
Abstract
Plasmodiophora brassicae is an important biotrophic eukaryotic plant pathogen and a member of the rhizarian protists. This biotrophic pathogen causes clubroot in cruciferous plants via novel intracellular mechanisms that are markedly different from those of other biotrophic organisms. To date, genomes from six single spore isolates of P. brassicae have been sequenced. An accurate description of the evolutionary status of this biotrophic protist, however, remains lacking. Here, we determined the draft genome of the P. brassicae ZJ-1 strain. A total of 10,951 protein-coding genes were identified from a 24.1 Mb genome sequence. We applied a comparative genomics approach to prove the Rhizaria supergroup is an independent branch in the eukaryotic evolutionary tree. We also found that the GPCR signaling pathway, the versatile signal transduction to multiple intracellular signaling cascades in response to extracellular signals in eukaryotes, is significantly enriched in P. brassicae-expanded and P. brassicae-specific gene sets. Additionally, treatment with a GPCR inhibitor relieved the symptoms of clubroot and significantly suppressed the development of plasmodia. Our findings suggest that GPCR signal transduction pathways play important roles in the growth, development, and pathogenicity of P. brassicae.
Similar content being viewed by others
Background
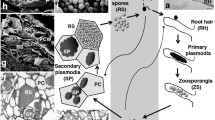
Plasmodiophora brassicae is a member of the protist supergroup Rhizaria (Burki et al. 2010), a group of biotrophic eukaryotic plant pathogen that causes clubroot in cruciferous plants. Clubroot accounts for a 10%–15% loss in cruciferous crop production and poses a major threat to the production of several cruciferous crops (Dixon 2009; Hwang et al. 2012). Two distinct infection stages, a primary phase in root hairs followed by a secondary phase in root cortex and adjacent tissues, were defined during the disease cycle of P. brassicae. The primary phase only lasts for a few days after P. brassicae invasion of the plant root hairs via primary zoospores released from the germinated resting spores. P. brassicae forms primary plasmodia and then secondary zoospores in the root hairs. The secondary zoospores are ultimately released by broken root hairs and then trigger the secondary phase of infection and the development of intracellular secondary plasmodia in root cortical cells (Ingram and Tommerup 1972; Mcdonald et al. 2014). The most characteristic symptom of clubroot is the formation of large galls on the plant roots that result from alterations to plant cell hypertrophy and hyperplasia during cortical infection. Although infected host cells are rapidly filled with cleaving secondary plasmodia or resting spores, there is no apparent host defense response and the plant cells themselves remain alive until late in the infection process (Kageyama and Asano 2009; Schwelm et al. 2015).
There are six to eight major branches in the eukaryotic tree of life (Adl et al. 2012). One of these, the Rhizaria supergroup, encompasses the majority of skeleton-building protists (Burki et al. 2010). Unlike the other branches, however, where the entire genomes have been sequenced for numerous species to allow for comparative analysis, only a few genomes of the Rhizaria branch are currently available. These include genomes for Bigelowiella natans (Curtis et al. 2012), Reticulomyxa filosa (Glöckner et al. 2014), P. brassicae (Schwelm et al. 2015; Rolfe et al. 2016), and Spongospora subterranean (Ciaghi et al. 2018). Given this, it is clear that the Rhizaria supergroup is the least studied of the major eukaryotic branches (Campo et al. 2014). A previous study found a clade grouping of Foraminifera and Acantharea in the evolution of Rhizaria using evidence from phylogenomic analyses of ESTs of three protist groups (Burki et al. 2010). Comparisons among the various genomes of the Rhizaria supergroup have been conducted for P. brassicae (strain e3), S. subterranean, and the non-pathogenic Rhizarian B. natans and R. filosa (Schwelm et al. 2015). The relationship between the Rhizaria supergroup and other eukaryotic species, however, has not been fully investigated and remains poorly understood.
Unlike other obligate biotrophic pathogens with diverse infection structures, P. brassicae is an intracellular parasite that spends most of its lifecycle in the intracellular environment of the host, similar to the lifecycle of endophytic microbes (Kageyama and Asano 2009). An effective molecular mechanism is therefore required for P. brassicae to adapt to the intracellular environment of its plant hosts. To date, draft genome sequences from single spore isolates of Pbe3 (Europe strain), Pb3, and Pb6 (Canadian strain) of P. brassicae have been obtained, and the genomes of three other pathotypes (Pb2, Pb5, and Pb8) have been resequenced (Schwelm et al. 2015; Rolfe et al. 2016). Although the genomes of these strains of P. brassicae have been sequenced and analyzed, the evolutionary status of Plasmodiophora and their relatives have not yet been investigated in detail.
The ability to sense and transduce extracellular signals into intracellular responses using a sophisticated molecular signaling system is crucial for living organisms. G-protein-coupled receptors (GPCRs), characterized by a seven transmembrane domain structure, are involved in a wide variety of physiological processes. Signals from a diverse range of extracellular signaling molecules, including hormones, neurotransmitters, ions, photons, and odorants, can be relayed by GPCRs to intracellular downstream signaling pathways to ultimately elicit cellular responses (Hilger et al. 2018). Based on their sequence homology and functional similarity, the superfamily of GPCRs can be generally divided into 6 classes (A, B, C, D, E, and F) (Qiu et al. 2009; Alexander et al. 2017). The signal transduction of GPCRs is dependent upon receptor-mediated activation of heterotrimeric G proteins consisting of three subunits, Gα, Gβ and Gγ (Kamp et al. 2016). As one of the main signaling pathways in eukaryotes, the signaling system of GPCRs is involved in numerous processes including developmental control, cell growth, migration, density sensing, and neurotransmission (Rosenbaum et al. 2009; de Mendoza et al. 2014). As important central sensors that activate eukaryotic signaling, GPCRs are the primary targets of human drugs (van den Hoogen et al. 2018). Despite this, the specific roles of these receptors and their downstream signaling pathways in protists remain poorly understood. In the current study, we conducted a comparative genomics analysis to determine the role of the GPCR signaling pathway in P. brassicae.
Results
Genome sequencing, assembly, and gene annotation of ZJ-1
For genomic analyses, we employed a whole-genome shotgun (WGS) sequencing strategy using the Illumina Genome Analyzer (GA) sequencing technology for genome sequencing. The draft genome sequence was assembled using high-quality sequenced clean reads. The final assembly was 24.1 Mb, and the N50 size of the assembly scaffolds was 510.27 kb. The overall GC-content of the genome was 59.4% (Table 1). Both the size and GC-content of the assembled ZJ-1 genome were similar to those of P. brassicae isolates Pbe3, Pb3, and Pb6 (Schwelm et al. 2015; Rolfe et al. 2016). Based on the conservative gene database for eukaryotes, we used CEGMA software to predict the core genes of P. brassicae. The assembly captured 94.35% of the total 248 ultra-conserved CEGs (core eukaryotic genes), ensuring the reliability of the genome structures (Table 1). A combination of gene prediction methods was used to predict gene models in the P. brassicae genome, which resulted in the identification of 10,951 genes with an average length of 1744 bp, a coding sequence size of 1364 bp per gene, and a total length of genes and CDS of 19,101.9 kb and 14,942.2 kb, respectively, comprising 76.93% and 60.17% of the assembly genome (Table 1). Evidence-based annotations using transcriptome sequencing (RNA-seq) data ensured high integrity and continuity of genome assembly. After discarding low-quality raw reads, we obtained 210 million clean reads from each of the three samples, and we then aligned these reads against the reference genome (Additional file 1: Table S1). The results of ZJ-1 genome assembly and gene annotation are consistent with those of Pbe3 and Pb3, where the total number of predicted genes from Pbe3 and Pb3 was 9730 and 10,851, respectively (Schwelm et al. 2015; Rolfe et al. 2016).
Low rate of repetitive regions and extensive rearrangements in the P. brassicae genome
The overall features of the ZJ-1 genome reported here, including genome size and GC-content, are similar to those of the recently published genomes of P. brassicae strains e3 and Pb3 (Table 2). The genomes among members of the representative Stramenopiles-Alveolata-Rhizaria (SAR) supergroup were compared. The P. brassicae genome (~ 24 Mb) is smaller than that of the free-living Rhizaria B. natans (~ 100 Mb), R. filosa (~ 320 Mb), or Stramenopiles Phytophthora infestans (~ 240 Mb), and is similar to the Alveolata P. falciparum genome (~ 22.8 Mb). The GC-content (58.5%) and gene density (genes per Mb) of P. brassicae are significantly higher than those of the other species within the SAR supergroup (Table 2). Additionally, the genome of P. brassicae shows a higher gene density compared to that of other representative plant pathogens (Additional file 2: Table S2). We used a combination of de novo repeat identification and homology analysis to identify repetitive DNA. The P. brassicae genome includes 1.917% repeated transposable elements (TEs), 0.086% simple repeats, and 0.004% satellite sequences (Table 3). Characterization of the observed transposable elements demonstrates that the majority of the repetitive sequences exist as retrotransposons (21.23%), while 3.64% of the transposable elements were DNA transposons (Table 3). The reported repeat content for Pb3 and Pb6 was less than 2% (Rolfe et al. 2016), demonstrating that P. brassicae species possess a small genome with few repetitive elements.
A comparative analysis of the entire genome reveals a high level of synteny, with 104 LCBs (locally collinear blocks) between the single spore isolates of ZJ-1 and Pbe3. Significant genome rearrangements (transposition and inversion) were identified (Fig. 1a). A total of 139,388 SNP sites were found upon comparison of the two sequenced genomes. These findings indicate that there was most likely a period of extensive rearrangement caused by recombination (Fig. 1b, c).

Whole genome comparison and analysis between Plasmodiophora brassicae strain ZJ-1 and Pbe3. a Simple global view of syntonic alignments from the scaffolds larger than 10 kb. The minimum weight parameter is set by default. The matching regions (minimum set of local collinear blocks) represent as connected blocks. Each sequence of identically colored blocks represents a collinear set of matching regions. One connecting line is drawn per collinear block. Blocks below a genome’s center line are inverted relative to the reference sequence. b Whole-genome dot-plot comparison between P. brassicae strain ZJ-1 and Pbe3. c The statistic distribution of SNP rates from different scaffolds between the two strains
Comparative genomics reveals the unique evolutionary status of Rhizaria, including P. brassicae, in the eukaryotic evolutionary tree
The exact status of P. brassicae within the eukaryotic evolutionary tree has yet to be fully elucidated. To better understand the phylogenetic relationship of P. brassicae with other eukaryotic species, the genomes of 18 eukaryotic model species were analyzed using OrthoMCL to build gene families. The maximum likelihood phylogenetic tree derived from the concatenated sequence alignment of 90 single-copy orthologs shared by P. brassicae and the 18 other model species showed that P. brassicae has a closer evolutionary relationship with other Rhizaria species (R. filosa and B. natans) as opposed to the other model species. Our phylogenomics analysis revealed that Rhizaria supergroup forms an independent branch in the phylogenetic tree, and our findings demonstrated that P. brassicae shares common ancestors with the other sequenced Rhizaria species (Fig. 2a). This is the first evidence obtained from comparative genome-wide analysis to demonstrate the important evolutionary status of the Rhizaria supergroup. A better understanding of P. brassicae may help us improve our understanding of eukaryotic evolution as a whole, as this species is a representative of the most poorly understood branch in the eukaryotic evolutionary tree.

Phylogeny and ortholog distribution in Plasmodiophora brassicae and other species. a The phylogeny was constructed by the maximum likelihood method from a concatenated alignment of 90 single-copy orthologs conserved across species. The scale bar of branch length represents 0.2 amino acid-substitutions/site, and the bootstrap values are indicated on nodes (the value of 1 was omitted). Different subphylum branches are highlighted as follows: Rhizaria, red; Chromalveolata, dark blue; plants, light green; amoebozoa, light purple; animals, dark orange; fungi, light blue. b The colored bars represent orthologous proteins identified using OrthoMCL in the species shown in A. The bars are divided into 3 categories: present in all species (green), absent in at least one other species (blue), species-specific (orange)
We defined 123,509 orthologous gene families, of these, 878 were shared by all tested species and 7960 can be found only in the P. brassicae genome. Additionally, 2984 families were unique to P. brassicae, and 2434 of these were single gene families (Additional file 3: Table S3). Of the 10,951 P. brassicae genes, 10.2% (1112) were included in clusters present in all species, 51.5% (5642) were absent in at least one species, and 38.3% (4197) were present only in P. brassicae (Fig. 2b). The species-specific set of P. brassicae contained a large number of proteins with no orthologues in other test species. Using the KOG-term annotation, it was found that a high proportion of the proteins within the Plasmodiophorids were poorly characterized, and the majority of the P. brassicae-specific genes possessed no assigned functional annotation (Schwelm et al. 2015). These findings demonstrate the unique evolutionary status of P. brassicae among eukaryotic organisms.
Comparative genomics reveals a significantly emphasized GPCR signaling pathway in P. brassicae
Cluster analysis of orthologous gene families containing at least 5 genes demonstrated that a group of gene families (shown as orange strips) were significantly expanded in P. brassicae (Fig. 3a). In total, 117 gene families (611 genes) were enriched in P. brassicae. For these specifically expanded gene families, we conducted Gene Ontology (GO) enrichment analysis and found specific enrichment for the downstream targets of the G-protein coupled receptor signaling pathway (GPCR) (69 genes), G-protein coupled glutamate receptor signaling pathway (26 genes), DNA integration, and chitin biosynthetic process (6 genes) at the level of Biological Process ontology. When analyzing Molecular Function ontology, we identified enrichment in G-protein coupled GABA receptor activity (26 genes), G-protein beta/gamma-subunit complex binding (13 genes), hydrolase activity, reactivity toward carbon-nitrogen (but not peptide) bonds, chitin binding, and chitin synthase activity, highlighting the importance of GPCRs and chitin-related pathways in P. brassicae (Additional file 3: Table S3 and Fig. 3b, c).

Orthologus families clustering analyses of Plasmodiophora brassicae-expanded genes and the enrichment of GO terms of the expanded orthologous genes. a Orthologus families clustering analysis of P. brassicae-expanded genes (at least 5 copies of orthologs in each family). The number of genes of every orthologus family was normalized by Z-Score. The orange strip represents the cluster of expanded orthologs in P. brassicae (mv: Mortierella verticillata, rd: Rhizopus delemar, plasb: Plasmodiophora brassicae, dd: Dictyostelium discoideum, bd: Batrachochytrium dendrobatidis, ri: Rhizophagus irregularis, fg: Fusarium graminearum, co: Capsaspora owczarzaki, sp.: Spizellomyces punctatus, ac: Acanthamoeba castellanii, pp.: Physcomitrella patens, at: Arabidopsis thaliana, phy: Phytophthora parasitica, sd: Saprolegnia declina, gt: Guillardia theta, cg: Crassostrea gigas, bd: Batrachochytrium dendrobatidis, rf: Reticulomyxa filosa, es: Ectocarpus siliculosus). b, c The enrichment of biology process ontology and molecular function ontology of expanded orthologs
We identified all significantly enriched GO terms (with a corrected P ≤ 0.05) for the gene set of 4197 P. brassicae-specific genes. For the Biological Process ontology, we found enrichment in single organism signaling, signal transduction, cell communication, the G-protein coupled receptor signaling pathway, and the cell surface receptor signaling pathway. Molecular Function ontology analysis revealed enrichment in G-protein beta/gamma-subunit complex binding, protein complex binding, protein binding, carbohydrate binding, GTPase activity, signal transducer activity, and molecular transducer activity (Additional file 3: Table S3).
GO enrichment analysis of the set of P. brassicae-expanded and P. brassicae-specific genes revealed several functional groups of particular interest for their possible role in growth, development, and pathogenesis in P. brassicae. For example, the enrichment of GPCR signaling pathway-associated genes was emphasized.
GPCR inhibitor treatment relieves the severity of clubroot
A comparative genomics analysis of P. brassicae indicated that the GPCR signaling pathway was significantly emphasized in the set of P. brassicae-expanded and P. brassicae-specific genes. Based on these findings, we focused on the genes involved in the GPCR signaling pathway of P. brassicae. As a result, 115 GPCR signaling pathway related genes were identified from the genome of P. brassicae strain ZJ-1, of which 30 encoded GPCRs, 38 encoded G proteins, and 47 encoded RGS (regulators of G protein signaling) (Additional file 4: Table S4). The transcripts of these genes involved in the GPCR signaling pathway were detected throughout growth and development, and the majority of them exhibited increased expression during the germinating resting spore stage (GS) and cortical infection stage (IN), suggesting that these genes may play a key role in plant-pathogen interactions (Fig. 4). Most of the GPCRs belong to class C in the P. brassicae ZJ-1 genome. Three class C-group II metabotropic glutamate receptors (PlasB_04548, PlasB_04549, and PlasB_09545) that are highly expressed during infection exhibited a 46.05%, 45.95%, and 46.25% similarity with their homologs in mammals, respectively (Additional file 4: Table S4). LY341495, the first known highly potent systemically active group II mGlu receptor antagonist in mammals, has been widely used to enable both in vitro and in vivo preclinical target validation studies (Kingston et al. 1998). We hypothesized that LY341495 affected the same potential target in both P. brassicae and mammals. Therefore, we applied LY341495 to evaluate the involvement of the GPCR signaling pathway in the interaction between P. brassicae and the host. Inhibitor treatment had no influence on the growth or development of rape plants. Upon comparison between mock and the inhibitor-treated group, the expression levels of the GPCR signaling pathway-related genes GPA1, AGB1, and AGG1 in B. napus exhibited no statistically significant differences (Fig. 5a, b). During the early infection stage, the application of inhibitor reduced the resting spore germination rate and the root hair infection rate of P. brassicae (Fig. 5c, d). At 21 and 28 days post-inoculation (dpi), the inhibitor-treated plants had a lower disease incidence (DI) than the mock-treated group, and suffered less severe symptoms (Fig. 6a, c). The transverse sections of root tissues were observed by scanning electron microscopy, and the results indicated that the cortical infection process of inhibitor-treated plants was blocked at the initial development stage of secondary plasmodia, while the mature resting spore had already been formed in the mock-treated plants, suggesting that inhibitor treatment relieved clubroot symptoms by reducing the secondary infection and delaying the cortical infection process of the pathogen (Fig. 6b). The relative accumulated amount of pathogens in the roots was evaluated using quantitative PCR (qPCR). An 8-fold increase in the amount of pathogens was detected in the mock-treated group compared to the inhibitor-treated group (Fig. 6d). The expression levels of PKA2 and Gβ, the primary downstream genes of GPCRs, were significantly reduced in the inhibitor-treated group (Fig. 6e). Our results indicated that GPCR inhibitor treatment reduced pathogen density and relieved the symptoms of infected roots by reducing the rates of resting spore germination and root hair infection during the early infection stage. Additionally, the development of secondary plasmodia was blocked during cortical infection.

Expression pattern of expanded and species-specific GPCR pathway related genes from Plasmodiophora brassicae. The expanded and species-specific GPCR pathway related genes show a specific expression pattern throughout growth and development of P. brassicae. For the expression values (Log2 (RPKM+ 1)), ranging from green (− 1.0) to red (1.0), a Pearson correlation was used for distance calculations. The red rectangles indicating expression pattern of these GPCR pathway related genes show a high level during germinating resting spore stage (GS) and cortical infection stage (IN) compared to the resting spore stage (RS)

Effect of GPCR inhibitor treatment on the growth, development of rapeseed plants, resting spore germination rate and root hair infection rate of Plasmodiophora brassicae. a Growth and development status of rape plants treated with GPCR inhibitor (right). Mock (DMSO) treatment served as control (left). The pictures were taken at 28 days after treatment. Bar = 1.5 cm. b At 28 days after treatment, the roots of mock- and inhibitor-treated plants were harvested. The expression levels of GPA1, AGB1, and AGG1 (downstream genes of GPCRs) in Brassica napus with mock and inhibitor treatment were quantified by qPCR. The actin gene of B. napus was used as control to normalize the expression level. Data represent the means and standard deviations. The expression level of mock-treated group was set as 1.0. Statistically significant difference of data between mock and inhibitor treated groups was compared, and the same letter in the graph indicates no significant differences at the level of P < 0.05. c, d Resting spore germination rate and root hair infection rate of P. brassicae were compared between H2O, mock and inhibitor treated groups. After 4 days, the treated spores were stained with orcein (Sigma-Aldrich Canada). The germination rate of spore was counted under microscope. At 7 dpi, the roots of rapeseed plant were stained with Trypan Blue, and then the root hair infection rate was counted with microscopic examination. The graphic data represent the means and standard deviations from three biological replicates. At the level of P < 0.05, statistically significant differences of data between H2O, mock and inhibitor treated groups were compared, different letters in the graph indicate statistically significant differences according to Student’s t test

The effect of GPCR inhibitor treatment on the symptom of clubroot. Clubroot symptoms (a, b, c) and quantification of Plasmodiophora brassicae DNA (d) in infected roots of the host plants treated or not with GPCR inhibitor. a Illustration of clubroot symptoms of GPCR inhibitor non-treated and treated plants at 28 dpi. Bar = 1 mm. b The transverse section of plant root tissues were observed by SEM. The red arrows indicate the typical infection structure of pathogen. Bar = 10 μm. c Clubroot symptoms of 70 infected roots from three biological repeats were evaluated by using percentage of plants in individual disease classes and DI at 21 and 28 dpi. Asterisks indicate statistically significant differences at the level of P < 0.01 according to Student’s t test. d At 28 dpi, the infected roots of mock-treated plants and inhibitor-treated plants were divided into two groups (A: severe; B: less severe). The relative accumulated amount of pathogen DNA was quantified by qPCR. Error bars represent the SE with three technical repeats. e The expression levels of the primary downstream genes of GPCR in cancer related pathway. Error bars represent the SD with three technical repeats
Discussion
The unique evolutionary status of Rhizaria: An independent branch in the eukaryotic phylogenetic tree
Divided into five kingdoms, eukaryotes contain most of phagotrophic Protozoa, which are the ancestors of four distinct clades, including osmotrophic Fungi, epithelial phagotrophic Animalia, photosynthetic Plantae, and Chromista (Cavalier-Smith et al. 2018). Rhizaria, the main heterotrophic infrakingdom in the protist groups, are markedly different from those of animals, fungi or plants. The eukaryotic supergroup Rhizaria also contains important plant pathogens, including the obligate biotrophic Plasmodiophorida, which belongs to the Phytomyxea of Rhizaria (Schwelm et al. 2018). Based on the partial transcriptome sequencing of eight Rhizaria genomes, a recent study revealed that Rhizaria is a sister of the Halvaria infrakingdom within the Harosa chromist subkingdom (Cavalier-Smith et al. 2018). Therefore, comparative genomics analyses may prove useful in determining the evolutionary status of the Rhizaria clade within the eukaryotic tree. In our study, comparative genomics analysis was performed among P. brassicae and 18 other available eukaryotic model species. Ninety genes were selected for phylogenetic analysis based on the conservation of these single-copy orthologs across species. The phylogenomic analysis of this large dataset strongly supported the unique evolutionary status of the Rhizaria. This comprehensive study of the most poorly understood eukaryotic branch will undoubtedly fill an important gap in our understanding of the eukaryotic tree of life. Though sharing an independent branch of the phylogenetic tree with common ancestors, the genome sizes of the species in Rhizaria group vary dramatically (P. brassicae ~ 24 Mb, B. natans ~ 100 Mb, and R. filose ~ 320 Mb). This may be a result of evolutionary genomic events (expansion or contraction). The genomes of P. brassicae exhibit a low level of similarity to known sequences from the other eukaryotic species (Pérez-López et al. 2018). Although it is extremely difficult to annotate P. brassicae genes, determining the key proteins involved in the interactions between this pathogen and its host will improve our understanding of eukaryotic plant pathogens, which we get so far predominantly from fungi, nematodes, and oomycetes.
P. brassicae maintains high genetic diversity despite low repetitive content and a smaller genome
Certain biotrophs, including obligate biotrophic powdery mildew and rust fungi, display marked increases in overall genome size compared to that of their close relatives either non-pathogenic or non-biotrophic. One possible explanation for this is that any increase is invariably the result of increased proliferation of transposable elements detectable as repetitive DNA (Spanu 2012); however, this is not the case with biotrophic P. brassicae. The genome size of P. brassicae (e3, Pb3, and ZJ-1) is small (24.1–25.5 Mb) (Schwelm et al. 2015; Rolfe et al. 2016), and smaller than that of most other members in the SAR supergroup, whereas the genome of P. brassicae has a higher gene density. Some of the predicted ORFs from the P. brassicae genome revealed a reduction in the length of the intergenic region. The overlapping of untranslated regions (UTRs) is one of the reasons for the compact genome structure of P. brassicae (Rolfe et al. 2016). The recently sequenced genome of S. subterranea, another Plasmodiophorida (Phytomyxea, Rhizaria) that causes potato powdery scab, also exhibits a similarly small size (28.08 Mb) (Ciaghi et al. 2018). The ZJ-1 genome only consists of approximately 2% repeated content. Transposable elements (TEs) are stretches of DNA that move throughout the genome and have significant influences on the organization and evolution of the genome (Medstrand et al. 2005). Inserting TEs into genes or gene regulatory regions can potentially disrupt the gene’s function and trigger chromosome rearrangements. Additionally, the mutational activities of TEs contribute to genetic diversity and innovation among organisms (Biémont and Vieira 2006). Despite of a low rate of TEs, the key players in evolution, P. brassicae strains possess a marked genetic diversity. This could be reflected in single-nucleotide polymorphisms (SNPs). SNP cluster analyses of the available P. brassicae genomes revealed a high degree of correlation between SNPs, host ranges, and regional origins, and 48,578 SNPs were identified within the genomes of the Pbe3 and Pb3 strains of P. brassicae (Rolfe et al. 2016). Our study also revealed that significant rearrangement events occurred in both the ZJ-1 and Pbe3 strains, suggesting that a rapid adaptation via dynamic regions within the genomes is not a universal phenomenon. Certain species, such as P. brassicae, have developed alternative strategies for maintaining genetic diversity and evolution that are independent of transposable elements. This confirms that different organisms possess diverse mechanisms for creating genetic diversity.
GPCR signal transduction pathways play important roles in P. brassicae
Our comparative genomics analysis demonstrated the presence of a set of significantly expanded and species-specific genes within the P. brassicae genome. GO enrichment analysis in this set of genes revealed that several functional groups, including GPCR signaling pathways, were significantly enriched in the P. brassicae.
Recent experimental data have shown that GPCRs, a large family of extremely versatile cell-surface receptors, are able to sense signaling molecules (such as biogenic amines, purines, odorants, ions, and pheromones) outside the cells, then activate signal transduction pathways inside the cells and, ultimately, elicit cellular responses (Prabhu et al. 2007). GPCRs play a central role in eukaryotic signal transduction and are involved in the regulation of many cell functions, including cell proliferation, survival, and motility (Dorsam and Gutkind 2007; Krishnan et al. 2014). The function of the GPCR signaling pathway in protists, however, remains largely unknown. Our transcriptome analysis revealed that these GPCR pathway-related genes are highly expressed during the infection process, including the germinating resting spore stage and the cortical infection stage. Our research also demonstrated that treatment with a GPCR inhibitor relieves the symptoms of clubroot disease, suggesting that a significantly enriched GPCR signaling pathway plays an important role in plant-P. brassicae interaction. Inhibitor treatment suppressed both the root hair and cortical infection, indicating that the GPCR signaling pathway of P. brassicae is involved in the perception and recognition of external signals, including chemical molecules (from host root exudate or soil environment) and signal stimulation from the intracellular space of the host. As a soil-borne obligate parasite, P. brassicae survives in soil in the form of resting spores. Precise sensing of signal molecules (such as calcium ions or other signals from the host plant) is important for resting spores to exit dormancy and begin germination, as initiation of the initial step of the life cycle must take place under suitable conditions. During the root hair infection stage and the cortical infection stage, P. brassicae acts as an intracellular parasite that takes up nutrients from the host and is able to sense the intracellular environment of the host cells (Kageyama and Asano 2009; Pérez-López et al. 2018). For P. brassicae, a close interaction with its host may be the driving force behind the evolution of enriched GPCR signaling pathways, as these are key molecular mechanisms for adapting to the intracellular environment. Given this, we propose that the GPCR signaling pathways are important regulators of growth, development, and pathogenicity of P. brassicae.
In summary, our findings indicate that the genome of P. brassicae is compact with a low rate of repetitive elements; however, this genome does exhibit extensive rearrangements. Our comparative genomics analysis highlighted the important evolutionary status of the Rhizaria group within the eukaryotic evolutionary tree. We also demonstrated the molecular basis by which P. brassicae adopts intracellular parasitic characteristics. We propose that the significantly emphasized GPCR signaling pathways in the genome of P. brassicae are important for the growth, development, and pathogenicity of this organism.
Conclusions
In this study, the draft genome of the P. brassicae ZJ-1 strain was analyzed using a comparative genomics approach. As a result, GPCR signal transduction pathways were found to play key roles in the growth, development, and pathogenicity of P. brassicae.
Methods
Collection of field populations and isolation of single spores
The clubroot galls of P. brassicae on oilseed rape (Brassica napus) were collected from an experimental field in Zhijiang City (ZJ) (Hubei province, N30°25′42.54″, E111°45′17.09″) in 2011 and then frozen at − 20 °C until use. The resting spores were extracted and purified according to the method described by Asano et al. (1999). For extraction of resting spores, the mature clubroot galls were surface-sterilized with 70% ethanol for 2 min and 10% hydrogen peroxide for 1 h, and then rinsed five times with sterile distilled water. The clubroot tissues were cut into small pieces, homogenized, filtered through a gauze (25 μm pore width), and centrifuged twice (2500×g, 10 min). After spore extraction, the spore suspension was further purified by centrifugation for 15 min at 2000×g in 50% (w/v) sucrose solution. The clean spores were harvested by centrifuging for 15 min at 2000×g, and then washed five times with sterile distilled water. Resting spores were suspended in a freshly prepared 2% chloramine-T solution (wt/vol) (Sigma-Aldrich) at room temperature for 20 min and then washed twice with sterile water by centrifugation, as described above. The resulting pellet was suspended in an antibiotic solution containing 1000 μg/mL colistin sulfate (Sigma-Aldrich), 1000 μg/mL vancomycin hydrochloride (Sigma-Aldrich), and 6000 μg/mL cefotaxime sodium (Sigma-Aldrich). After incubated at 25 °C under dark for 24 h, the suspension was washed five times with sterile water. The clean spore suspension was counted and diluted to 5.0 × 107 spores/mL, then suspended in 1/10-strength modified Hoagland’s solution (5 mM Ca(NO3)2, 5 mM KNO3, 2 mM MgSO4, and 2 mM KH2PO4; pH 5.8) with 100 μg/mL cefotaxime sodium. Single spore production was performed according to Fahling et al. (2004). The virulence of the single-spore isolate ZJ-1 was tested on the differential hosts of the Williams system. It was shown that the isolate was derived from the pathotype 1 of Williams’ clubroot differential set (Williams 1966). According to the method described above, the resting spore materials were extracted from plant clubroots (single spore isolate ZJ-1).
DNA and RNA extraction
DNA was extracted from resting spores of strain ZJ-1 using a modified CTAB method (Russell and Bulman 2004) and stored at − 20 °C. For transcriptome expression analysis, samples from three specific infection stages were collected. Resting spore stage sample (RS) was prepared from plant clubroots (single spore isolate ZJ-1). For germinating resting spore stage sample (GS), the purified resting spores were thawed at 4 °C for 48 h followed by 24 h at room temperature in root exudates solution for germinating under a dark environment. Then the root tissue was immersed in 2.0 mL EP tubes with treated germinating spores (107 spores/mL). Samples were harvested after 24 h when the germinating resting spores aggregated to and were adsorbed in root tissues under microscope. To prepare the early cortical infection stage sample (IN), seeds of oilseed rape (Brassica napus) were surface sterilized in 1% NaClO for 5 min, washed with distilled water and germinated on moistened filter paper for 6 days in a growth chamber maintained at 22 °C/20 °C (day/night) with a 14 h photoperiod and 80% relative humidity. Parts of the seeds were transplanted to autoclaved potting mix in 60-cell (4 cm × 3.5 cm × 6 cm) plastic pot trays in a controlled environment growth chamber (HP300GS-C; Ruihua Instrument, Wuhan, China) at a constant 20 °C with 14 h photoperiod and 80% relative humidity and watered once daily. Nineteen days after sowing, the plants were inoculated with resting spore suspension (107 spores/mL). After inoculation, the plants grew under glasshouse conditions as described above. Root masses were collected from at least 50 plants at 21 dpi, when signs of secondary infection were visible. Roots were extensively washed in tap water and then inspected with microscope to ensure the absence of successful infection of pathogen and trimmed away fine roots tissues. The total RNA samples of three biological replicates for each stage were prepared. Total RNA was extracted with RNAiso Plus (TaKaRa, Dalian, China) according to the manufacturer’s protocols and stored at − 80 °C. RNA quantity and quality was assessed using a Nanodrop (NanoDrop Technologies, Inc. Wilmington, DE USA) and checked by running a gel electrophoresis in 1.5% (w/v) agarose gels.
Genome sequencing and assembly
The entire genome of the P. brassicae ZJ-1 strain was de novo sequenced using the Illumina MiSeq, a next generation sequencing technology. DNA libraries PE400 bp, PE700 bp, and MP 8 kb insert fragments were constructed and paired-end sequenced using the Illumina Genome Analyzer at Shanghai Personal Biotechnology Co., Ltd. (Shanghai, China). Low-quality data with q-values below 20 consisting of short reads (length < 35 bp) were discarded. High-quality data were then subjected to assembly. The genome sequence was assembled using Newbler v2.8 and GapCloser. Finally, using Blastn search, scaffolds with high hits against the B. napus genome sequence were removed from the assembly.
Transcriptome expression analysis
A RNA-seq library was prepared according to manufacturer’s instructions (Illumina, San Diego, CA), and then sequenced using an Illumina NextSeq 500 (paired-end 2 × 150 bp) at the Shanghai Personal Biotechnology Co., Ltd. (Shanghai, China). Firstly, the raw reads of the RNA-Seq library were processed to obtain high quality reads by removing the adapter sequences and low-quality bases at the 3′ end and then trimming any low-quality bases (Q < 20) from the 3′ to 5′ ends of the remaining reads. Reads filtering out the poor quality and with a length greater than 50 bp were considered for further analysis. The filtered reads were mapped to the P. brassicae genome using Tophat v2.0.9 (http://ccb.jhu.edu/software/tophat/index.shtml) (Trapnell et al. 2009). HTSeq (http://htseq.readthedocs.io/) (Anders et al. 2015), and DESeq (http://www-huber.embl.de/users/anders/DESeq) (Anders and Huber 2010) for the transcriptome differential expression analysis.
Gene prediction and annotation
To obtain a more accurate gene set, we performed an integrated prediction method that included de novo gene prediction programs, homology-based methods, and RNA-Seq data. CEGMA software was used to predict the core genes of the P. brassicae ZJ-1 strain based on the eukaryotes conservative gene database (Parra et al. 2007). The core gene set was used as the training set to predict genes using a de novo prediction software, Augustus (Stanke et al. 2004). We aligned the predictions to the nr database to obtain the conserved sequences. These sequences were then used as the training set of Augustus, GlimmerHMM (Majoros et al. 2004), and SNAP (Korf 2004). This constructed model was used for gene prediction. Additionally, we used the homolog-based software Exonerate (version 2.2.0) for gene prediction, with B. natans and R. filosa as the reference sequences. BLAST (E ≤ 1e− 5) was also used, where matches with a length coverage of > 30% to the homologous proteins were considered gene-model candidates. Clean RNA-Seq reads were obtained by matching to the sequence of the B. napus genome, where any high hit reads were removed. Then, the RNA-Seq data were de novo assembled using Trinity (version r20140717) (Haas et al. 2013), and aligned assembled against the P. brassicae genome using PASA (version r2014-04-17). Finally, EvidenceModeler (version r2012-06-25) software was employed to integrate the above results to generate a weighted consensus gene set (Haas et al. 2008). Partial genes and small genes (< 100 bp) were filtered. The result of the RNA-Seq de novo assembly was used for boundary correction of the predicted gene set model with PASA (version r20140417), which is able to identify splice junctions between exons. Cufflinks v2.1.1 (Trapnell et al. 2010) was used to determine the transcript structures. To eliminate false positives (genes only supported by de novo methods), novel genes with reads per kilobase of gene model per million mapped reads (RPKM) ≤ 1 were removed.
Gene functions were predicted by comparing the NCBI NR protein database with the GO (Ashburner et al. 2000), KEGG (Kanehisa and Goto 2000), and eggnog (Powell et al. 2013) databases. The protein domain was predicted using the InterPro and Pfam databases. The criteria used to assign a function to CDSs consisted of a minimum cut off of 30% identity and 70% coverage of the protein length, as well as at least two high hits in the NCBI, GO, KEGG, and eggNOG databases (Blake and Cohen 2001). We annotated the predicted genes by searching against the NCBI NR database using blastall (Altschul et al. 1990) with an E-value cut-off of 1e− 6. Using BLAST2GO (Conesa and Götz 2008), the descriptions of the gene products were presented by Gene Ontology (with a blast E-value of 1e− 6). We also mapped the reference genes to KEGG pathway maps using KAAS (the KEGG Automatic Annotation Server) (http://www.genome.jp/tools/kaas/) with the non-organism specific option (Moriya et al. 2007). The reference genes were eggNOG annotated by searching against eggNOG (V4) using blast (E ≤ 1e− 6). For the repetitive sequences of the P. brassicae ZJ-1 genome, a combination of several prediction programs was used. We identified known repeat sequences using RepeatMasker (v3.3.0). The database version and the search engine were 20,140,418 and rmblastn 2,2,27+, respectively (Tempel 2012). We constructed a de novo repeat library using RepeatMolder. We ran RepeatMasker on the genome sequences, using the RepeatMolder consensus sequence as the library.
Genomic data collection for comparative genomic analysis
Genome sequence of P. brassicae strain e3 was downloaded from ENA (European Nucleotide Archive) database under the projects PRJEB8376 (Accessions CDSF01000001-CDSF01000165). We selected genomes of 18 other available representative eukaryotic model species for comparative analysis. Capsaspora owczarzaki (Opisthokonta unicellular amoeba), Crassostrea gigas (Opisthokonta animal), Batrachochytrium dendrobatidis (Opisthokonta fungi), Spizellomyces punctatus (Opisthokonta fungi), Mortierella verticillata (Opisthokonta early diverging fungal lineages), Rhizopus delemar (Opisthokonta fungi, Mucorales), Rhizophagus irregularis (Opisthokonta Glomeromycota, mycorrhizal fungus), Fusarium graminearum (Opisthokonta fungi), Physcomitrella patens (Plantae, Streptophyta, mosses), Arabidopsis thaliana (Plantae, plant), Phytophthora parasitica (Chromalveolata oomycetes), Saprolegnia declina (Chromalveolata oomycetes), Ectocarpus siliculosus (Chromalveolata Stramenopiles, brown algae), Guillardia theta (Chromalveolata Cryptophyta), Acanthamoeba castellanii (Amoebozoa), Dictyostelium discoideum (Amoebozoa), Reticulomyxa filosa (Rhizaria), Bigelowiella natans (Rhizaria). The predicted proteomes of these model species were downloaded from Broad Institute (http://www.broadinstitute.org/science/projects/projects.) and DOE Joint Genome Institute (JGI) site (http://genome.jgi-psf.org/). Fungi with different trophic types were chosen for comparative analysis with P. brassicae. Saprophytic fungi Saccharomyces cerevisiae, Trichoderma reesei and Neurospora crassa, hemibiotrophic fungi Magnaporthe oryzae and Fusarium graminearum, necrotrophic fungi Verticillium dahliae, Botrytis cinerea, Sclerotinia sclerotiorum and Colletotrichum graminicola; biotrophic fungi Blumeria graminis, Sphacelotheca reilianum, Melampsora larici-populina, Sphacelotheca reilianum and Ustilago maydis were selected. Genome sequences of these fungi used in this study were downloaded from Broad Institute and JGI database.
Evolutionary and comparative analysis
Gene families (ortholog, co-ortholog, and inparalog pairs) for P. brassicae and 18 other species were built using OrthoMCL (http://orthomcl.org/orthomcl/) with a Markov inflation of 1.5 and a maximum e-value of 1e− 5 (Fischer et al. 2011). The 90 single-copy genes from the gene family analysis were aligned using MUSCLE (Edgar 2004), and the alignments were concatenated as a single data set. To determine the gene evolution rates, we calculated the tree branch lengths for 90 clusters of 1:1 orthologs for all the species. Phylogeny was inferred using PhyML 3.0 with 100 bootstrap replicates (LG model of amino acids substitution) (Guindon et al. 2010). The tree branch lengths indicated different evolutionary rates for these ortholog groups. Scaffolds larger than 10 kb from the P. brassicae ZJ-1 strain and Pbe3 genomes were compared. Whole genome alignment was analyzed using Mauve version 2.4.0 (Darling et al. 2010). Whole-genome dot-plot comparison between two genomes was conducted using r2cat (related reference contig arrangement tool) (Husemann and Stoye 2009).
Enrichment analysis of gene function and hierarchical clustering analysis
By using Gene Ontology (GO) term enrichment analysis, all genes for testing were initially mapped against GO terms in the GO database (http://www.geneontology.org/), the number of genes was calculated for each term, and then hypergeometric testing was used to identify significantly enriched GO terms in the testing group by comparing them to the genome background. Significantly enriched pathways of testing genes were identified using KEGG pathway enrichment analysis. The calculation formula is the same as that for the GO analysis. To analyze gene expression data and genes possessing similar functional cluster groups, hierarchical clustering methods have been widely applied based on transcriptome expression data. To compare the expression patterns of each gene among the different samples, the abundance of each transcript was normalized by RPKM (Mortazavi et al. 2008). The heat map of the clustered genes and samples was generated using the clustering affinity search technique (CAST) assay with the MultiExperiment Viewer v4.9 software package (Saeed et al. 2003; Lombard et al. 2014). An average linkage hierarchical clustering method was used, and Pearson’s correlation coefficient (the distance metric is the default) was employed to measure the similarity between expressed genes.
Quantitative RT-PCR (qRT-PCR) validation
qRT-PCR primers were designed to generate amplicons for validating the RNA-seq data (Additional file 5: Table S5). For RT-PCR and qRT-PCR, 5 μg of total RNA was reverse transcribed into first-strand cDNA using oligo (dT) primer and M-MLV Reverse Transcriptase according to the manufacturer’s instructions (TransScript, Beijing, China). All qRT-PCR experiments were run in a SYBR Green Real-Time PCR Master Mix (Bio-Rad, California, USA) in 20 μL reactions with the CFX96™ real-time PCR detection system (Bio-Rad). The actin gene of P. brassicae was used as an internal control to normalize the expression data. Data were acquisited and analyzed by using the Bio-Rad CFX Manager™ Software (version 2.0). The relative expression level of each target gene was quantified by the comparative CT method (2 -△△Ct) of quantification (Livak and Schmittgen 2001). Similar results were obtained from two biological replicates, each with three technical repeats. The relative expression levels of each gene were then validated for the RNA-seq data.
Symptom quantification of the treatment of GPCR antagonist LY341495
Twenty-eight days after GPCR inhibitor treatment, the growth and development status of B. napus was verified. A DMSO treated group was used as the control. The root-tissues of treated plants were harvested, and the expression levels of the downstream genes of GPCRs (Gα GPA1-XM_013808529.1; Gβ AGB1-XM_013795701.1; Gγ AGG1-XM_013833980.1) from B. napus were quantified using qRT-PCR according to method described above. The effect of GPCR inhibitor treatment on the germination rate of the resting spores and the infection rate of the root hairs were analyzed according to the method described by Feng et al. (2010) and Chen et al. (2016). Ten days after germination, the prepared plants were inoculated with the pretreated resting spores (diluted to 107 spores/mL with modified 1/2 Hoagland nutrient solution and inhibitor solution) (LY341495: 2000 nM, MedChemExpress) in 10 mL EP tubes. As a parallel control, 1/2 Hoagland nutrient solution and equivalent DMSO treatment was administered to the mock group under the same condition. The CK group (not inoculated) was placed in EP tubes with equal parts 1/2 Hoagland nutrient solution and inhibitor solution. The solution of the EP tubes was refreshed every 7 days. The DI was calculated according to Klewer et al. (2001). At 28 dpi, all seedlings were harvested for investigation of clubroot symptoms via phytopathological analysis or scanning electron microscope (SEM). For each biological experiment, at least 20 plants were analyzed. Results were obtained from three independent biological experiments. The plants were cut at the top of the hypocotyl towards the roots. For quantitative estimations, genomic DNA was prepared from a pool of root samples (severe and less severe groups). In the mock group, the A-severe group consists of the roots belonging to classes 3 and 4, and the B-less severe group consists of the roots belonging to classes 1 and 2. In the inhibitor-treated group, the A-severe group consists of the roots belonging to classes 2 and 4, and the B-less severe group consists of the roots belonging to classes 0 and 1. The expression levels of the actin gene of P. brassicae, which represents the amount of the pathogen accumulated in the roots, were normalized to the actin gene of B. napus. Results were obtained from three biological replicates, with three technical repeats each.
Abbreviations
- CEGs:
-
Core eukaryotic genes
- DI:
-
Disease index
- GPCR:
-
G-protein coupled receptor signaling pathway
- GS:
-
Germinating resting spore stage
- IN:
-
Infection stage
- qPCR:
-
Quantitative PCR
- SAR:
-
Stramenopiles-Alveolata-Rhizaria
- TEs:
-
Transposable elements
- WGS:
-
Whole-genome shotgun
References
Adl SM, Simpson AGB, Lane CE, Lukeš J, Bass D, Bowser SS, et al. The revised classification of eukaryotes. J Eukaryot Microbiol. 2012;59(5):429–514.
Alexander SP, Christopoulos A, Davenport AP, Kelly E, Marrion NV, Peters JA, et al. The concise guide to PHARMACOLOGY 2017/18: G protein-coupled receptors. Br J Pharmacol. 2017;174(Suppl 1):S17–129. https://doi.org/10.1111/bph.13878.
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215(3):403–10.
Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11:R106 https://doi.org/10.1186/gb-2010-11-10-r106.
Anders S, Pyl PT, Huber W. HTSeq—a python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31(2):166–9.
Asano T, Kageyama K, Hyakumachi M. Surface disinfestation of resting spores of Plasmodiophora brassicae used to infect hairy roots of Brassica spp. Phytopathology. 1999;89(4):314–9.
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, et al. Gene ontology: tool for the unification of biology. Nat Genet. 2000;25(1):25–9.
Biémont C, Vieira C. Genetics: junk DNA as an evolutionary force. Nature. 2006;443(7111):521–4.
Blake JD, Cohen FE. Pairwise sequence alignment below the twilight zone. J Mol Biol. 2001;307(2):721–35.
Burki F, Kudryavtsev A, Matz MV, Aglyamova GV, Bulman S, Fiers M, et al. Evolution of Rhizaria: new insights from phylogenomic analysis of uncultivated protists. BMC Evol Biol. 2010;10(1):377.
Campo JD, Sieracki ME, Molestina R, Keeling P, Massana R, Ruiz-Trillo I. The others: our biased perspective of eukaryotic genomes. Trends in Ecol Evolut. 2014;29(5):252–9.
Cavalier-Smith T, Chao EE, Lewis R. Multigene phylogeny and cell evolution of chromist infrakingdom Rhizaria: contrasting cell organisation of sister phyla Cercozoa and Retaria. Protoplasma. 2018;255(5):1517–74.
Chen T, Bi K, He Z, Gao Z, Zhao Y, Fu Y, et al. Arabidopsis mutant bik1 exhibits strong resistance to Plasmodiophora brassicae. Front Physiol. 2016;7:402. https://doi.org/10.3389/fphys.2016.00402.
Ciaghi S, Neuhauser S, Schwelm A. Draft genome resource for the potato powdery scab pathogen Spongospora subterranea. Mol Plant-Microbe Interact. 2018;31(12):1227–9.
Conesa A, Götz S. Blast2GO: a comprehensive suite for functional analysis in plant genomics. Int J Plant Genomics. 2008;2008:619832. https://doi.org/10.1155/2008/619832.
Curtis BA, Tanifuji G, Burki F, Gruber A, Irimia M, Maruyama S, et al. Algal genomes reveal evolutionary mosaicism and the fate of nucleomorphs. Nature. 2012;492(7427):59–65.
Darling AE, Mau B, Perna NT. ProgressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One. 2010;5(6):e11147.
de Mendoza A, Sebé-Pedrós A, Ruiz-Trillo I. The evolution of the GPCR signaling system in eukaryotes: modularity, conservation, and the transition to metazoan multicellularity. Genome Biol Evol. 2014;6(3):606–19.
Dixon GR. The occurrence and economic impact of Plasmodiophora brassicae and clubroot disease. J Plant Growth Regul. 2009;28(3):194–202.
Dorsam RT, Gutkind JS. G-protein-coupled receptors and cancer. Nat Rev Cancer. 2007;7(2):79–94.
Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32(5):1792–7.
Fahling M, Graf HJ, Siemens J. Characterization of a single-spore isolate population of Plasmodiophora brassicae resulting from a single club. J Phytopathol. 2004;152(7):438–44.
Feng J, Hwang R, Hwang SF, Strelkov SE, Gossen BD, Zhou QX, et al. Molecular characterization of a serine protease Pro1 from Plasmodiophora brassicae that stimulates resting spore germination. Mol Plant Pathol. 2010;11(4):503–12.
Fischer S, Brunk BP, Chen F, Gao X, Harb OS, Iodice JB, et al. Using OrthoMCL to assign proteins to OrthoMCL-DB groups or to cluster proteomes into new ortholog groups. Curr Protoc Bioinformatics. 2011; Chapter 6:Unit 6.12.1–19. doi:https://doi.org/10.1002/0471250953.bi0612s35.
Glöckner G, Hülsmann N, Schleicher M, Noegel AA, Eichinger L, Gallinger C, et al. The genome of the foraminiferan Reticulomyxa filosa. Curr Biol. 2014;24(1):11–8.
Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol. 2010;59(3):307–21.
Haas BJ, Papanicolaou A, Yassour M, Grabherr M, Blood PD, Bowden J, et al. De novo transcript sequence reconstruction from RNA-seq using the trinity platform for reference generation and analysis. Nat Protoc. 2013;8(8):1494–512.
Haas BJ, Salzberg SL, Zhu W, Pertea M, Allen JE, Orvis J, et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the program to assemble spliced alignments. Genome Biol. 2008;9(1):R7 https://doi.org/10.1186/gb-2008-9-1-r7.
Hilger D, Masureel M, Kobilka BK. Structure and dynamics of GPCR signaling complexes. Nat Struct Mol Biol. 2018;25(1):4–12.
Husemann P, Stoye J. r2cat: synteny plots and comparative assembly. Bioinformatics. 2009;26(4):570–1.
Hwang SF, Strelkov SE, Feng J, Gossen BD, Howard RJ. Plasmodiophora brassicae: a review of an emerging pathogen of the Canadian canola (Brassica napus) crop. Mol Plant Pathol. 2012;13(2):105–13.
Ingram DS, Tommerup IC. The life history of Plasmodiophora brassicae Woron. R Soc Lond B. 1972;180(1058):103–12.
Kageyama K, Asano T. Life cycle of Plasmodiophora brassicae. J Plant Growth Regul. 2009;28(3):203–11.
Kamp ME, Liu Y, Kortholt A. Function and regulation of heterotrimeric G proteins during chemotaxis. Int J Mol Sci. 2016;17(1):90. https://doi.org/10.3390/ijms17010090.
Kanehisa M, Goto S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28(1):27–30.
Kingston AE, Ornstein PL, Wright RA, Johnson BG, Mayne NG, Burnett JP, et al. LY341495 is a nanomolar potent and selective antagonist of group II metabotropic glutamate receptors. Neuropharmacology. 1998;37(1):1–12.
Klewer A, Luerben H, Graf H, Siemens J. Restriction fragment length polymorphism markers to characterize Plasmodiophora brassicae single-spore isolates with different virulence patterns. J Phytopathol. 2001;149(3/4):121–7.
Korf I. Gene finding in novel genomes. BMC Bioinformatics. 2004;5(1):59 https://doi.org/10.1186/1471-2105-5-59.
Krishnan A, Dnyansagar R, Almén MS, Williams MJ, Fredriksson R, Manoj N, et al. The GPCR repertoire in the demosponge Amphimedon queenslandica: insights into the GPCR system at the early divergence of animals. BMC Evol Biol. 2014;14(1):270 https://doi.org/10.1186/s12862-014-0270-4.
Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) method. Methods. 2001;25(4):402–8.
Lombard V, Golaconda Ramulu H, Drula E, Coutinho PM, Henrissat B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 2014;42(Database issue):D490–5.
Majoros WH, Pertea M, Salzberg SL. TigrScan and GlimmerHMM: two open source ab initio eukaryotic gene-finders. Bioinformatics. 2004;20(16):2878–9.
Mcdonald MR, Sharma K, Gossen BD, Deora A, Feng J, Hwang SF. The role of primary and secondary infection in host response to Plasmodiophora brassicae. Phytopathology. 2014;104(10):1078–87.
Medstrand P, van de Lagemaat LN, Dunn CA, Landry JR, Svenback D, Mager DL. Impact of transposable elements on the evolution of mammalian gene regulation. Cytogenet Genome Res. 2005;110(1–4):342–52.
Moriya Y, Itoh M, Okuda S, Yoshizawa AC, Kanehisa M. KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 2007; 35 (Web Server issue): W182–5. doi: https://doi.org/10.1093/nar/gkm321.
Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5(7):621–8.
Parra G, Bradnam K, Korf I. CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics. 2007;23(9):1061–7.
Pérez-López E, Waldner M, Hossain M, Kusalik AJ, Wei Y, Bonham-Smith PC, et al. Identification of Plasmodiophora brassicae effectors-a challenging goal. Virulence. 2018;9(1):1344–53.
Powell S, Forslund K, Szklarczyk D, Trachana K, Roth A, Huerta-Cepas J, et al, eggNOG v4. 0: nested orthology inference across 3686 organisms. Nucleic Acids Res. 2013; (Database issue): D231–9. doi: https://doi.org/10.1093/nar/gkt1253.
Prabhu Y, Müller R, Anjard C, Noegel AA. GrlJ, a dictyostelium GABAB-like receptor with roles in post-aggregation development. BMC Dev Biol. 2007;7:44 https://doi.org/10.1186/1471-213X-7-44.
Qiu JD, Huang JH, Liang RP, Lu XQ. Prediction of G-protein-coupled receptor classes based on the concept of Chou’s pseudo amino acid composition: an approach from discrete wavelet transform. Anal Biochem. 2009;390(1):68–73.
Rolfe SA, Strelkov SE, Links MG, Clarke WE, Robinson SJ, Djavaheri M, et al. The compact genome of the plant pathogen Plasmodiophora brassicae is adapted to intracellular interactions with host Brassica spp. BMC Genomics. 2016;17:272. https://doi.org/10.1186/s12864-016-2597-2.
Rosenbaum DM, Rasmussen SG, Kobilka BK. The structure and function of G-protein-coupled receptors. Nature. 2009;459(7245):356–63.
Russell J, Bulman S. The liverwort marchantia foliacea forms a specialized symbiosis with arbuscular mycorrhizal fungi in the genus glomus. New Phytol. 2004;165(2):567–79.
Saeed AI, Sharov V, White J, Li J, Liang W, Bhagabati N, et al. TM4: a free, open-source system for microarray data management and analysis. Biotechniques. 2003;34(2):374–8.
Schwelm A, Badstöber J, Bulman S, Desoignies N, Etemadi M, Falloon RE, et al. Not in your usual top 10: protists that infect plants and algae. Mol Plant Pathol. 2018;19(4):1029–44.
Schwelm A, Fogelqvist J, Knaust A, Jülke S, Lilja T, Bonillarosso G, et al. The Plasmodiophora brassicae genome reveals insights in its life cycle and ancestry of chitin synthases. Sci Rep. 2015;5:11153. https://doi.org/10.1038/srep11153.
Spanu PD. The genomics of obligate (and nonobligate) biotrophs. Annu Rev Phytopathol. 2012;50:91–109.
Stanke M, Steinkamp R, Waack S, Morgenstern B. AUGUSTUS: a web server for gene finding in eukaryotes. Nucleic Acids Res. 2004;32(Web Server issue):W309–12. https://doi.org/10.1093/nar/gkh379.
Tempel S. Using and understanding RepeatMasker. Methods Mol Biol. 2012;859:29–51. https://doi.org/10.1007/978-1-61779-603-6_2.
Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25(9):1105–11.
Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28(5):511–5.
van den Hoogen DJ, Meijer HJG, Seidl MF, Govers F. The ancient link between G-protein-coupled receptors and C-terminal phospholipid kinase domains. MBio. 2018;9(1):e02119–7. https://doi.org/10.1128/mBio.02119-17.
Williams PH. A system for the determination of races of Plasmodiophora brassicae that infect cabbage and rutabaga. Phytopathology. 1966;56(6):624–6.
Acknowledgements
We would like to acknowledge the anonymous reviewers for their work on this paper.
Funding
This research was financially supported by the National Key R & D Program of China (2017YFD0200600) and the earmarked fund for China Agriculture Research System (CARS-13).
Availability of data and materials
The datasets used and analyzed during the current study are available from the corresponding author on reasonable request.
Author information
Authors and Affiliations
Contributions
KB, TC and DJ conceived and designed the experiment. KB, HL and TC analyzed the genomic sequence. ZH, ZG and YZ performed the experiment. JC, JX and DJ checked all the data. KB, YF and DJ wrote the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional files
Additional file 1:
Table S1. Statistical overview of RNASeq Map events (DOCX 15 kb)
Additional file 2:
Table S2. Comparative analysis of gene density of genome between Plasmodiophora brassicae ZJ-1 and other plant pathogens (DOCX 29 kb)
Additional file 3:
Table S3. The enrichment of GO terms of Plasmodiophora brassicae-expanded and P. brassicae-specific genes (DOCX 32 kb)
Additional file 4:
Table S4. Gene information of GPCR signaling pathway in Plasmodiophora brassicae ZJ-1 (XLSX 17 kb)
Additional file 5:
Table S5. Information of primers used in this study (DOCX 18 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Bi, K., Chen, T., He, Z. et al. Comparative genomics reveals the unique evolutionary status of Plasmodiophora brassicae and the essential role of GPCR signaling pathways. Phytopathol Res 1, 12 (2019). https://doi.org/10.1186/s42483-019-0018-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s42483-019-0018-6