constructed independently for each organism based on coverage information only, this strategy is often unreliable. To address this problem, we develop an algorithm to allow direct comparisons between paths in the two intermediate de Bruijn graph structures by an iterative extension strategy (see Figure 1). Since sequence similarity information is often more reliable, this strategy allows the direct extraction of shared transcripts based on evolutionary support.
Methods
De Bruijn graph
Given a set of reads and a parameter k, a de Bruijn graph is constructed by taking each k-mer that appears within the reads as a vertex. Two k-mers are connected by a directed edge if the (k − 1)-suffix of the first k-mer is the same as the (k − 1)-prefix of the second k-mer [5, 6]. The de Bruijn graph implicitly assembles the reads by linking together the overlapping parts, and it is employed as the main intermediate structure by most short read assembly algorithms [8–12]. To obtain a more compact structure, each linear sequence of vertices that have no branches is collapsed into a single node that corresponds to contigs.
Iterative extension
Given de Bruijn graphs G1 and G2 that correspond to transcriptome assemblies of two related organisms, we first apply BLAST to obtain similarity scores between each pair of nodes u from G1 and v from G2. We then start the iterative extension process as follows. For each node u from G1, we extract its most similar node v from G2 with e-value below a cutoff. If such a node v exists, we retain u as a single-node path. We extend u by one node along all its outgoing edges into multiple paths, and apply BLAST from each of these extended paths from u against v. If at least one of these extended paths gives a better e-value against v, we retain all the paths that have better e-values and continue to extend the top path that gives the best e-value. We repeat the procedure starting from this new path until the e-value no longer improves. Note that only one best direction is chosen since extending in more than one direction is very time-consuming. By starting from each node u in G1 independently, the probability of missing the real best path is reduced a lot.
After the above procedure, we have retained u and all the extended paths from u that have improved e-values, with the top path that gives the best e-value being fully extended. We then retain v as a single-node path and perform a similar extension process starting from v by extending it by one node along all its outgoing edges into multiple paths. We apply BLAST from each of these extended paths from v against all the retained paths from u. If at least one of these extended paths gives a better e-value, we retain all the paths that have better e-values and continue to extend the top path that gives the best e-value. Similar to above, we repeat the procedure starting from this new path until the e-value no longer improves to obtain a fully extended path and a set of retained paths from v that have improved e-values (see Figure 2).

Illustration of the iterative extension procedure. The paths that are fully extended from u in G1 and from v in G2 are marked in bold, while the other retained paths with improved e-value are not marked.
We then repeat the entire extension procedure in turn in G1 and G2 by replacing u by the fully extended path from u and comparing against all the retained paths from v, and replacing v by the fully extended path from v and comparing against all the retained paths from u. The entire process is repeated until no more improvements can be made, and the algorithm is applied again by switching the role of G1 and G2 and repeating all the steps.
To obtain longer paths, we consider the retained paths from each node u and the retained paths from its twin node u′, in which u′ represents the reverse complementary sequence of u on the opposite strand. We merge the twin paths that are complementary to the retained paths from u′ with the retained paths from u, and keep those paths with improved e-values.
Extraction of predicted transcripts
We consider all the retained paths in G1 as predicted transcripts in the first organism and all the retained paths in G2 as predicted transcripts in the second organism. Since the collection of all these retained paths can be very big, we only keep a path if it contains a node in the de Bruijn graph that is not covered by another path with a better e-value according to the top BLAST alignment. In this condition, a node is covered by a path if it contains the node itself or its twin node. To avoid a large number of incorrectly predicted isoforms, we remove paths with worse e-values so that each node in the de Bruijn graph along with its twin node appears at most 10 times within the final set of paths.
Extraction of predicted shared transcripts
To obtain predicted shared transcripts that have correspondences between the two organisms, we apply BLAST from each predicted transcript in one organism against the set of all predicted transcripts in the other organism as database. We retain a predicted transcript as a predicted shared transcript if it appears both as a query with BLAST hits from one direction and as a subject BLAST hit in the other direction.
Results and discussion
Validation
We implement our algorithm Mutual as a postprocessing module of Velvet [10], which is a popular sequence assembly algorithm that returns a set of contigs along with the de Bruijn graph. We compare our performance to Oases [3], which uses output from Velvet to construct predicted transcripts. We validate our algorithm by applying it to simultaneously recover transcripts in mouse against rat and in mouse against human from publicly available RNA-Seq libraries at the sequence read archive [13], including two libraries from mouse in [14] (SRX017794), one library from rat in [15] (SRX076903), and four libraries from human in [16] (SRX011545). We perform quality trimming by removing all positions including and to the right of the first position that has a quality score of less than 15, resulting in a size of 1.3 G for the mouse libraries, 2.5 G for the rat libraries and 1.1 G for the human libraries.
We apply each algorithm over k = 25, 31, and over k-mer coverage cutoff c = 3, 5, 10. In our algorithm Mutual, iterative extension is applied twice with an e-value cutoff of 0.1 using the bl2seq (BLAST 2 Sequences) variant of BLAST, once with translated BLAST and once with nucleotide BLAST. Velvet and Oases are applied independently in each organism. Since Oases applies the coverage cutoff itself to obtain a de Bruijn graph by modifying Velvet's original de Bruijn graph without coverage cutoff, Mutual is applied on the two de Bruijn graphs given by Oases to obtain predicted transcripts.
To obtain predicted shared transcripts for both Oases and Mutual, we apply both translated BLAST and nucleotide BLAST with an e-value cutoff of 10−7 or 10−20 from each predicted transcript in one organism against the set of all predicted transcripts in the other organism as database. The predicted transcripts that appear both as a query with BLAST hits from one direction and as a subject BLAST hit in the other direction are retained as predicted shared transcripts. To evaluate the accuracy of the predicted shared transcripts, we apply nucleotide BLAST to compare them against known mouse, rat or human transcriptome databases using the same e-value cutoff as the one used to obtain the transcripts, which is 10−7 or 10−20. To assess the extent of translocated transcripts, we apply GMAP [17] to map the predicted shared transcripts to known mouse, rat or human genomes.
Predicted transcripts
Tables 1 and 2 show that Mutual constructed less predicted transcripts than Oases. Note that the predicted transcripts from Mutual are obtained by extending similar paths that appear in the two organisms through iterative BLAST, while the predicted transcripts from Oases are obtained independently in each organism. The similarity constraints in Mutual ensure that a predicted transcript in one organism has a similar counterpart in the other organism, albeit with a loose e-value cutoff. The later reciprocal BLAST is needed to enforce more stringent e-value cutoffs. On the other hand, the predicted transcripts from Oases have no such constraints, and reciprocal BLAST is used to obtain shared transcripts.
Predicted shared transcripts
When compared to Tables 1 and 2, Tables 3 and 4 show that only a small percentage of predicted transcripts were shared in the two organisms, with a smaller decrease by Mutual than by Oases. The decrease by Mutual is due to more stringent e-value cutoffs, while the decrease by Oases is due to imposing similarity constraints between the two organisms. While the actual amount of predicted shared transcripts that can be recovered depends on the size of libraries, the evolutionary distance between the two organisms and the experimental conditions, Tables 3 and 4 show that Mutual recovered more predicted shared transcripts than Oases. Almost all these predicted shared transcripts are found in the corresponding known transcriptome database, with comparable percentages between Mutual and Oases. The percentages are lower in rat, probably due to the fact that the rat genome is less well annotated. The number of predicted shared transcripts decreases as the assembly parameters become more stringent, but these transcripts are of higher quality.
Top BLAST hits to databases
By applying BLAST from each set of predicted shared transcripts in each organism to its known transcriptome database, Tables 5 and 6 show that Mutual recovered more shared transcripts than Oases, with many more shared transcripts recovered when the assembly parameters are less stringent. When compared to Tables 3 and 4, there is an effect of diminishing returns since a few thousand more predicted shared transcripts are needed to recover a few hundred more shared transcripts.
Length distribution of transcripts
Figures 3 and 4 show that the lengths of predicted shared transcripts recovered by Mutual were comparable to the ones recovered by Oases, which are slightly shorter in mouse but have slightly higher medians in rat. These transcripts are generally longer when the k-mer coverage cutoff c increases.

Length distribution of predicted shared transcripts in the test on mouse against rat from Oases and from Mutual over different values of k and k -mer coverage cutoff c (represented by k_c ) and over different e -value cutoffs 10−7 and 10−20 . The width of each box is proportional to the square root of the size of each group, while outliers are ignored.

Length distribution of predicted shared transcripts in the test on mouse against human. Notations are the same as in Figure 3.
Recovery of full length transcripts
The situation is different when considering predicted shared transcripts that are close to full length. Tables 7 and 8 show that Mutual recovered more or a comparable number of 80% full length transcripts as Oases when the assembly parameters are more stringent, and less 80% full length transcripts than Oases when the assembly parameters are less stringent. Although Mutual performs worst in rat that recovers less 80% full length transcripts than Oases, its predicted shared transcripts have slightly higher median lengths when considering all the transcripts together (see Figure 3), instead of just the ones that are 80% full length transcripts.
Presence of translocated transcripts
As reported by GMAP, Tables 9 and 10 show that Mutual recovered a much larger number of predicted shared transcripts that are uniquely mapped than Oases, while at the same time returning more translocated transcripts that can be considered to be errors due to their rare occurrences [18]. The ratio of the number of translocated transcripts to the number of uniquely mapped transcripts is at most about twice as much for Mutual when compared to Oases. This ratio increases when k decreases or when the k-mer coverage cutoff c increases.
Accuracy of transcript reconstruction
By investigating the fitness of the alignment between the predicted shared transcripts and the known transcriptome database sequences, Figures 5 and 6 show that with respect to the accuracy of shared transcript reconstruction, there are tradeoffs between precision and recall by Mutual when compared to Oases. Mutual has slightly lower F-scores than Oases in most cases.

Precision, recall and F -score with respect to the accuracy of shared transcript reconstruction in the test on mouse against rat from Oases and from Mutual over different values of k and k -mer coverage cutoff c (represented by k _ c ) and over different e -value cutoffs 10−7 and 10−20 . Precision is defined to be the fraction of query positions from predicted shared transcripts that are included in BLAST alignments from each organism to its known transcriptome database. Recall is defined to be the fraction of subject positions from database sequences that are included in BLAST alignments from each organism to its known transcriptome database. F-score is the harmonic mean of precision and recall.

Precision, recall and F -score with respect to the accuracy of shared transcript reconstruction in the test on mouse against human. Notations are the same as in Figure 5.
Availability
A software program that implements our algorithm (Mutual) is available at http://faculty.cse.tamu.edu/shsze/mutual.
Conclusions
We have developed an algorithm that makes use of evolutionary information to simultaneously recover significantly more shared transcripts from RNA-Seq data in two related organisms that may be missed by traditional de novo approaches. While more shared transcripts are recovered due to the smaller evolutionary distance between mouse and rat, our algorithm can be applied to related organisms that are evolutionarily farther away, such as between mouse and human.
While known transcriptomes are used as databases during validation, one important characteristic of our algorithm is that no reference transcriptomes or a closely related model organism are needed. Our algorithm can be used to recover shared transcripts that are specific to two closely related non-model organisms, which may not be present in a related model organism that is evolutionarily farther away.
Depending on the size of the de Bruijn graphs, our algorithm can take many processor-hours to run. It takes more than 600 processor-hours to obtain all the predicted transcripts in mouse against rat or in mouse against human for the least stringent values of k and the k-mer coverage cutoff c. Although our algorithm can take much more computational time than the de novo algorithms Velvet or Oases, the iterative BLAST searches can be run independently in parallel on a computing cluster. While an additional 60 processor-hours are needed to obtain predicted shared transcripts from the predicted transcripts, a similar procedure is also needed for Oases. No special memory requirement is needed after the de Bruijn graphs are obtained.
One drawback of our algorithm is that only a weak recovery of non-coding regions of mRNA is possible since these regions may not be conserved. Due to the use of similarity information between two related organisms to extend transcripts, our algorithm cannot identify extended transcripts that are not shared between the two organisms.
References
Robertson G, Schein J, Chiu R, Corbett R, Field M, Jackman SD, Mungall K, Lee S, Okada HM, Qian JQ, Griffith M, Raymond A, Thiessen N, Cezard T, Butterfield YS, Newsome R, Chan SK, She R, Varhol R, Kamoh B, Prabhu AL, Tam A, Zhao Y, Moore RA, Hirst M, Marra MA, Jones SJM, Hoodless PA, Birol I: De novo assembly and analysis of RNA-seq data. Nat Methods. 2010, 7: 909-912.
Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q, Chen Z, Mauceli E, Hacohen N, Gnirke A, Rhind N, di Palma F, Birren BW, Nusbaum C, Lindblad-Toh K, Friedman N, Regev A: Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011, 29: 644-652.
Schulz MH, Zerbino DR, Vingron M, Birney E: Oases: robust de novo RNA-seq assembly across the dynamic range of expression levels. Bioinformatics. 2012, 28: 1086-1092.
Xie Y, Wu G, Tang J, Luo R, Patterson J, Liu S, Huang W, He G, Gu S, Li S, Zhou X, Lam TW, Li Y, Xu X, Wong GKS, Wang J: SOAPdenovo-Trans de novo transcriptome assembly with short RNA-Seq reads. Bioinformatics. 2014, 30: 1660-1666.
Pevzner PA: l-tuple DNA sequencing: computer analysis. J Biomol Struct Dyn. 1989, 7: 63-73.
Idury RM, Waterman MS: A new algorithm for DNA sequence assembly. J Comput Biol. 1995, 2: 291-306.
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ: Basic local alignment search tool. J Mol Biol. 1990, 215: 403-410.
Butler J, MacCallum I, Kleber M, Shlyakhter IA, Belmonte MK, Lander ES, Nusbaum C, Jaffe DB: ALLPATHS de novo assembly of whole-genome shotgun microreads. Genome Res. 2008, 18: 810-820.
Chaisson MJ, Pevzner PA: Short read fragment assembly of bacterial genomes. Genome Res. 2008, 18: 324-330.
Zerbino DR, Birney E: Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008, 18: 821-829.
Birol I, Jackman SD, Nielsen CB, Qian JQ, Varhol R, Stazyk G, Morin RD, Zhao Y, Hirst M, Schein JE, Horsman DE, Connors JM, Gascoyne RD, Marra MA, Jones SJM: De novo transcriptome assembly with ABySS. Bioinformatics. 2009, 25: 2872-2877.
Li R, Zhu H, Ruan J, Qian W, Fang X, Shi Z, Li Y, Li S, Shan G, Kristiansen K, Li S, Yang H, Wang J, Wang J: De novo assembly of human genomes with massively parallel short read sequencing. Genome Res. 2010, 20: 265-272.
Sayers EW, Barrett T, Benson DA, Bolton E, Bryant SH, Canese K, Chetvernin V, Church DM, DiCuccio M, Federhen S, Feolo M, Geer LY, Helmberg W, Kapustin Y, Landsman D, Lipman DJ, Lu Z, Madden TL, Madej T, Maglott DR, Marchler-Bauer A, Miller V, Mizrachi I, Ostell J, Panchenko A, Pruitt KD, Schuler GD, Sequeira E, Sherry ST, Shumway M, Sirotkin K, Slotta D, Souvorov A, Starchenko G, Tatusova TA, Wagner L, Wang Y, Wilbur WJ, Yaschenko E, Ye J: Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2010, 38: 5-16.
Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L: Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010, 28: 511-515.
Coon SL, Munson PJ, Cherukuri PF, Sugden D, Rath MF, Møller M, Clokie SJH, Fu C, Olanich ME, Rangel Z, Werner T, NISC Comparative Sequencing Program, Mullikin JC, Klein DC: Circadian changes in long noncoding RNAs in the pineal gland. Proc Natl Acad Sci USA. 2012, 109: 13319-13324.
Heap GA, Yang JHM, Downes K, Healy BC, Hunt KA, Bockett N, Franke L, Dubois PC, Mein CA, Dobson RJ, Albert TJ, Rodesch MJ, Clayton DG, Todd JA, van Heel DA, Plagnol V: Genome-wide analysis of allelic expression imbalance in human primary cells by high-throughput transcriptome resequencing. Hum Mol Genet. 2010, 19: 122-134.
Wu TD, Watanabe CK: GMAP: a genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics. 2005, 21: 1859-1875.
Chu HT, Hsiao WWL, Chen JC, Yeh TJ, Tsai MH, Lin H, Liu YW, Lee SA, Chen CC, Tsao TTH, Kao CY: EBARDenovo: highly accurate de novo assembly of RNA-Seq with efficient chimera-detection. Bioinformatics. 2013, 29: 1004-1010.
Acknowledgements
We thank the anonymous reviewers for many invaluable suggestions that significantly improve the paper. Computations were done on the Whole Systems Genomics Initiative Cluster and the Brazos Cluster at Texas A&M University. AMT was supported by startup funds from the College of Agriculture and Life Sciences at Texas A&M University and Texas Agrilife Research. This work was supported in part by NIJ grant 2012-DN-BX-K024. Points of view in this document are those of the authors and do not necessarily represent the official position or policies of the U.S. Department of Justice.
Declarations
Publication costs for this work were funded by the Open Access to Knowledge (OAK) Fund at the Texas A&M University Libraries.
This article has been published as part of BMC Genomics Volume 16 Supplement 11, 2015: Selected articles from the Fourth IEEE International Conference on Computational Advances in Bio and medical Sciences (ICCABS 2014): Genomics. The full contents of the supplement are available online at http://www.biomedcentral.com/bmcgenomics/supplements/16/S11.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
AMT and S-HS designed the computational work. SF developed the algorithm and software, did the computational experiments and analyzed the data. All authors wrote, read and approved the final manuscript.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Fu, S., Tarone, A.M. & Sze, SH. Heuristic pairwise alignment of de Bruijn graphs to facilitate simultaneous transcript discovery in related organisms from RNA-Seq data. BMC Genomics 16 (Suppl 11), S5 (2015). https://doi.org/10.1186/1471-2164-16-S11-S5
Published:
DOI: https://doi.org/10.1186/1471-2164-16-S11-S5