
Abstract
Background
Nitrogen heterocyclic rings and sulfonamides have attracted attention of several researchers.
Results
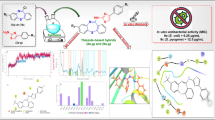
A series of regioselective imidazole-based mono- and bis-1,4-disubstituted-1,2,3-triazole-sulfonamide conjugates 4a–f and 6a–f were designed and synthesized. The first step in the synthesis was a regioselective propargylation in the presence of the appropriate basic catalyst (Et3N and/or K2CO3) to afford the corresponding mono-2 and bis-propargylated imidazoles 5. Second, the ligation of the terminal C≡C bond of mono-2 and/or bis alkynes 5 to the azide building blocks of sulfa drugs 3a–f using optimized conditions for a Huisgen copper (I)-catalysed 1,3-dipolar cycloaddition reaction yielded targeted 1,2,3-triazole hybrids 4a–f and 6a–f. The newly synthesized compounds were screened for their in vitro antimicrobial and antiproliferative activities. Among the synthesized compounds, compound 6a emerged as the most potent antimicrobial agent with MIC values ranging between 32 and 64 µg/mL. All synthesized molecules were evaluated against three aggressive human cancer cell lines, PC-3, HepG2, and HEK293, and revealed sufficient antiproliferative activities with IC50 values in the micromolar range (55–106 μM). Furthermore, we conducted a receptor-based electrostatic analysis of their electronic, steric and hydrophobic properties, and the results were in good agreement with the experimental results. In silico ADMET prediction studies also supported the experimental biological results and indicated that all compounds are nonmutagenic and noncarcinogenic.
Conclusion
In summary, we have successfully synthesized novel targeted benzimidazole-1,2,3-triazole-sulfonamide hybrids through 1,3-dipolar cycloaddition reactions between the mono- or bis-alkynes based on imidazole and the appropriate sulfonamide azide under the optimized Cu(I) click conditions. The structures of newly synthesized sulfonamide hybrids were confirmed by means of spectroscopic analysis. All newly synthesized compounds were evaluated for their antimicrobial and antiproliferative activities. Our results showed that the benzimidazole-1,2,3-triazole-sulfonamide hybrids inhibited microbial and fungal strains within MIC values from 32 to 64 μg/mL. The antiproliferative evaluation of the synthesized compounds showed sufficient antiproliferative activities with IC50 values in the micromolar range (55–106 μM). In conclusion, compound 6a has remarkable antimicrobial activity. Pharmacophore elucidation of the compounds was performed based on in silico ADMET evaluation of the tested compounds. Screening results of drug-likeness rules showed that all compounds follow the accepted rules, meet the criteria of drug-likeness and follow Lipinski’s rule of five. In addition, the toxicity results showed that all compounds are nonmutagenic and noncarcinogenic.

Similar content being viewed by others

Background
Currently, a steady increase in the incidences of infectious diseases has occurred due to increasing drug resistance in microbial strains, which has become a major global public health issue [1]. This problem has challenged researchers to develop new antimicrobial agents that will be more potent, more selective and less toxic for combating drug-resistant pathogens. Thus, nitrogen-containing heterocycles, in particular 1,2,3-triazoles [2], have attracted a great deal of interest from medicinal chemists in the design of potential drug candidates owing to their high biocompatibility and various pharmacological actions such as antibacterial [3], antiviral [4], antifungal [5], antimalarial [6], anti-HIV [7], antiallergic [8], antitubercular [9], CNS depressant [10], analgesic [11], anticonvulsant [12], antihypertensive [13] and antiproliferative activities [14].
In addition, 1,2,3-triazoles, attractive linkers that can tether two pharmacophores to provide innovative bifunctional drugs, have become increasingly useful and important in constructing bioactive and functional compounds [15,16,17,18,19,20].
On the other hand, benzimidazoles represent an important category of active therapeutic agents because their structures are well-suited for biological systems [21]. Their derivatives show various biological activities including antiviral [22], antifungal [23], antiproliferative [24], antihypertensive [25], analgesic [26], anti-inflammatory [27], antibacterial [28] and anthelmintic activities [29].
Sulfonamides, known as sulfa drugs (Fig. 1), are the oldest drugs commonly employed and systematically used as preventive and chemotherapeutic agents against various diseases [30, 31]. Generally, these compounds are easy to prepare, stable and bioavailable, which may explain why such a large number of drugs contain this functionality [32,33,34].

Structure of some sulfa drugs
Among their most important effects, they have been reported to exhibit antiproliferative [35], antibacterial [36], antiviral [37], antiprotozoal [38], antifungal [39], and anti-inflammatory [40] properties. Some important sulfonamide derivatives are also effective for the treatment of urinary diseases, intestinal diseases, rheumatoid arthritis [41], obesity [42] and Alzheimer’s disease [43].
Based on the aforementioned data and as an extension of our studies on the development of novel bioactive 1,2,3-triazoles [44,45,46,47,48,49], we report herein the design of compounds containing 1,2,3-triazole, benzimidazole and sulfonamides moieties in one scaffold via a Cu(I)-catalysed 1,3-dipolar cycloaddition reaction of sulfa drug azides with propargylated benzimidazoles derivatives and the synergistic effects of the moieties. The newly designed 1,2,3-triazole hybrids have been examined for their antimicrobial and antiproliferative activities.
Results and discussion
Chemistry
The target 1,2,3-triazole hybrids (4a–f and 6a–f) were synthesized by using commercially available 2-mercaptobenzothiazole (1) as the starting material as depicted in Schemes 1, 2, 3 and 4. First, the thiol functionality in the 2-position of compound 1 was regioselectively alkylated with propargyl bromide in the presence of triethylamine as a basic catalyst in refluxing ethanol for 1 h to afford target thiopropargylated benzimidazole 2 in 94% yield (Scheme 1). It should be noted that the regioselective synthesis of the thiopropargylated benzimidazole 2 has been previously described using different reaction conditions (NaOH/H2O, K2CO3, H2O) [50,51,52].

Synthesis of thiopropargylated benzimidazole 2

Synthesis of mono-1,4-disubstituted-1,2,3-triazole tethered benzimidazole-sulfonamide conjugates 5a–f

Synthesis of S,N-Bispropargylated benzimidazole 5

Synthesis of S,N-bis(1,2,3-triazole-sulfonamide)-benzimidazole hybrids 6a–f
The structure of compound 2 was assigned based on its spectral data. The IR spectrum confirmed that 1 had been monopropargylated based on the characteristic NH absorption band at 3390 cm−1. The spectrum also revealed the presence of two sharp bands at 3390 and 2140 cm−1 related to the acetylenic hydrogen (≡C–H) and the C≡C group, respectively.
The 1H NMR analysis clearly confirmed one propargyl side chain had been incorporated at the sulfur atom of 1 based on the presence of one exchangeable proton in the downfield region (δH12.65 ppm) attributable to the triazolyl NH proton. The propargyl sp-CH and SCH2 protons were assigned to the two singlets at δH 3.20 and 4.16 ppm, respectively. The four benzimidazole protons were observed at their appropriate chemical shifts (7.14–7.51 ppm). The 13C NMR analysis confirmed the incorporation of a propargyl residue by the appearance of diagnostic carbon signals at δC 20.6, 74.5 and 80.5 ppm, which were attributed to the alkyne SCH2 and C≡C groups, respectively. The signals observed at δC 110.9–148.8 ppm were associated with aromatic and C=N carbons.
An azide–alkyne Huisgen cycloaddition reaction was carried out by simultaneously mixing thiopropargylated benzimidazole 2 with the appropriate sulfa drug azide (4a–f), copper sulfate and sodium ascorbate in DMSO/H2O to regioselectively furnish target mono-1,4-disubstituted-1,2,3-triazole tethered benzimidazole-sulfonamide conjugates 5a–f in 85–90% yields after 6–8 h of heating at 80 °C (Scheme 2). The sulfonamide azides were prepared via the diazotization of the appropriate sulfa drugs in a sodium nitrite solution in acidic media followed by the addition of sodium azide.
The formation of compounds 4a–f was confirmed based on their spectroscopic data (IR, 1H NMR and 13C NMR). Their IR spectra revealed the disappearance of peaks belonging to C≡C at 2140 cm−1 and ≡C–H at 3310 cm−1, confirming their involvement in the cycloaddition reaction.
The 1H NMR spectra of compounds 4a–f revealed the disappearance of the signal attributed to the ≡C–H proton at δH 3.20 ppm of the precursor S-alkyne 2 and the appearance of one singlet at δH 8.81–8.87 ppm, which was assigned to the 1,2,3-triazole CH proton. Instead of a signal for the triazolyl CH-proton, the spectra showed two singlets at δH 4.70–4.81 and 10.85–12.08 ppm due to the SCH2 protons and the imidazolic NH proton, respectively. Additionally, the signals of two sp-carbons at 74.5 and 80.5 ppm and the SCH2-carbon at 20.2–26.3 ppm had disappeared from the 13C NMR spectra. New signals were also observed in the aromatic region, and they were assigned to the sp2 carbons of the sulfa drug moiety.
The strategy for synthesizing target S,N-bis-1,2,3-triazoles 6a–f was based on the regioselective alkylation of 1 with two equivalents of propargyl bromide in the presence of two equivalents of potassium carbonate as a basic catalyst according to our reported procedure [53]. Thus, propargylation of compound 5 by propargyl bromide in the presence of K2CO3 in DMF afford S,N-bispropargylated benzimidazole 5 in 91% yield after stirring at room temperature overnight (Scheme 3).
The absence of the SH and NH stretching bands in the IR spectrum of compound 5, and the appearance of the characteristic C≡C and ≡C–H bands at 2150 and 3320 cm−1, respectively, confirmed the incorporation of two alkyne side chains.
In the 1H NMR spectrum of compound 5, the absence of the SH and NH protons confirmed the success of the bis-alkylation reaction. The terminal hydrogens of the two ≡C–H groups appeared as singlets at δH 2.29 and 2.40 ppm. The thiomethylene protons (–SCH2) resonated as a distinct upfield singlet at δH 4.14 ppm. The 1H NMR spectrum also revealed the presence of a singlet at δH 4.93 ppm that integrated to two protons attributable to the NCH2 group. In the 13C NMR spectrum of compound 5, the signals characteristic of the sp C≡C carbons resonated at δC 72.3–78.5 ppm, while the SCH2 and NCH2 carbons appeared at δC 21.8 and 33.6 ppm, respectively. Additional signals were also observed in the aromatic region (δC 109.3–149.1 ppm), and these were attributed to the carbons in the benzimidazole ring.
The S,N-bis(1,2,3-triazole-sulfonamide)-benzimidazole hybrids (6a–f) were synthesized using the same click procedure as described above (Scheme 4). However, the synthesis was conducted using two equivalents of sulfa drug azides 3a–f by a copper-mediated Huisgen 1,3-dipolar cycloaddition reaction in the presence of copper sulfate and sodium ascorbate, and this reaction generated 1,4-disubstituted 1,2,3-triazoles 6a–f in 82–88%.
The structures of S,N-bis(1,2,3-triazoles) 6a–f were established on the basis of their spectral data, which indicated the presence of two 1,2,3-triazole moieties based on the absence of the signals for C≡C and ≡C–H at 2150 and 3320 cm−1, respectively.
The 1H NMR spectra of compounds 6a–f confirmed the presence of the two alkyne linkages between the two 1,2,3-triazole rings based on the disappearance of the sp-carbon signals and the appearance of two triazolyl CH-protons at δH 8.85–8.93 ppm. The SCH2 and NCH2 protons were assigned to the two singlets at δH 4.77–4.80 and 5.54–5.58 ppm, respectively. The aromatic protons of the sulfa drug moieties appeared in the appropriate aromatic region. The chemical structures of compounds 6a–f were further elucidated from their 13C NMR spectra, which revealed the presence of SCH2 and NCH2 carbon signals at δC 26.6–27.2 and 40.1–42.3 ppm, respectively. In the cyclization of 5 to 6a–f, the terminal sp carbons disappeared, and new signals that could be assigned to the sulfa drug moieties appeared in the downfield region.
Biological study
Antimicrobial screening
An antimicrobial screening against a group of pathogenic microorganisms, including Gram-positive bacteria, Gram-negative bacteria, and fungi, was carried out for the newly synthesized compounds, and the results are summarized in Table 1. Antimicrobial activities are presented as the minimum inhibitory concentrations (MICs), which is the lowest concentration of the examined compound that resulted in more than 80% growth inhibition of the microorganism [54, 55]. In general, the mono-1,2,3-triazole derivatives (4a–f) exhibited less potent antimicrobial activities than their bis-1,2,3-triazoles (6a–f) counterparts; this could be attributed to the synergistic effect of the sulfonamoyl and tethered heterocyclic components in addition to the improved lipophilicity of the bis-substituted derivatives.
Antiproliferative screening
The newly synthesized compounds were examined for their in vitro antiproliferative activity against a human prostate cancer cell line (PC-3), a human liver cancer cell line (HepG2), and a human kidney cancer cell line (HEK293). The correlation between the percentage of proliferating cells and the drug concentration was plotted to generate the proliferation curves of the cancer cell lines. The IC50 values were calculated and were defined as the response parameter that corresponds to the concentration required for 50% inhibition of cell proliferation. The results are presented in Table 2.
Sulfonamides are a valuable chemical scaffold with numerous pharmacological activities including antibacterial, anticarbonic anhydrase, diuretic, hypoglycaemic, and antithyroid activity [56,57,58]. Notably, structurally novel sulfonamide analogues have been shown to possess significant antitumour activities both in vitro and in vivo. Several mechanisms, such as an anti-angiogenesis effect via matrix metalloproteinase inhibition, carbonic anhydrase inhibition, cell cycle arrest and the disruption of microtubule assembly, have been proposed to explain this interesting activity [59,60,61].
Interestingly, the newly synthesized compounds exhibited considerable antiproliferative activities against the three cancer cell lines used in this study with IC50 values ranging from 55 to 106 μM. Further investigation should shed light on the exact mechanism through which the antiproliferative activity is exerted.
POM analysis
Prediction of pharmacologically relevant inhibition
POM theory is robust and available method to confirm the reliability of experimental data. In actuality, the benefit of POM theory is the ability to predict the biological activities of molecules and easily establish the relationship between steric and electrostatic properties and biological activity. Evaluation of in silico physicochemical properties or ADMET (adsorption, distribution, metabolism, excretion and toxicity) is a robust tool to confirm the potential of a drug candidate [62]. Drug-likenesses of a library of compounds were evaluated by Lipinski’s rule of five, and 90% of orally active compounds follow Lipinski’s rule of five [63]. As per Lipinski’s rule of five, an orally administered drug should have a log P ≤ 5, a molecular weight (MW) < 500 Daltons and an HBD ≤ 5 [63] to be in the acceptable range. Results have shown that all compounds have in good agreement in term of HBD, except compound 6a. This set of criteria is also called Veber’s rule. However, compounds that meet the criteria, i.e., topological polar surface area (TPSA) ≤ 140 Å, are expected to have appropriate oral bioavailability [64]. TPSA is a parameter used to predict the transport properties of drugs in passive molecular transport [64]. The compounds that showed good oral bioavailability or cell permeability were those having TPSA values between 118 and 155 for 4a–f and 197–271 for 6a–f (Table 3).
As shown in Table 3, the drug likeness values of the synthesized compounds are larger than that of the standard. The overall drug score (DS) values calculated for sulfonamides 4a–f and 6a–f used ciprofloxacin and fluconazole as the standard drugs, as shown in Table 3. Better drug scores indicate that the compound is more likely to become a drug candidate.
In silico bioavailability prediction and cLogP
The hydrophilicity and cLogP values are correlated because hydrophilicity depends on and is expressed in term of the cLogP value. As cLogP increases above 5, absorption and permeability decrease. From Table 4, it is clear that our synthesized all sulfa drugs are in the accepting range i.e., lower than 5 (between 0.75 and 4.41) and are potentially active against various biotargets (GPCRL: GPCR ligand; ICM: ion channel modulator; KI: kinase inhibitor; NRL: nuclear receptor ligand; PI: protease inhibitor; and EI: enzyme inhibitor), which confirm the good permeability of all tested molecules. To confirm the reliability of the cLogP values and the agreement of these values with the bioavailability, we determined four combine parameters, i.e., the Lipinski, Ghose [65] and Veber rules [66] and the bioavailability score [67], and the results are summarized in Table 4. It is clear from Table 4 that only sulfa drugs 4a–f follow Lipinski rule. Likewise, only sulfa drugs 4a–h follow the Ghose’s rule. In contrast, the screening process showed that none of the sulfa drugs follow Veber’s rule in term of agreement with the in silico bioavailability.
In silico pharmacokinetic analysis of the synthesized sulfonamides
Due to poor pharmacokinetics, most drugs fail to move into clinic trials in the discovery process. Pharmacokinetics determine the human therapeutic use of compounds, and these properties depend on the absorption, distribution, metabolism, excretion, and toxicity (ADMET) properties [68, 69], which is why in silico pharmacokinetic studies are necessary to minimize the possibility of failure of any drug in clinical trials. In silico pharmacokinetic has explained in term of ADME/T and toxicity. Further analyzed in silico data has been correlated and found in good agreement (Table 5).
In silico toxicity analysis
In silico carcinogenicity has been evaluated and tabulated in Table 6. It was found that all the synthesized sulfonamides were noncarcinogenic. In Table 6, the green colour indicates drug-like behaviour. For further investigation of the in vivo antimicrobial activity, the computed LD50 in rat from the acute toxicity model seems to be sufficiently safe (2.29–2.41 mol/kg).
Materials and methods
General methods
Melting points were measured on a melt-temp apparatus (SMP10) and are uncorrected. TLC analyses were performed on silica gel-coated aluminium plates (Kieselgel, 0.25 mm, 60 F254, Merck, Germany), and spots were visualized by ultraviolet (UV) light absorption using a developing solvent system of ethyl acetate/hexane. The IR spectra were measured in a KBr matrix using a SHIMADZU FTIR-8400S spectrometer. 1H NMR spectra were recorded using an Advance Bruker NMR spectrometer at 400–600 MHz, whereas 13C NMR spectra were recorded on the same instrument at 100–150 MHz using tetramethylsilane (TMS) as the internal standard. High-resolution mass spectrometry (HRMS) was carried out using an LC–MS/MS impact II.
Synthesis and characterization of 2-(prop-2-yn-1-ylthio)-1H-benzo[d]imidazole (2)
To a solution of 2-mercaptobenzimidazole (1) (10 mmol) in ethanol (40 mL) and triethylamine (Et3N) (12 mmol) was added propargyl bromide (12 mmol) with stirring, and the solution was heated to reflux for 1 h. The excess solvent was removed under reduced pressure, and the resulting crude product was washed with water and recrystallized from ethanol to afford compound 2 in 94% yield as colourless crystals, mp: 163–164 °C (lit. 164–165 °C [50, 51]); IR (KBr) υmax/cm−1 1580 (C=C), 1615 (C=N), 2140 (C≡C), 2950 (C–H al), 3070 (C–H Ar), 3310 cm−1 (≡CH), 3390 cm−1 (N–H). 1H NMR (400 MHz, DMSO-d6) δH = 3.20 (s, 1H, ≡CH), 4.16 (s, 2H, SCH2), 7.14–7.16 (m, 2H, Ar–H), 7.46–7.51 (m, 2H, Ar–H), 12.65 (s, 1H, NH). 13C NMR (100 MHz, DMSO-d6) δC = 20.6 (SCH2); 74.5, 80.5 (C≡C); 110.9, 118.0, 122.1, 122.6, 135.9, 144.1, 148.8 (Ar–C, C=N). HRMS (ESI): 188.0410 [M+].
Synthesis of 1,4-disubstituted mono-1,2,3-triazoles 4a–f
To a solution of compound 2 (1 mmol) in a 1:1 mixture of dimethyl sulfoxide (DMSO) and water (20 mL), CuSO4 (0.10 g) were added Na ascorbate (0.15 g) and the appropriate sulfonamide azide (3a–f, 1 mmol) with stirring. The resulting mixture was stirred at 80 °C for 6–8 h. The consumption of the starting materials was monitored using TLC. The reaction mixture was quenched with water, and the solid thus formed was collected by filtration, washed with a saturated solution of sodium chloride and recrystallized from ethanol to give the desired 1,2,3-triazoles (4a–f).
4-(4-((1H-Benzo[d]imidazol-2-ylthio)methyl)-1H-1,2,3-triazol-1-yl)-N-(4,6-dimethylpyrimidin-2-yl)benzenesulfonamide (4a). White solid; Yield: 90%; mp: 153–154 °C; IR (KBr) υmax/cm−1 1580 (C=C), 1620 (C=N), 2935 (C–H al), 3045 (C–H Ar), 3340–3385 cm−1 (N–H). 1H NMR (400 MHz, DMSO-d6) δH = 2.26 (s, 6H, 2 × CH3), 4.76 (s, 2H, SCH2), 6.73 (bs, 1H, Ar–H), 7.13 (bs, 2H, Ar–H), 7.44–7.54 (m, 2H, Ar–H), 7.89–8.13 (m, 4H, Ar–H), 8.86 (bs, 1H, CH-1,2,3-triazole), 12.04 (bs, 1H, NH), 12.86 (s, 1H, NH). 13C NMR (100 MHz, DMSO-d6) δC = 20.2 (CH3), 24.7 (SCH2), 110.8, 116.1, 117.4, 120.1, 122.3, 122.5, 123.7, 130.0, 138.9, 139.9, 140.2, 142.8, 143.3, 154.0, 164.2 (Ar–C, C=N). HRMS (ESI): 492.1296 [M+].
4-(4-((1H-Benzo[d]imidazol-2-ylthio)methyl)-1H-1,2,3-triazol-1-yl)-N-(pyrimidin-2-yl)benzenesulfonamide (4b). White solid; Yield: 87%; mp: 165–166 °C; IR (KBr) υmax/cm−1 1585 (C=C), 1625 (C=N), 2910 (C–H al), 3065 (C–H Ar), 3330–3395 cm−1 (N–H). 1H NMR (400 MHz, DMSO-d6) δH = 4.74 (s, 2H, SCH2), 7.06–7.13 (m, 3H, Ar–H), 7.50 (bs, 2H, Ar–H), 8.11–8.16 (bs, 4H, Ar–H), 8.52 (bs, 2H, Ar–H), 8.87 (s, 1H, CH-1,2,3-triazole), 12.08 (bs, 1H, NH), 12.69 (bs, 1H, NH). 13C NMR (100 MHz, DMSO-d6) δC = 26.3 (SCH2), 110.6, 116.1, 117.2, 120.6, 122.0, 122.4, 125.9, 129.9, 139.6, 140.1, 140.4, 142.6, 143.5, 155.2, 163.9 (Ar–C, C=N). HRMS (ESI): 464.1272 [M+].
4-(4-(((1H-Benzo[d]imidazol-2-yl)thio)methyl)-1H-1,2,3-triazol-1-yl)-N-(pyridin-2-yl)benzenesulfonamide (4c). White solid; Yield: 85%; mp: 216–218 °C; IR (KBr) υmax/cm−1 1575 (C=C), 1610 (C=N), 2930 (C–H al), 3040 (C–H Ar), 3290–3365 cm−1 (N–H). 1H NMR (600 MHz, DMSO-d6) δH = 4.70 (s, 2H, SCH2), 6.84 (bs, 1H, Ar–H), 7.11–7.21 (m, 3H, Ar–H), 7.40–7.54 (m, 2H, Ar–H), 7.75 (m, 1H, J = 6 Hz, Ar–H), 7.84–7.95 (m, 2H, Ar–H), 7.95–8.10 (m, 4H, Ar–H), 8.81 (s, 1H, CH-1,2,3-triazole), 12.59 (bs, 1H, NH), 12.63 (bs, 1H, NH). 13C NMR (100 MHz, DMSO-d6) δC = 25.8 (SCH2), 110.4, 117.5, 120.3, 121.2, 121.8, 122.0, 125.6, 128.2, 134.2, 135.5, 138.5, 141.8, 144.8, 149.1, 163.5 (Ar–C, C=N). HRMS (ESI): 463.0975 [M+].
4-(4-((1H-Benzo[d]imidazol-2-ylthio)methyl)-1H-1,2,3-triazol-1-yl)-N-(thiazol-2-yl)benzenesulfonamide (4d). White solid; Yield: 89%; mp: 148–150 °C; IR (KBr) υmax/cm−1 1590 (C=C), 1610 (C=N), 2925 (C–H al), 3055 (C–H Ar), 3315–3380 cm−1 (N–H). 1H NMR (400 MHz, DMSO-d6) δH = 4.72 (s, 2H, SCH2), 6.86–7.27 (m, 6H, Ar–H), 7.99 (bs, 4H, Ar–H), 8.86 (s, 1H, CH-1,2,3-triazole), 12.52 (bs, 2H, 2 × NH). 13C NMR (100 MHz, DMSO-d6) δC = 20.2 (SCH2), 110.8, 114.2, 116.1, 117.4, 122.3, 122.5, 123.7, 130.0, 135.4, 138.9, 139.9, 140.2, 142.8, 143.3, 154.5, 161.3 (Ar–C, C=N). HRMS (ESI): 469.0896 [M+].
4-(4-(((1H-Benzo[d]imidazol-2-yl)thio)methyl)-1H-1,2,3-triazol-1-yl)-N-(3,4-dimethylisoxazol-5-yl)benzenesulfonamide (4e). White solid; Yield: 88%; mp: 204–206 °C; IR (KBr) υmax/cm−1 1570 (C=C), 1620 (C=N), 2975 (C–H al), 3080 (C–H Ar), 3300–3395 cm−1 (N–H). 1H NMR (600 MHz, DMSO-d6) δH = 2.21 (s, 3H, CH3), 2.55 (s, 3H, CH3), 4.81 (s, 2H, SCH2), 7.09–7.16 (m, 2H, Ar–H), 7.49–7.54 (m, 2H, Ar–H), 7.77–7.84 (m, 2H, Ar–H), 7.98–8.03 (m, 2H, Ar–H), 8.87 (s, 1H, CH-1,2,3-triazole), 10.85 (bs, 1H, NH), 13.36 (bs, 1H, NH). 13C NMR (150 MHz, DMSO-d6) δC = 21.0 (CH3), 23.2 CH3), 26.3 (SCH2), 111.0, 114.0, 117.5, 119.7, 120.5, 122.5, 127.0, 129.5, 135.8, 138.5, 139.7, 140.8, 143.1, 148.9, 162.5 (Ar–C, C=N). HRMS (ESI): 481.0934 [M+].
4-(4-(((1H-Benzo[d]imidazol-2-yl)thio)methyl)-1H-1,2,3-triazol-1-yl)-N-(diaminomethylene)benzenesulfonamide (4f). White solid; Yield: 90%; mp: 244–246 °C;IR (KBr) υmax/cm−1 1570 (C=C), 1615 (C=N), 2980 (C–H al), 3025 (C–H Ar), 3265–3380 cm−1 (N–H). 1H NMR (600 MHz, DMSO-d6) δH = 4.73 (s, 2H, SCH2), 6.70 (bs, 4H, 2 × NH2), 7.13 (dd, 2H, J = 6, 12 Hz, Ar–H), 7.48 (bs, 2H, Ar–H), 7.92–8.01 (m, 4H, Ar–H), 8.81 (s, 1H, CH-1,2,3-triazole), 12.57 (bs, 2H, 2 × NH). 13C NMR (150 MHz, DMSO-d6) δC = 25.9 (SCH2), 120.2, 121.60, 122.0, 127.4, 135.7, 138.1, 144.3, 144.8, 148.8, 158.2 (Ar–C, C=N). HRMS (ESI): 428.0841 [M+].
Synthesis and characterization of 1-(prop-2-yn-1-yl)-2-(prop-2-yn-1-ylthio)-1H-benzo[d]imidazole (5)
A mixture of 2-mercaptobenzimidazole (1) (10 mmol), dimethylformamide (DMF) (20 mL) and potassium carbonate (22 mmol) were stirred at room temperature for 2 h. Then, propargyl bromide (24 mmol) was added, and the mixture was stirred overnight at room temperature. The consumption of the starting materials was monitored using TLC. The reaction mixture was poured into crushed ice. The product was collected by filtration, washed with water and recrystallized from ethanol to afford compound 5 in 91% yield as colourless crystals. mp: 72–73 °C (lit. 70–71 °C [53]); 1585 (C=C), 1610 (C=N), 2150 (C≡C), 2930 (C–H al), 3045 (C–H Ar), 3320 cm−1 (≡CH). 1H NMR (400 MHz, DMSO-d6) δH = 2.29 (s, 1H, ≡CH), 2.40 (s, 1H, ≡CH), 4.14 (s, 2H, SCH2), 4.93 (s, 2H, NCH2), 7.27–7.31 (m, 2H, Ar–H), 7.42–7.45 (m, 1H, Ar–H), 7.73–7.77 (m, 1H, Ar–H). 13C NMR (100 MHz, DMSO-d6) δC = 21.8 (SCH2); 33.6 (NCH2); 72.3, 73.8, 76.3, 78.5 (C≡C); 109.3, 118.9, 122.5, 122.7, 135.5, 143.4, 149.1 (Ar–C, C=N). HRMS (ESI): 226.0569 [M+].
Synthesis of 1,4-disubstituted bis-1,2,3-triazoles 6a–f
To a solution of compound 5 (1 mmol) in a 1:1 mixture of dimethyl sulfoxide (DMSO) and water (20 mL) were added CuSO4 (0.20 g), Na ascorbate (0.30 g) and sulfonamide azide (3a–f, 2 mmol) with stirring. The resulting mixture was stirred at 80 °C for 8–12 h. The consumption of the starting materials was monitored using TLC. The reaction mixture was quenched with water, and the solid thus formed was collected by filtration, washed with a saturated solution of sodium chloride and recrystallized from ethanol to give the desired 1,2,3-triazoles (6a–f).
N-(4,6-Dimethylpyrimidin-2-yl)-4-(4-((1-((1-(4-(N-(4,6-dimethylpyrimidin-2-yl)sulfamoyl)-phenyl)-1H-1,2,3-triazol-4-yl)methyl)-1H-benzo[d]-imidazol-2-ylthio)methyl)-1H-1,2,3-triazol-1-yl)benzenesulfonamide (6a). White solid; Yield: 87%; mp: 176–178 °C; IR (KBr) υmax/cm−1 1595 (C=C), 1630 (C=N), 2915 (C–H al), 3070 (C–H Ar), 3310–3370 cm−1 (N–H). 1H NMR (400 MHz, DMSO-d6) δH = 2.55 (s, 6H, 2 x CH3), 4.78 (s, 2H, SCH2), 5.56 (s, 2H, NCH2), 6.72 (bs, 2H, Ar–H), 7.20 (bs, 2H, Ar–H), 7.64–7.66 (m, 2H, Ar–H), 8.06–8.15 (m, 8H, Ar–H), 8.87 (s, 1H, CH-1,2,3-triazole), 8.95 (s, 1H, CH-1,2,3-triazole), 12.21 (s, 2H, NH). 13C NMR (100 MHz, DMSO-d6) δC = 27.2 (SCH2), 40.2 (NCH2), 23.0 (CH3), 110.5, 116.3, 117.5, 120.1, 122.3, 122.4, 122.5, 122.7, 130.2, 135.3, 139.0, 140.3, 142.6, 143.9, 149.4, 154.2, 156.2, 164.6 (Ar–C, C=N). HRMS (ESI): 834.2319 [M+].
N-(Pyrimidin-2-yl)-4-(4-((1-((1-(4-(N-pyrimidin-2-ylsulfamoyl)phenyl)-1H-1,2,3-triazol-4-yl)-methyl)-1H-benzo[d]imidazol-2-ylthio)methyl)-1H-1,2,3-triazol-1-yl)benzenesulfonamide (6b). White solid; Yield: 83%; mp: 199–201 °C; IR (KBr) υmax/cm−1 1580 (C=C), 1610 (C=N), 2925 (C–H al), 3040 (C–H Ar), 3320–3375 cm−1 (N–H). 1H NMR (400 MHz, DMSO-d6) δH = 4.78 (s, 2H, SCH2), 5.57 (s, 2H, NCH2), 7.06 (s, 2H, Ar–H), 7.20 (bs, 2H, Ar–H), 7.63 (bs, 2H, Ar–H), 8.07–8.17 (m, 8H, Ar–H), 8.51 (bs, 4H, Ar–H), 8.88 (s, 2H, 2 × CH-1,2,3-triazole), 12.11 (s, 2H, 2 × NH). 13C NMR (100 MHz, DMSO-d6) δC = 26.6 (SCH2), 40.4 (NCH2), 109.8, 116.5, 118.0, 120.1, 122.0, 122.2, 122.6, 123.1, 129.4, 130.4, 135.6, 139.1, 140.2, 142.8, 143.4, 144.5, 149.8, 154.1, 156.6, 165.0 (Ar–C, C=N). HRMS (ESI): 778.12077 [M+].
N-(Pyridin-2-yl)-4-(4-(((1-((1-(4-(N-(pyridin-2-yl)sulfamoyl)phenyl)-1H-1,2,3-triazol-4-yl)-methyl)-1H-benzo[d]imidazol-2-yl)thio)methyl)-1H-1,2,3-triazol-1-yl)benzenesulfonamide (6c). White solid; Yield: 82%; mp: 220–222 °C; IR (KBr) υmax/cm−1 1580 (C=C), 1630 (C=N), 2985 (C–H al), 3025 (C–H Ar), 3280–3350 cm−1 (N–H). 1H NMR (600 MHz, DMSO-d6) δH = 4.77 (s, 2H, SCH2), 5.54 (s, 2H, NCH2), 6.85 (bs, 2H, Ar–H), 7.19–7.26 (m, 4H, Ar–H), 7.61–7.64 (m, 2H, Ar–H), 7.75–7.77 (m, 2H, Ar–H), 7.88–7.92 (m, 2H, Ar–H), 7.97–8.04 (m, 8H, Ar–H), 8.84 (s, 1H, CH-1,2,3-triazole), 8.93 (s, 1H, CH-1,2,3-triazole), 12.41 (s, 2H, NH). 13C NMR (150 MHz, DMSO-d6) δC = 26.7 (SCH2), 40.4 (NCH2), 110.1, 117.9, 119.5, 120.3, 120.3, 121.8, 122.0, 122.1, 122.2, 128.2, 128.5, 135.9, 138.4, 142.9, 144.4, 150.3, 154.8, 156.4, 164.7 (Ar–C, C=N). HRMS (ESI): 776.2614 [M+].
N-(Thiazol-2-yl)-4-(4-((1-((1-(4-(N-thiazol-2-ylsulfamoyl)phenyl)-1H-1,2,3-triazol-4-yl)methyl)-1H-benzo[d]imidazol-2-ylthio)methyl)-1H-1,2,3-triazol-1-yl)benzenesulfonamide (6d). White solid; Yield: 85%; mp: 158–160 °C; IR (KBr) υmax/cm−1 1580 (C=C), 1625 (C=N), 2945 (C–H al), 3030 (C–H Ar), 3325–3370 cm−1 (N–H). 1H NMR (400 MHz, DMSO-d6) δH = 4.78 (s, 2H, SCH2), 5.56 (s, 2H, NCH2), 6.87 (bs, 2H, Ar–H), 7.20–7.29 (m, 4H, Ar–H), 7.61–7.65 (m, 2H, Ar–H), 7.80–8.05 (m, 8H, Ar–H), 8.86 (s, 1H, CH-1,2,3-triazole), 8.95 (s, 1H, CH-1,2,3-triazole), 12.86 (s, 2H, 2 × NH). 13C NMR (100 MHz, DMSO-d6) δC = 27.2 (SCH2), 41.1 (NCH2), 109.0, 110.5, 118.3, 119.9, 120.8, 120.9, 122.3, 122.4, 122.5, 122.6, 125.1, 128.0, 128.2, 139.0, 139.1, 142.5, 142.6, 143.4, 143.9, 144.9, 150.8, 154.3, 169.5 (Ar–C, C=N). HRMS (ESI): 788.0685 [M+].
N-(3,4-Dimethylisoxazol-5-yl)-4-(4-(((1-((1-(4-(N-(3,4-dimethylisoxazol-5-yl)sulfamoyl)phenyl)-1H-1,2,3-triazol-4-yl)methyl)-1H-benzo[d]imidazol-2-yl)thio)-methyl)-1H-1,2,3-triazol-1-yl)benzenesulfonamide (6e). White solid; Yield: 85%; mp: 238–240 °C; IR (KBr) υmax/cm−1 1580 (C=C), 1610 (C=N), 2955 (C–H al), 3045 (C–H Ar), 3315–3370 cm−1 (N–H). 1H NMR (600 MHz, DMSO-d6) δH = 2.08 (s, 6H, 2 × CH3), 2.57 (s, 3H, CH3), 4.80 (s, 2H, SCH2), 5.58 (s, 2H, NCH2), 7.21–7.23 (m, 2H, Ar–H), 7.53–7.63 (m, 4H, Ar–H), 7.95–8.14 (m, 6H, Ar–H), 8.92 (bs, 2H, 2 × CH-1,2,3-triazole), 10.75 (bs, 1H, NH), 11.17 (bs, 1H, NH). 13C NMR (150 MHz, DMSO-d6) δC = 18.5 (CH3), 21.0 (CH3), 26.7 (SCH2), 42.3 (NCH2), 109.8, 110.1, 117.9, 120.6, 121.9, 122.0, 122.7, 122.8, 128.6, 129.6, 135.9, 139.6, 142.8, 143.6, 144.2, 150.3, 155.0, 168.8 (Ar–C, C=N). HRMS (ESI): 812.1731 [M+].
N-(Diaminomethylene)-4-(4-(((1-((1-(4-(N-(diaminomethylene)sulfamoyl)phenyl)-1H-1,2,3-triazol-4-yl)methyl)-1H-benzo[d]imidazol-2-yl)thio)methyl)-1H-1,2,3-triazol-1-yl)benzenesulfonamide (6f). White solid; Yield: 88%; mp: 276–278 °C; IR (KBr) υmax/cm−1 1575 (C=C), 1620 (C=N), 2950 (C–H al), 3040 (C–H Ar), 3260–3350 cm−1 (N–H). 1H NMR (600 MHz, DMSO-d6) δH = 4.79 (s, 2H, SCH2), 5.58 (s, 2H, NCH2), 6.80 (bs, 2H, 2 × NH2), 7.19–7.20 (m, 2H, Ar–H), 7.63–7.67 (m, 2H, Ar–H), 7.91–7.98 (m, 8H, Ar–H), 8.85 (s, 1H, CH-1,2,3-triazole), 8.92 (s, 1H, CH-1,2,3-triazole), 12.40 (bs, 2H, NH). 13C NMR (150 MHz, DMSO-d6) δC = 26.7 (SCH2), 40.1 (NCH2), 110.1, 111.3, 117.8, 120.1, 120.2, 122.1, 122.5, 122.8, 127.2, 128.3, 135.4, 137.9, 143.2, 144.3, 149.3, 158.0 (Ar–C, C=N). HRMS (ESI): 706.1343 [M+].
Biological activity
Antimicrobial activity
Minimal inhibitory concentration (MIC) determination
The microdilution susceptibility tests were carried out in Müller–Hinton broth (Oxoid) and Sabouraud liquid medium (Oxoid) for the assessment of antibacterial and antifungal activity, respectively. The newly synthesized compounds were evaluated for their antimicrobial activity against four pathogenic bacterial strains [Gram-positive: Bacillus cereus (ATTC 10876) and Staphylococcus aureus (ATTC 25923) and Gram-negative: Escherichia coli (ATTC 25922) and Pseudomonas aeruginosa (ATTC 27853)] and two fungal strains [(Candida albicans (ATTC 50193) and Aspergillus brasiliensis (ATTC 16404)].
The examined compounds were dissolved in DMSO, and stock solutions were prepared at a concentration of 10 mM before being further diluted to the desired concentrations. Ciprofloxacin and fluconazole were used as standard antimicrobial agents. A 10 µL aliquot of the medium containing approximately 106 CFU/mL of each bacterial species or 104 CFU/mL of the test fungus was added to each well of a 96-well microtiter plate. The wrapped microplates were incubated at 37 °C for 24 h to measure antibacterial activity and at 25 °C for 48 h for antifungal activity in a humidified atmosphere. Optical densities were measured at 600 nm (OD600) using a microplate reader (Palo Alto, CA, USA). The minimal inhibitory concentrations (MICs) were determined at the end of the incubation period and were defined as the lowest concentration at which more than 80% of the microbial growth was inhibited. MIC assessments were carried out in triplicate and repeated three times for each microorganism, and the SD values never exceeded 5%. Control experiments with the standard antimicrobial agents (positive control) and the uninoculated media (negative control) were performed parallel to the examined compounds and under the same conditions.
Antiproliferative activity
MTT assay
Logarithmically growing cells were washed with PBS, detached from the surface with trypsin, and then transferred to fresh cultured medium containing 10% FBS, 100 U/mL penicillin and 0.1 mg/mL streptomycin. Cells were plated at a density of 1 × 104 cell/well into 96-well culture plates and incubated at 37 °C in an atmosphere containing 5% CO2 for 24 h to allow for adhesion. Stock solutions (1 mM) of the newly synthesized compounds were freshly prepared in dimethyl sulfoxide (DMSO) prior to the experiment and applied on the cells at concentrations ranging from 1 to 300 µM. The highest DMSO concentration in the medium (0.1%) did not have any significant effect on cell proliferation. Cells were incubated with the tested compounds for 48 h. Control wells were treated with 0.1% DMSO in medium or the standard antiproliferative agent doxorubicin. At the end of the exposure time (48 h), the medium was removed, and the wells were washed with 200 µL of PBS. Afterward, 100 µL of freshly prepared MTT [(3-[4,5-dimethylthiazole-2-yl]-2,5-diphenyl-tetrazolium bromide)] reagent was added to each well, and the plates were incubated at 37 °C for 4 h. Then, the supernatant was aspirated, and 100% DMSO was added to solubilize the formed formazan crystals. The optical density (OD) was obtained by reading the absorbance using an ELISA plate reader at 540 nm and 670 nm. Cell proliferation percentages were plotted against the examined compound concentrations to determine the IC50 values. The human prostate cancer cell line (PC-3), the human liver cancer cell line (HepG2), and the human kidney cancer cell line (HEK293) were used in this study. Each concentration of the examined compounds was tested in triplicate, and IC50 values, i.e., concentration of the compound at which 50% inhibition of cell proliferation occurred, was calculated based on the mean value of triplicate readings.
Conclusions
In summary, we have successfully synthesized novel targeted benzimidazole-1,2,3-triazole-sulfonamide hybrids through 1,3-dipolar cycloaddition reactions between the mono- or bis-alkynes based on imidazole and the appropriate sulfonamide azide under the optimized Cu(I) click conditions. The structures of newly synthesized sulfonamide hybrids were confirmed by means of spectroscopic analysis. All newly synthesized compounds were evaluated for their antimicrobial and antiproliferative activities. Our results showed that the benzimidazole-1,2,3-triazole-sulfonamide hybrids inhibited microbial and fungal strains within MIC values from 32 to 64 μg/mL. The antiproliferative evaluation of the synthesized compounds showed sufficient antiproliferative activities with IC50 values in the micromolar range (55–106 μM). In conclusion, compound 6a has remarkable antimicrobial activity. Pharmacophore elucidation of the compounds was performed based on in silico ADMET evaluation of the tested compounds. Screening results of drug-likeness rules showed that all compounds follow the accepted rules, meet the criteria of drug-likeness and follow Lipinski’s rule of five. In addition, the toxicity results showed that all compounds are nonmutagenic and noncarcinogenic.
References
Yoneyama H, Katsumata R (2006) Antibiotic resistance in bacteria and its future for novel antibiotic development. Biosci Biotechnol Biochem 70:1060–1075. https://doi.org/10.1271/bbb.70.1060
Dheer D, Singh V, Shankar R (2017) Medicinal attributes of 1,2,3-triazoles: current developments. Bioorg Chem 71:30–54. https://doi.org/10.1016/j.bioorg.2017.01.010
Faidallah HM, Girgis AS, Tiwari AD, Thomas Honkanadavar HH, Samir A et al (2018) Synthesis, antibacterial properties and 2D-QSAR studies of quinolone-triazole conjugates. Eur J Med Chem 143:1524–1534. https://doi.org/10.1016/j.ejmech.2017.10.042
Jordao AK, Ferreira VF, Souza TM, Faria GG, Machado V, Abrantes JL et al (2011) Synthesis and anti-HSV-1 activity of new 1,2,3-triazole derivatives. Bioorg Med Chem 19:1860–1865. https://doi.org/10.1016/j.bmc.2011.02.007
Dai ZC, Chen YF, Zhang M, Li SK, Yang TT, Shen L et al (2015) Synthesis and antifungal activity of 1,2,3-triazole phenylhydrazone derivatives. Org Biomol Chem 13:477–486. https://doi.org/10.1039/C4OB01758G
Boechat N, Ferreira Mde L, Pinheiro LC, Jesus AM, Leite MM, Júnior CC et al (2014) New compounds hybrids 1H-1,2,3-triazole-quinoline against Plasmodium falciparum. Chem Biol Drug Des 84:325–332. https://doi.org/10.1111/cbdd.12321
Whiting M, Muldoon J, Lin YC, Silverman SM, Lindstron W, Olson AJ et al (2006) Inhibitors of HIV-1 protease by using in situ click chemistry. Angew Chem Int Ed 45:1435–1439. https://doi.org/10.1002/anie.200502161
Buckle DR, Outred DJ, Rockell CJM, Smith H, Spicer BA (1983) Studies on v-Triazoles. 7. Antiallergic 9-Oxo-1H, 9H-benzopyrano [2,3-d]-v–triazoles. J Med Chem 26:251–254. https://doi.org/10.1021/jm00356a025
Zhang S, Xu Z, Gao C, Ren QC, Chang L, Lv ZS et al (2017) Triazole derivatives and their anti-tubercular activity. Eur J Med Chem 138:501–513. https://doi.org/10.1016/j.ejmech.2017.06.051
Shukla JD, Khan PMAA, Deo K (2015) Synthesis and pharmacological evaluation of novel 1-(2,6-difluorobenzyl)-1H-1,2,3-triazole derivatives for CNS depressant and anticonvulsant profile. Am J Pharm Tech Res 5:423–433
Rajasekaran A, Rajagopal KA (2009) Synthesis of some novel triazole derivatives as anti-nociceptive and anti-inflammatory agents. Acta Pharm 59:355–364. https://doi.org/10.2478/v10007-009-0026-7
Nassar EM, Abdelrazek FM, Ayyad RR, El-Farargy AF (2016) Synthesis and some reactions of 1-aryl-4-acetyl-5-methyl-1,2,3-triazole derivatives with anticonvulsant activity. Mini Rev Med Chem 16:926–936. https://doi.org/10.2174/1389557516666160118105505
Reddy MK, Kumar KS, Sreenivas P, Krupadanam GLD, Reddy KJ (2011) Synthesis of novel 1,4-disubstituted-1,2,3-triazole semi synthetic analogues of forskolin by click reaction. Tetrahedron Lett 52:6537–6540. https://doi.org/10.1016/j.tetlet.2011.08.154
Penthala NR, Madhukuri L, Thakkar S, Madadi NR, Lamture G, Eoff RL et al (2015) Synthesis and anti-cancer screening of novel heterocyclic-(2H)- 1,2,3-triazoles as potential anti-cancer agents. Med Chem Commun 6:1535–1543. https://doi.org/10.1039/C5MD00219B
Kant R, Singh V, Nath G, Awasthi SK, Agarwal A (2016) Design, synthesis and biological evaluation of ciprofloxacin tethered bis-1,2,3-triazole conjugates as potent antibacterial agents. Eur J Med Chem 124:218–228. https://doi.org/10.1016/j.ejmech.2016.08.031
Gregorić T, Sedić M, Grbčić P, Paravić AT, Pavelić SK, Cetina M et al (2017) Novel pyrimidine-2,4-dione–1,2,3-triazole and furo[2,3-d]pyrimidine-2-one-1,2,3-triazole hybrids as potential anti-cancer agents: synthesis, computational and X-ray analysis and biological evaluation. Eur J Med Chem 125:1247–1267. https://doi.org/10.1016/j.ejmech.2016.11.028
Ouahrouch A, Ighachane H, Taourirte M, Engels JW, Sedra MH, Lazrek HB (2014) Benzimidazole-1,2,3-triazole hybrid molecules: synthesis and evaluation for antibacterial/antifungal activity. Arch Pharm 347:748–755. https://doi.org/10.1002/ardp.201400142
Xu Z, Zhang S, Song X, Qiang M, Lv Z (2017) Design, synthesis and in vitro anti-mycobacterial evaluation of gatifloxacin-1H-1,2,3-triazole-isatin hybrids. Bioorg Med Chem Lett 27:3643–3646. https://doi.org/10.1016/j.bmcl.2017.07.023
Sharma S, Saquib M, Verma S, Mishra NN, Shukla PK, Srivastava R et al (2014) Synthesis of 2,3,6-trideoxy sugar triazole hybrids as potential new broad spectrum antimicrobial agents. Eur J Med Chem 83:474–548. https://doi.org/10.1016/j.ejmech.2014.06.048
Yan X, Lv Z, Wen J, Zhao S, Xu Z (2018) Synthesis and in vitro evaluation of novel substituted isatin-propylene-1H-1,2,3-triazole-4-methylene-moxifloxacin hybrids for their anti-mycobacterial activities. Eur J Med Chem 143:899–904. https://doi.org/10.1016/j.ejmech.2017.11.090
Akhtar W, Khan MF, Verma G, Shaquiquzzaman M, Rizvi MA, Mehdi SH et al (2017) Therapeutic evolution of benzimidazole derivatives in the last quinquennial period. Eur J Med Chem 138:705–753. https://doi.org/10.1016/j.ejmech.2016.12.010
Evans CW, Atkins C, Pathak A, Gilbert BE, Noah JW (2015) Benzimidazole analogs inhibit respiratory syncytial virus G protein function. Antiviral Res 121:31–38. https://doi.org/10.1016/j.antiviral.2015.06.016
Ates-Alagoz Z (2016) Antimicrobial activities of 1H-benzimidazole-based molecules. Curr Top Med Chem 16:2953–2962. https://doi.org/10.2174/1568026616666160506130226
Sharma P, Reddy TS, Kumar NP, Senwar KP, Bhargava SK, Shankaraiah N (2017) Conventional and microwave-assisted synthesis of new 1H-benzimidazole-thiazolidinedione derivatives: a potential antiproliferative scaffold. Eur J Med Chem 138:234–245. https://doi.org/10.1016/j.ejmech.2017.06.035
Sharma MC, Sharma S, Sahu NK, Kohli DV (2013) 3D QSAR kNN-MFA studies on 6-substituted benzimidazoles derivatives as nonpeptide angiotensin II receptor antagonists: a rational approach to anti-hypertensive agents. J Saudi Chem Soc 17:167–176. https://doi.org/10.1016/j.jscs.2011.03.005
Gabaa M, Gabaa P, Uppala D, Dhingrac N, Bahiad MS, Silakarid O et al (2015) Benzimidazole derivatives: search for GI-friendly anti-inflammatory analgesic agents. Acta Pharm Sin B5:337–342. https://doi.org/10.1016/j.apsb.2015.05.003
El-Feky SAH, El-Samii ZKA, Osman NA, Lashine J, Kamel MA, Thabet HK (2015) Synthesis, molecular docking and anti-inflammatory screening of novel quinoline incorporated pyrazole derivatives using the Pfitzinger reaction II. Bioorg Chem 58:104–116. https://doi.org/10.1016/j.bioorg.2014.12.003
Picconi P, Hind C, Jamshidi S, Nahar K, Clifford M, Wand ME et al (2017) Triarylbenzimidazoles as a new class of antibacterial agents against resistant pathogenic microorganisms. J Med Chem 60:6045–6059. https://doi.org/10.1021/acs.jmedchem.7b00108
Lingala S, Nerella R, SambasivaRao KRS (2011) Synthesis, antimicrobial and anthelmintic activity of some novel benzimidazole derivatives. Der Pharma Chemica 3:344–352
Supuran CT (2017) Special issue: sulfonamides. Molecules 22:1642–1646. https://doi.org/10.3390/molecules22101642
Ajeet A, Mishra AK, Kumar A (2015) Recent advances in development of sulfonamide derivatives and their pharmacological effects—a review. Am J Pharmacol Sci 3:18–24. https://doi.org/10.12691/ajps-3-1-4
Lal J, Sushil K, Gupta D (2013) Biological activity, design, synthesis and structure activity relationship of some novel derivatives of curcumin containing sulfonamides. Eur J Med Chem 64:579–588. https://doi.org/10.1016/j.ejmech.2013.03.012
Carta F, Supuran CT, Scozzafava A (2014) Sulfonamides and their isosters as carbonic anhydrase inhibitors. Future Med Chem 6:1149–1165. https://doi.org/10.4155/fmc.14.68
Winum JY, Scozzafava A, Montero JL, Supuran CT (2006) The sulfamide motif in the design of enzyme inhibitors. Expert Opin Ther Pat 16:27–47. https://doi.org/10.1517/13543776.16.1.27
Mun J, Jabbar AA, Devi NS, Yin S, Wang Y, Tan C et al (2012) Design and in vitro activities of N-alkyl-N-[(8-R-2,2-dimethyl-2H-chromen-6-yl)methyl] hetero aryl sulfonamides, novel, small-molecule hypoxia inducible factor-1 pathway inhibitors and antiproliferative agents. J Med Chem 55:6738–6750. https://doi.org/10.1021/jm300752n
Ibrahim HS, Eldehna WM, Abdel-Aziz HA, Elaasser MM, Abdel-Aziz MM (2014) Improvement of antibacterial activity of some sulfa drugs through linkage to certain phthalazin-1(2H)-one scaffolds. Eur J Med Chem 85:480–486. https://doi.org/10.1016/j.ejmech.2014.08.016
Kumar M, Ramasamy K, Manic V, Mishra RK, Abdul Majeed A, DeClercq E et al (2014) Synthesis, antimicrobial, antiproliferative, antiviral evaluation and QSAR studies of 4-(1-aryl-2-oxo-1,2-dihydro-indol-3-ylidene amino)-N-substituted benzene sulfonamides. Arab J Chem 7:396–408. https://doi.org/10.1016/j.arabjc.2012.12.005
Chibale K, Haupt H, Kendrick H, Yardley V, Saravanamuthu A, Fairlamb AH et al (2001) Antiprotozoal and cytotoxicity evaluation of sulfonamide and urea analogues of quinacrine. Bioorg Med Chem Lett 11:2655–2657. https://doi.org/10.1016/S0960-894X(01)00528-5
Camoutsis C, Geronikaki A, Ciric A, Soković M, Zoumpoulakis P, Zervou M (2010) Sulfonamide-1,2,4-thiadiazole derivatives as antifungal and antibacterial agents: synthesis, biological evaluation, lipophilicity, and conformational studies. Chem Pharm Bull 58:160–167. https://doi.org/10.1248/cpb.58.160
Potey LC, Marathe R, Sable P (2017) In vitro anti-inflammatory activity of quinoxalinsulfonamides. Int J Chemtech Res 10:726–734
Pandit SS, Kulkarni MR, Pandit YB, Lad NP, Khedk VM (2018) Synthesis and in vitro evaluations of 6-(hetero)-aryl-imidazo[1,2-b]pyridazine-3-sulfonamide’s as an inhibitor of TNF-α production. Bioorg Med Chem Lett 28:24–30. https://doi.org/10.1016/j.bmcl.2017.11.026
Hu B, Ellingboe J, Han S, Largis E, Lim K, Malamas M et al (2001) Novel (4-piperidin-1-yl)-phenyl sulfonamides as potent and selective human b3 agonists. Bioorg Med Chem 8:2045–2059. https://doi.org/10.1016/S0968-0896(01)00114-6
Roush WR, Gwaltney SL, Cheng J, Scheidt KA, McKerrow JH, Hansell E (1998) Vinyl sulfonate esters and vinyl sulfonamides: potent, irreversible inhibitors of cysteine proteases. J Am Chem Soc 120:10994–10995. https://doi.org/10.1021/ja981792o
Aouad MR, Mayaba MM, Naqvi A, Bardaweel SK, Alblewi FF, El Ashry EH et al (2017) Design, synthesis, in silico and in vitro antimicrobial screenings of novel 1,2,4-triazoles carrying 1,2,3-triazole scaffold with lipophilic side chain tether. Chem Cent J 11:117–129. https://doi.org/10.1186/s13065-017-0347-4
Rezki N, Mayaba MM, Alblewi FF, El Ashry EH, Aouad MR (2017) Click 1,4-regioselective synthesis, characterization, and antimicrobial screening of novel 1,2,3-triazoles tethering fluorinated 1,2,4-triazole and lipophilic side chain. Res Chem Intermed 43:995–1011. https://doi.org/10.1007/s11164-016-2679-4
Aouad MR (2015) Efficient eco-friendly solvent-free click synthesis and antimicrobial evaluation of new fluorinated 1,2,3-triazoles and their conversion into Schiff Bases. J Braz Chem Soc 10:2105–2115. https://doi.org/10.5935/0103-5053.20150196
Aouad MR (2016) Synthesis and antimicrobial screening of novel thioglycosidesanalogs carrying 1,2,3-triazole and 1,3,4-oxadiazole moieties. Nucleosides Nucleotides Nucleic Acids 35:1–15. https://doi.org/10.1080/15257770.2015.1109098
Aouad MR (2017) Click synthesis and antimicrobial screening of novel isatin-1,2,3-triazoles with piperidine, morpholine or piperazine moieties. Org Prep Proc Int 49:216–227. https://doi.org/10.1080/00304948.2017.1320515
Rezki N, Aouad MR (2017) Green ultrasound-assisted three-component click synthesis of novel 1H-1,2,3-triazole carrying benzothiazoles and fluorinated-1,2,4-triazole conjugates and their antimicrobial evaluation. Acta pharm 67:309–324. https://doi.org/10.1515/acph-2017-0024
Faraji L, Shahkarami S, Nadri H, Moradi A, Saeedi M, Foroumadi A, Ramazani A, Haririan I, Ganjali MR, Shafiee A, Khoobi M (2017) Synthesis of novel benzimidazole and benzothiazole derivatives bearing a 1,2,3-triazole ring system and their acetylcholinesterase inhibitory activity. J Chem Res 41(1):30–35. https://doi.org/10.3184/174751917X14836231670980
Youssif BGM, Mohamed YAM, Salim MTA, Inagaki F, Mukai Ch, Abdu-Allah HHM (2016) Synthesis of some benzimidazole derivatives endowed with 1,2,3-triazole as potential inhibitors of hepatitis C virus. Acta Pharm 66(2):219–231. https://doi.org/10.1515/acph-2016-0014
Yaroshenko TI, Nakhmanovich AS, Larina LI, Elokhina VN, Amosova SV (2008) Interaction of benzimidazole-2-thion with propargyl bromide and 1,3-dibromopropyne. Chem Heterocycl Compd 44:1129–1134. https://doi.org/10.1007/s10593-008-0166-6
Rezki N (2017) Green microwave synthesis and antimicrobial evaluation of novel triazoles. Org Prep Proc Int 49:525–541. https://doi.org/10.1080/00304948.2017.1384262
European Committee for Antimicrobial Susceptibility Testing (EUCAST) of the European Society of Clinical Microbiology and Infectious Diseases (ESCMID) (2000) Determination of minimum inhibitory concentrations (MICs) of antibacterial agents by agar dilution. Clin Microbiol Infect 6:509–515
National Committee for Clinical Laboratory Standards (2000) Methods for dilution, antimicrobial susceptibility tests for bacteria that grow aerobically approved standard M7-A5, 5th edn. Wayne, NCCLS
Supuran CT, Scozzafava A (2001) Carbonic anhydrase inhibitors. Curr Med Chem Imm Endoc Metab Agents 1:61–97. https://doi.org/10.2174/1568013013359131
Maren TH (1976) Relations between structure and biological activity of sulphonamides. Annu Rev Pharmacol Toxicol 16:309–327. https://doi.org/10.1146/annurev.pa.16.040176.001521
Supuran CT, Conroy CW, Maren TH (1996) Carbonic anhydrase inhibitors. Synthesis and inhibitory properties of 1,3,4-thiadiazole-2,5-bissulfonamide. Eur J Med Chem 31:843–846. https://doi.org/10.1016/S0223-5234(97)89847-9
Chegwidden WR, Spencer IM (1995) Sulphonamide inhibitors of carbonic anhydrase inhibit the growth of human lymphoma cells in culture. Inflamm pharmacol 3:231–239. https://doi.org/10.1007/BF02659120
Supuran CT, Scozzafava A (2001) Matrix metalloproteinase inhibitors. In proteinase and peptidase inhibition: recent potential targets for drug development. Smith HJ, Simons C, eds. 1st ed. London: Francis & Taylor; p. 35–52
Branda RF, McCormack JJ, Perlmutter CA (1988) Cellular pharmacology of chloroquinoxaline sulfonamide and a related compound in murine B16 melanoma cells. Biochem Pharmacol 37:4557–4564
Biswas D, Roy S, Sen S (2006) A simple approach for indexing the oral drug likeness of a compound: discriminating drug like compounds from nondrug like ones. J Chem Inf Model 46:1394–1401
Lipinski CA, Lombardo F, Dominy BW, Feene PJ (2001) Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev 46:3–26
Ertl P, Rohde B, Selzer P (2000) Fast calculation of molecular polar surface area as a sum of fragment-based contributions and its application to the prediction of drug transport properties. J Med Chem 43:3714–3717
Ghose AK, Viswanadhan VN, Wendoloski JJ (1999) A knowledge-based approach in designing combinatorial or medicinal chemistry libraries for drug discovery. 1. A qualitative and quantitative characterization of known drug databases. J Comb Chem 1:55–68
Veber DF, Johnson SR, Cheng HY, Smith BR, Ward KW, Kopple KD (2002) Molecular properties that influence the oral bioavailability of drug candidates. J Med Chem 45:2615–2623
Martin YC (2005) A bioavailability score. J Med Chem 48:3164–3170. https://doi.org/10.1021/jm0492002
Ekins S, Andreyev S, Ryabov A, Kirillov E, Rakhmatulin EA, Sorokina S et al (2006) A combined approach to drug metabolism and toxicity assessment. Drug Metab Dispos 34:495–503. https://doi.org/10.1124/dmd.105.008458
Norinder U, Bergström CAS (2006) Prediction of ADMET properties. Chem Med Chem 1:920–937. https://doi.org/10.1002/cmdc.200600155
Feixiong C, Weihua L, Yadi Z, Jie S, Zengrui W, Guixia L (2012) Admetsar: a comprehensive source and free tool for assessment of chemical ADMET properties. J Chem Inf Model 52:3099–3105
Authors’ contributions
MRA, NR, and ESH gave the concepts of this work. NR, FFA and MAA, carried out the experimental work and cooperated in the preparation of the manuscript. SKB and PS performed the biological part. MRA, NR, MM and FFA collected data, interpreted the results and prepared the manuscript. All authors discussed the results, wrote and commented on the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Al-blewi, F.F., Almehmadi, M.A., Aouad, M.R. et al. Design, synthesis, ADME prediction and pharmacological evaluation of novel benzimidazole-1,2,3-triazole-sulfonamide hybrids as antimicrobial and antiproliferative agents. Chemistry Central Journal 12, 110 (2018). https://doi.org/10.1186/s13065-018-0479-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13065-018-0479-1