
Abstract
Background
In the majority of cases, trigeminal neuralgia (TN) is a unilateral condition with ultra-short stabbing pain located along one or more branches of the trigeminal nerve. Although prophylactic pharmacological treatment is first choise, considering of insufficient effect or unacceptable side effects, neurosurgical treatment or lesion treatment should be considered. In addition to all these procedures mentioned above, one approach has been based on local intradermal and/or submucosal injections of Botulinum Toxin Type A (BTX-A).
Methods
We conducted a randomized, double-blind, placebo-controlled since November 2012, and adopted local multi-point injection in 84 cases of classical TN with different doses of BTX-A. Eighty four patients were randomized into following groups: placebo (n = 28); BTX-A 25U (n = 27); BTX-A 75U (n = 29). Follow-up visits were conducted every week after the injection, and the overall duration of the study for each patient were 8 weeks to observe the pain severity, efficacy and adverse reactions at endpoint.
Results
The visual analogue scale (VAS) scores of 25U and 75U groups reduced significantly compared to placebo as early as week 1, and sustained until week 8 throughout the study. There was no significant difference in VAS between 25U and 75U groups throughout the study. The response rates of 25U group (70.4%) and 75U group (86.2%) were significantly higher than placebo group (32.1%) at week 8, and there was no significant difference between 25U and 75U groups. Evaluation of the Patient Global Impression of Change (PGIC) demonstrated that 66.7% (25U group) and 75.9% (75U group) of the patients reported that their pain symptoms were ‘much improved’ or ‘very much improved’ versus 32.1% of the placebo group, and there was also no significant difference between 25U and 75U groups. All adverse reactions were graded as mild or moderate.
Conclusions
BTX-A injection in TN is safe and efficient. It is a useful treatment for refractory TN. Lower dose (25U) and high dose (75U) were similar in efficacy in short-term.
Similar content being viewed by others

Background
In the majority of cases, trigeminal neuralgia (TN) is a unilateral condition with ultra-short stabbing pain located along one or more branches of the trigeminal nerve. The pain is often triggered by stimuli such as chewing, washing of the face, speech and tooth-brushing [1]. It usually affects one side of the face, and occurs more often in the elderly and has a profound effect on quality of life. According to epidemiological studies, approximately 4–28.9/100,000 persons worldwide experience TN [2–4].
Trigeminal neuralgia is primarily caused by compression of the nerve near its origin at the pons. The treatment consists primarily of prophylactic pharmacological treatment with anti-epileptics. In case of insufficient effect or unacceptable side effects, neurosurgical treatment using microvascular decompression or lesion treatment should be considered [5]. In addition to all these procedures mentioned above, one approach has been based on local intradermal and/or submucosal injections of Botulinum Toxin Type A (BTX-A) [6].
BTX-A is one of the most potent neurotoxins from natural toxins and synthetic toxicant known, which blocks neuromuscular transmission through decreased acetylcholine release, resulting in muscle relaxation. It is also used in treating blepharospam and hemifacial spasm [7]. Additionally it is reported to be effective in the treatment of chronic migraine and may be a promising choice in headache treatment [8–11]. In some reports, BTX-A has been shown to be a new possible choice of treatment for TN, and may be an efficient, safe and novel strategy for TN treatment [6, 12–19]. This therapy is simple and can be performed as an outpatient procedure without anesthesia, and the toxin does not cause skin sensory loss or dysesthesia. However, there is no large randomized controlled trial and relevant data except our study in 2012 [6], and the dose has not been standardised. The aim of the present study was to define an effective and safe dose of BTX-A for the treatment.
For this purpose, we conducted a randomized, double-blind, placebo-controlled since November 2012, and adopted local multi-point injection in 84 cases of classical TN with different doses of BTX-A. The primary objective was to further clarify the efficacy, safety and tolerability of intradermal and/or submucosal administration of different doses of BTX-A in patients with TN.
Methods
Study design
The study was a randomized, double-blind, placebo-controlled and parallel-group clinical study of BTX-A in the management of patients with TN. The overall duration of the study for each patient was 9 weeks, including a 1-week observation period to establish baseline pain symptoms, followed by a 8-week placebo run-in period. Follow-up visits were conducted every week after the injection.
The trial was approved by the local ethics committee of the First Affiliated Hospital of Zhengzhou University. The goal, procedure and possible adverse reactions of the study were explained to each patient before the study, and they were informed of the possibility of transient attendant risk of weakness and disfigurement produced by localized administration of BTX-A. Written informed consent was obtained from each patient. Patients were free to discontinue the trial at any time during the double-blind period.
The Patients were randomized into 3 groups: placebo groups received sterile isotonic saline injection and treatment group received 25U/1 mL (25U group) and 75U/1 mL (75U group) of BTX-A in the dermatome and/or mucosa (if the oral mucosa was involved) where pain was experienced. Patients were assigned to a computer-generated randomisation list, and randomization data were kept strictly confidential, accessible only by authorized persons. The data was locked and verified and unblended only when the trial was completed.
Study participants
All included patients were recruited from the outpatients and inpatients who attended the neurology department of the First Affiliated Hospital of Zhengzhou University from Nov 2012 to Aug 2013. Each patient underwent MRI or CT to rule out the presence of structural pathology. All patients underwent detailed physical examination and data registration, including the history of present illness, past history, the auxiliary examination results, nervous system examination. Complete blood count (CBC), ECG, Liver function tests, renal function tests and other diagnostic tests should be done before the trial, to exclude coagulopathy, severe heart, liver, kidney and other organ dysfunction. According to the current version of the International Classification of Headache Disorders (ICHD-II) [20], and all patients were diagnosed with classical TN.
At baseline, patients usually received medications (e.g. carbamazepine, gabapentin, or opioids) to alleviate their pain. These medications remained unchanged during the course of the study. There were no new analgesic therapies at any time during the baseline or placebo run-in period.
Participants(>18 years), who were failure of recent treatment for TN at baseline (pain intensity mean score ≥ 4; mean attack frequency ≥ 4 per day; course > 4 months), were required to be in good general health, and understanding the possible complications such as transient facial weakness.
Exclusion criteria included: any disease that might put patients at increased risk if exposed to BTX-A (e.g. myasthenia gravis, motor neuron disease or Lambert-Eaton syndrome); an infection or skin problem at any of the injection sites; use of drug that damage the neuromuscular junction within 7 days before study entry (e.g. quinine, aminoglycosides or penicillamine); significant unstable medical disease or a history of a significant mental disorder; history of substance dependence or abuse. Furthermore, women who were pregnant, nursing, or planning a pregnancy during the study, or who were unable or unwilling to use a reliable form of contraception during the study were also excluded.
Patients who continued to meet all inclusion/exclusion criteria at the end of the baseline phase were randomized and proceeded to the double-blind period.
Treatment
All of the treatment was administered in the treatment room at the Department of Neurology, the First Affiliated Hospital of Zhengzhou University, which is equipped with all the necessary facilities to ensure the safety of possible severe reactions or emergencies.
BTX-A (100U of Clostridium botulinum type A neurotoxin complex, 5 mg gelatin, 25 mg dextran, and 25 mg saccharose) was obtained from Lanzhou Biological Products Institute, China. Patients lay still in a supine position on a bed during the injections.
Patients were randomly assigned to receive treatment with placebo or total doses of 25Uor 75U of botulinum toxin type A (placebo group, 25U group, or 75U group) in a double blind prospective study design. The way to blind the study was to supply the study medication for each patient in three individual vials. Each vial contained either active botulinum toxin type A (25U or 75U) or matching placebo. All three vials were identical in appearance and were reconstituted with 1 ml saline solution (0.9%). For treatment 1 ml was drawn from vials, and the injections were administered intradermally and/or submucosally using a 1 mL syringe with a 0.45 × 16 mm needle.
The BTX-A or the same volume of isotonic saline were applied at 20 points, (0.05 ml) per point, between the epidermis and dermis of the skin where pain was experienced according to the patient’s description. The injections were conducted submucosally in the oral mucosa if the pain involved the oral mucosa. During the procedure, injection in deeper structures such as the muscles was avoided to prevent unwanted effects.
Efficacy and safety measures
On awakening each morning, patients were required to provide a verifiable diary of their pain symptoms covering provoking factors, frequency of TN attacks, severity of pain (according to an 11-point visual analogue scale, VAS) and adverse reaction experienced during the previous 24 hours from baseline to endpoint. During the baseline phase, patient demographics, gender, and age were also recorded.
The following items were assessed during each follow up visit:
-
Assessment of pain severity and pain attack frequency from baseline to endpoint.
-
Global assessment of the overall response to treatment on the basis of the Patient Global Impression of Change (PGIC) scale. The PGIC is a self-evaluation of the patient’s overall change since the start of the study according to a seven-point scale (1, very much improved; 2, much improved; 3, minimally improved; 4, no change; 5, minimally worse; 6, much worse; 7, very much worse).
-
The proportion of responders, defined as patient with ≥ 50% reduction in mean pain score from baseline to endpoint, was also used to assess the efficacy of the treatment.
-
Safety was measured as the occurrence of adverse events and recorded with information including the date of onset, severity, duration, frequency, treatment required (if any), relationship to BTX-A treatment, and outcome.
Statistical analysis
All analyses were performed on the intent-to-treat population and all statistical testing was two-sided. The quantitative data was assessed using the median value. Between groups comparisons were evaluated by means of the Kruskall-Wallis analysis to compare the age, duration of diseases, and pain intensity. Chi-square test was performed to assess the differences of the gender, PGIC distribution, and the proportion of responders among 3 groups. The SPSS 20.0 software package was used for statistical evaluation; p < 0.05 was considered statistically significant.
Results
Patient disposition
The recruitment period was between November 2012 and August 2013, with an 8-week follow-up period after the last patient was enrolled. A total of 95 patients were screened, and 84 were randomized into the following groups: placebo (n = 28); BTX-A 25U (n = 27); BTX-A 75U (n = 29). Four patients (one from 25U group, one from 75U group and two from placebo group) withdrew from the study owing to a lack of efficacy, leaving data on 80 patients for the final analysis. Overall, the groups were well matched for age, sex, duration of symptoms, pain severity, and pain attack frequency, and there were no significant differences at baseline (Table 1).
Pain severity-VAS score
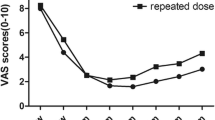
At baseline, each group was well matched for severity of their VAS scores (6.96 ± 1.97, 6.24 ± 2.13, and 7.18 ± 2.21 for placebo, 25U, and 75U groups respectively). After injections, The VAS scores of 25U and 75U groups reduced significantly compared to placebo as early as week 1, and sustained until week 8 throughout the study (*P < 0.017, **P < 0.017) (Figure 1). There was no significant difference between 25U and 75U groups throughout the study (P > 0.05) (Figure 1, Table 2).

Weekly mean scores as measured by VAS. At baseline, each group was well matched for severity of their VAS scores. After injections, The VAS scores of 25U and 75U groups reduced significantly compared to placebo as early as week 1, and sustained until week 8 throughout the study (*P < 0.017, **P < 0.017) . There was no significant difference between 25U and 75U groups throughout the study (P > 0.05).
Efficacy results
The proportion of responders, defined as patient with ≥ 50% reduction in mean pain score from baseline to endpoint, was used to assess the efficacy. The response rates of 25U group (70.4%) and 75U group (86.2%) were significantly higher than placebo group (32.1%) at week 8(*P < 0.017). There was no significant difference between 25U and 75U groups (P > 0.05) (Figure 2).

The proportion of responders. The response rates of 25U group (70.4%) and 75U group (86.2%) were significantly higher than placebo group (32.1%) at week 8 (*P < 0.017). There was no significant difference between 25U and 75U groups (P > 0.05).
Evaluation of the PGIC demonstrated that 66. 7% of the patients in 25U group reported that their pain symptoms were ‘much improved’ or ‘very much improved’ versus 32.1% of the placebo group (*P < 0.017). In addition, there was a much higher proportion in 75U group (*P < 0.017) (75.9%), but there was no significant difference between 25U and 75U groups (P > 0.05) (Figure 3).

Patient Global Impression of Change (PGIC) results. Evaluation of the PGIC demonstrated that 66.7% of the patients in 25U group reported that their pain symptoms were ‘much improved’ or ‘very much improved’ versus 32.1% of the placebo group (*P < 0.017). In addition, there was a much higher proportion in 75U group (75.9%) compare to the placebo group (*P < 0.017), but there was no significant difference between 25U and 75U groups (P > 0.05).
Safety and adverse reaction
Three patients (two in 25U group, one in 75U group) experienced short term facial asymmetry in the injection area during dynamic movement, which disappeared within 6 weeks. Transient edema in the injection area was observed in two patients (both in 25U group), and disappeared within 5 days. All adverse reactions were graded as mild or moderate, and none led to discontinuation of study.
Discussion and conclusions
The International Association for the Study of Pain defines trigeminal neuralgia (TN) as “sudden, usually unilateral, severe, brief, stabbing, recurrent episodes of pain in the distribution of one or more branches of the trigeminal nerve”. Medical management is the first choice, including Carbamazepine, Oxcarbazepine, Bacloten, Lamotrigine, Gabapentin and ropivacaine. When drug treatments fail due to pharmacoresistance or intolerability of side effects, surgical treatments need to be considered. Surgical treatments include peripheral techniques (such as cryosurgery, neurectomy, laser, acupuncture, thermocoagulation, injections of streptomycin, alcohol and phenol), Gasserian ganglion radiofrequency thermocoagulation, glycerol, balloon compression, Gamma knife and microvascular decompression. All the surgical treatments can result in nerve damage except microvascular decompression, which limits the application of these techniques. We faced with the challenge when some elder patients cannot tolerant surgical treatment.
In 2002, analgesic effect of BTX-A in TN was reported [14]. Some open studies [17] and a randomized controlled trial (RCT) [6] showed that BTX-A could significantly relieve pain in TN. The subcutaneously injection usually was adopted in many points[9, 21],or muscle injection in region of the zygomatic arch [18] or in masseter [19]. But the dose has not been standardised. Based on our previous experience, the highest dose is 170U in a woman with the third branch injection, and the pain was completely relieved for 2 years [6, 13]. In the present study, the VAS scale between 25U and 75U group were no significant difference from the first week to eighth week. However, the high dose led to facial weakness with high risk in our study.
Pain relief was observed in several minutes after injection [7], usually several days or as long as 20 days after injection [13]. In this study, the VAS scale was different between BTX-A and control group in the first week. The pain was relieved rapidly. The effect of BTX-A on TN usually lasted for several months [18, 22]. Based on our previous experience, it may last for from 8 weeks to 2 years, but it was still unknown whether effect duration was associated with dose [6, 13]. It was felt that the duration of pain relief sometimes was longer than the period of muscle weakness in our study. Because of this, it was considered that the antinociceptive effect of BTX-A may not associate with inhibiting the release of acetylcholine from the neuromuscular junction.
More patients in 25U and 75U group presented that they were “much improved” or “very much improved” than control in PGIC, but it was similar between 25U and 75U group at the endpoint. The effective rate was not significant different between 25U and 75U group, but they were different with control group. Complications induced by BTX-A were mild, including regional weakness, facial asymmetry during voluntary movement and edema in injection sites. Regional weakness and edema were transient and usually recovered within several weeks, and there were no systemic complications. Recent studies showed that BTX-A may reduce neurogenic inflammation by inhibiting the release of glutamate, substance P and calcitonin gene-related peptide (CGRP) in sensory terminal [23]. BTX-A may also reduce the release of substance P in dorsal root ganglia [24]. In other animal studies, BTX-A inhibits CGRP release in trigeminal sensory neurons of brainstem [25]. Diminishing peripheral sensory nerve chemical stimuli may contribute to decreased peripheral and central nervous system responsiveness [26]. More research is needed to explore the mechanism of action of BTX-A on pain in TN. BTX-A or its degradation product could achieve analgesic effect through indirect effects on spinal cord by reducing the transmission of the sympathetic nervous system or diminishing the inhibition of inhibitory interneuron renshaw cell [27]. Other Study of BTX-A treatment of migraine indicated that the analgesic effect of BTX-A not only associated with inhibiting the release of acetylcholine, but also associated with blocking the parasympathetic nervous system [28].
Taken together, our study suggested that BTX-A injection in TN was safe and efficient. It is a useful treatment for refractory TN. Lower dose (25U) and high dose (75U) were similar in efficacy in short-term. But we cannot claim that they were same in the long term. More studies are needed to clarify it in the following study.
Research highlights
-
1.
BTX-A injection in TN is safe and efficient.
-
2.
BTX-A is a useful treatment for refractory TN.
-
3.
Lower dose (25U) and high dose (75U) were similar in efficacy in short-term.
References
Headache Classification Subcommittee of the International Headache Society: The International Classification of Headache Disorders: 2nd edition. Cephalalgia 2004, 24(suppl 1):9–160.
Campos WK, Linhares MN: A prospective study of 39 patients with trigeminal neuralgia treated with percutaneous balloon compression. Arq Neuropsiquiatr 2011, 69: 221–226. 10.1590/S0004-282X2011000200016
Turp JC, Gobetti JP: Trigeminal neuralgia--an update. Compend Contin Educ Dent 2000, 21: 279–282. 284, 287–8 passim;quiz 292
Zakrzewska JM, McMillan R: Trigeminal neuralgia: the diagnosis and management of this excruciating and poorly understood facial pain. Postgrad Med J 2011, 87: 410–416. 10.1136/pgmj.2009.080473
Bendtsen L, Birk S, Kasch H, Aegidius K, Sorensen PS, Thomsen LL, Poulsen L, Rasmussen MJ, Kruuse C, Jensen R, Danish Headache Society: Reference programme: diagnosis and treatment of headache disorders and facial pain. Danish Headache Society, 2nd Edition. J Headache Pain 2012, 13(Suppl 1):S1-S29.
Wu CJ, Lian YJ, Zheng YK: Botulinum toxin type A for the treatment of trigeminal neuralgia: results from a randomized, double-blind, placebo-controlled trial. Cephalalgia 2012, 32: 443–450. 10.1177/0333102412441721
Wu CJ, Shen JH, Chen Y, Lian YJ: Comparison of two different formulations of botulinum toxin A for the treatment of blepharospasm and hemifacial spasm. Turk Neurosurg 2011, 21: 625–629.
Aurora SK, Dodick DW, Turkel CC, DeGryse RE, Silberstein SD, Lipton RB: OnabotulinumtoxinA for treatment of chronic migraine: results from the double-blind, randomized, placebo-controlled phase of the PREEMPT 1 trial. Cephalalgia 2010, 30: 793–803. 10.1177/0333102410364676
Diener HC, Dodick DW, Aurora SK, Turkel CC, DeGryse RE, Lipton RB: OnabotulinumtoxinA for treatment of chronic migraine: results from the double-blind, randomized, placebo-controlled phase of the PREEMPT 2 trial. Cephalalgia 2010, 30: 804–814. 10.1177/0333102410364677
Schoenen J: Botulinum toxin in headache treatment: finally a promising path. Cephalalgia 2010, 30: 771–773. 10.1177/0333102410373154
Torgovnick J: OnabotulinumtoxinA for the treatment of chronic migraine not quite there. Cephalalgia 2011, 31: 377. author reply 378–9 10.1177/0333102410388529
Allam N, Brasil-Neto JP, Brown G, Tomaz C: Injections of botulinum toxin type a produce pain alleviation in intractable trigeminal neuralgia. Clin J Pain 2005, 21: 182–184. 10.1097/00002508-200503000-00010
Shen JH, Lian YJ, Zheng YK: Effect of botulinum toxin type a on clssic trigeminal neuralgia. Chin J Rehab Med 2011, 26: 483–484.
Micheli F, Scorticati MC, Raina G: Beneficial effects of botulinum toxin type a for patients with painful tic convulsif. Clin Neuropharmacol 2002, 25: 260–262. 10.1097/00002826-200209000-00006
Ngeow WC, Nair R: Injection of botulinum toxin type A (BOTOX) into trigger zone of trigeminal neuralgia as a means to control pain. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 2010, 109: e47-e50.
Piovesan EJ, Leite LS, Teive HG, Kowacs PA, Mulinari RA, Radunz V: Botulinum toxin type-A effect as a preemptive treatment in a model of acute trigeminal pain: a pre-clinical double-blind and placebo-controlled study. Arq Neuropsiquiatr 2011, 69: 56–63. 10.1590/S0004-282X2011000100012
Piovesan EJ, Teive HG, Kowacs PA, Della CMV, Werneck LC, Silberstein SD: An open study of botulinum-A toxin treatment of trigeminal neuralgia. Neurology 2005, 65: 1306–1308. 10.1212/01.wnl.0000180940.98815.74
Turk U, Ilhan S, Alp R, Sur H: Botulinum toxin and intractable trigeminal neuralgia. Clin Neuropharmacol 2005, 28: 161–162. 10.1097/01.wnf.0000172497.24770.b0
Zuniga C, Diaz S, Piedimonte F, Micheli F: Beneficial effects of botulinum toxin type A in trigeminal neuralgia. Arq Neuropsiquiatr 2008, 66: 500–503. 10.1590/S0004-282X2008000400012
Olesen J, Steiner TJ: The International classification of headache disorders, 2nd edn (ICDH-II). J Neurol Neurosurg Psychiatry 2004, 75: 808–811. 10.1136/jnnp.2003.031286
Borodic GE, Acquadro MA: The use of botulinum toxin for the treatment of chronic facial pain. J Pain 2002, 3: 21–27. 10.1054/jpai.2002.27142
Bohluli B, Motamedi MH, Bagheri SC, Bayat M, Lassemi E, Navi F: Use of botulinum toxin A for drug-refractory trigeminal neuralgia: preliminary report. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 2011, 111: 47–50. 10.1016/j.tripleo.2010.04.043
Aoki KR: Review of a proposed mechanism for the antinociceptive action of botulinum toxin type A. Neurotoxicology 2005, 26: 785–793. 10.1016/j.neuro.2005.01.017
Ishikawa H, Mitsui Y, Yoshitomi T, Mashimo K, Aoki S, Mukuno K: Presynaptic effects of botulinum toxin type A on the neuronally evoked response of albino and pigmented rabbit iris sphincter and dilator muscles. Jpn J Ophthalmol 2000, 44: 106–109. 10.1016/S0021-5155(99)00197-5
Meng J, Ovsepian SV, Wang J, Pickering M, Sasse A, Aoki KR: Activation of TRPV1 mediates calcitonin gene-related peptide release, which excites trigeminal sensory neurons and is attenuated by a retargeted botulinum boxin with anti-nociceptive potential. J Neurosic 2009, 29: 4981–4992. 10.1523/JNEUROSCI.5490-08.2009
Woolf CJ, Costigan M: Transcriptional and posttranslational plasticity and the generation of inflammatory pain. Proc Natl Acad Sci 1999, 96: 7723–7730. 10.1073/pnas.96.14.7723
Guyer BM: Mechanism of botulinum toxin in the relief of chronic pain. Curr Rev Pain 1999, 3: 427–431. 10.1007/s11916-999-0069-y
Binder WJ, Brin MF, Blitzer A: Botulinum toxin type A (BOTOX) for treatment of migraine. Dis Mon 2002, 48: 323–335. 10.1053/mda.2002.24423
Acknowledgement
The study was supported by the National Natural Science Foundation of China (grant number 81371438), the Provincial Natural Science Research Foundation of the Education Department of HeNan Province (2011A320038) and a project of the HeNan Health Department (201203006).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interest
The authors declare that they have no competing interest.
Authors' contribution
Y-JL - Study concept and design. H-FZ - carried out the treatment and studies, drafted the manuscript Y-QM - acquisition of data. C-HH - participated in the sequence alignment, analysis and interpretation. DYC - revise the manuscript. N-CX, C-JW - study supervision. All authors read and approved the final manuscript.
Haifeng Zhang, Yunqing Ma contributed equally to this work.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made.
The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
To view a copy of this licence, visit https://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, H., Lian, Y., Ma, Y. et al. Two doses of botulinum toxin type A for the treatment of trigeminal neuralgia: observation of therapeutic effect from a randomized, double-blind, placebo-controlled trial. J Headache Pain 15, 65 (2014). https://doi.org/10.1186/1129-2377-15-65
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1129-2377-15-65