
Abstract
Background
Ankle sprain is a common musculoskeletal injury. In Korean medicine, blood stasis is thought to be the main cause of pain and swelling in patients with ankle sprain. Dangguixu-san (DS), a herbal extract, is widely used in Korean medicine for the treatment of traumatic ecchymosis and pain by promoting blood circulation and relieving blood stasis. However, the effects of DS on ankle sprain have not been evaluated in a randomized clinical trial. Here, we describe the protocol for a randomized controlled trial that will evaluate the efficacy and safety of DS for the treatment of ankle sprain.
Methods/design
In this randomized, double-blinded, placebo-controlled, parallel-arm clinical trial with a 1:1 allocation ratio, participants (n = 48) with acute lateral ankle sprain (ALAS) that occurred within 72 h before enrollment will be randomly assigned to a DS (n = 24) or a placebo (n = 24) group. Both groups will receive acupuncture treatment once a day for 5 days a week (excluding Saturday and Sunday) and the trial medication (DS/placebo capsule) three times a day for seven consecutive days. The primary outcome measure will be pain relief evaluated using a Visual Analog Scale (VAS). Secondary outcome measures will include Foot and Ankle Outcome Scores (FAOS), edema, European Quality of Life Five-Dimension-Five-Level Scale (EQ-5D-5 L) scores, and the number of recurrent ankle sprains. VAS, FAOS, edema, and EQ-5D-5 L scores will be recorded before, at the end of, and at 4 weeks after treatment completion. EQ-5D-5 L scores will be additionally recorded at 26 weeks after treatment completion. The number of recurrent ankle sprains will be recorded at 4, 8, 12, and 26 weeks after treatment completion.
Discussion
This study is expected to provide evidence regarding the efficacy, safety, and usefulness of DS for the treatment of ALAS.
Trial registration
cris.nih.go.kr, registration number: KCT 0002374. Registered on 11 July, 2017 and approved by the Ministry of Food and Drug Safety (registration number, 31244).
Similar content being viewed by others

Background
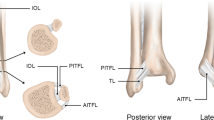
Ankle sprain is one of the more common musculoskeletal injuries and is associated with a high prevalence of persistent problems that lead to an increased burden of chronic health issues in the community [1,2,3,4,5]. The annual incidence rate of ankle sprain ranges from 2.15 per 1000 individuals in the USA (resulting in a cumulative healthcare cost of US$4.5 billion per annum) [1, 2] to 5.3–7.0 per 1000 individuals in Europe [3, 6]. Ankle sprain is commonly considered a benign injury that resolves quickly [7]. However, at least a third of individuals who develop an acute ankle sprain develop chronic ankle instability (CAI) [8,9,10], which is characterized by residual symptoms, including a feeling of “giving way” and instability, recurrent ankle sprain, and functional loss after acute ankle sprain [11]. CAI not only limits physical activity but also leads to articular degeneration of the ankle joint and an increased risk of osteoarthritis [12]. The three major types of treatment for ankle sprain are surgical treatment, conservative treatment involving immobilization with a plaster cast or splint, and functional conservative treatment with a tape or a semi-rigid or lace-up brace [13].
Blood stasis is an important pathological concept in traditional East Asian medicine (TEAM) since it was first documented in Huangdi’s Inner Classic. Generally, blood stasis is a significant pathological product of blood stagnation [14, 15]. Blood stasis occurring within the body is termed blood stasis syndrome (BSS), which is characterized by symptoms such as pain in a fixed position, nyctalgia, dark-purple coloring of the tongue and face, infraorbital darkness, sublingual varicosis, blood spots under the skin or tongue, and an astringent pulse [16]. According to TEAM, many diseases, including ischemic heart disease, cerebrovascular events, diabetes mellitus, chronic gastritis, chronic renal failure, chronic hepatitis, trauma, and dysmenorrhea, could be related to BSS [17, 18]. This phenomenon is termed EoHyeol in Korean, Yu Xue in Chinese, and Oketsu in Japanese [19].
In Korean medicine, because it is considered that pain and swelling associated with ankle sprain could be caused by blood stasis, activation of blood circulation is the main treatment principle for ankle sprain [20, 21]. In addition to conventional treatments, complementary and alternative treatment modalities, such as herbal medicine, have been thought to relieve pain, reduce swelling, and help the body restore damaged tissues [22]. Traditional Korean medications usually contain many compounds that act on multiple targets [23]. The combination of multiple drugs is thought to maximize the therapeutic efficacy by facilitating synergistic actions and preventing potential adverse events (AEs). Dangguixu-san (DS), which is composed of Angelicae gigantis radix, Paeoniae radix rubra, Linderae radix, Cyperi rhizoma, Sappan lignum, Carthami flos, Persicae semen, Cinnamomum cassia, and Glycyrrhizae radix, promotes blood circulation and relieves blood stasis; therefore, it is the most frequently prescribed herbal formula for the treatment of traumatic ecchymosis and pain [24] and is recommended for the treatment of ankle sprain [25]. This formula is also known as Dangkwisoo-san in Korean and Tokishusan in Japanese [26, 27].
Acupuncture is one of the more commonly used therapeutic modalities for painful conditions in complementary and alternative medicine, including Korean medicine [28]. A survey reported that 76% of responding American physicians used acupuncture for ankle sprain, and 90% of them considered it to be somewhat effective [29]. In 2009, approximately 2.8 million Korean individuals were diagnosed with an ankle injury, which was the fifth most common reason for visits to Korean medicine clinics. Of these, 1.2 million individuals sought acupuncture treatment [30]. Considering that DS is mainly used with acupuncture in clinical situations, the present study will use acupuncture as the basic treatment.
Although DS is used for the treatment of ankle sprain in Korean medicine, evidence regarding its efficacy is insufficient. Therefore, we designed a randomized, double-blinded, placebo-controlled, parallel-arm clinical trial for investigating the efficacy and safety of DS for the treatment of ankle sprain. The results of this study are expected to provide evidence regarding the usefulness of DS for the treatment of ankle sprain.
Method/design
Objective
The objective of this study is to compare the efficacy of DS treatment combined with acupuncture with that of placebo combined with acupuncture for pain reduction in patients with acute lateral ankle sprain (ALAS) in order to investigate the efficacy of DS for the treatment of ankle sprain.
Hypothesis
Our null hypothesis is that the pain control effects of DS are not superior to those of placebo in patients with ALAS.
Study design
The present study design is in accordance with the Standard Protocol Items: Recommendations for Interventional Trials (SPIRIT) and Consolidated Standards Of Reporting Trials (CONSORT) 2010 guidelines [31, 32] (see Additional file 1). The study is a randomized, double-blinded, placebo-controlled, parallel-arm, single-center (Semyung University Korean Medicine Hospital in Chungju, Republic of Korea) clinical trial with a 1:1 allocation ratio. A total of 48 participants who meet the inclusion and exclusion criteria will be randomly allocated to a DS (n = 24) or a placebo (n = 24) group. Patients in both groups will receive acupuncture treatment once a day for 5 days a week (excluding Saturday and Sunday) and the trial medication (DS/placebo capsule) three times a day for seven consecutive days. The primary outcome measure will be pain relief evaluated using a Visual Analog Scale (VAS). The secondary outcome measures will be Foot and Ankle Outcome Scores (FAOS), edema, European Quality of Life Five-Dimension-Five-Level Scale (EQ-5D-5 L) scores, and the number of recurrent ankle sprains. VAS, FAOS, edema, and EQ-5D-5 L scores will be assessed before, at the end of, and at 4 weeks after treatment completion. EQ-5D-5 L scores will be additionally recorded at 26 weeks after treatment completion. The number of recurrent ankle sprains will be recorded at 4, 8, 12, and 26 weeks after treatment completion.
This study protocol complies with the principles of the Declaration of Helsinki and Korean Good Clinical Practice guidelines and has been approved by the Ministry of Food and Drug Safety (registration number, 20160318110). The trial has been registered at cris.nih.go.kr (registration number, KCT 0002374). The study design is summarized in Table 1 and Fig. 1.

Study design flow chart
Participant recruitment
Participants will be recruited at Semyung University Korean Medicine Hospital in Chungju, Republic of Korea. The study will be advertised through local newspapers, the Internet, and posters in communities and hospitals. Participants will be provided with an explanation about the study by the clinical research coordinator (CRC) and will be requested to voluntarily sign an informed consent before participation. The CRC will continuously monitor the medical condition of the enrolled participants to ensure adherence to the intervention protocols.
Inclusion criteria
Patients aged older than 19 years who have sustained a grade I or grade II ALAS within 72 h before enrollment and are willing to voluntarily sign an informed consent form will be considered for enrollment. A grade I ankle sprain will be diagnosed when there is no loss of function or ligament laxity (i.e., negative anterior drawer and talar tilt test results), little or no hemorrhaging, no point tenderness, decreased total ankle motion by ≤ 5°, and a swelling of ≤ 0.5 cm. Patients with some loss of function, positive anterior drawer test findings (indicating anterior talofibular ligament involvement), negative talar tilt test findings (indicating no calcaneofibular ligament involvement), hemorrhaging, point tenderness, decreased total ankle motion by > 5° but < 10°, and a swelling of > 0.5 cm but < 2.0 cm will be diagnosed with grade II ALAS [33].
Exclusion criteria
Subjects with a poor general condition and those who are not fit for acupuncture or DS treatment will be excluded. Other exclusion criteria are as follows: fracture confirmed on x-rays or grade III ankle sprain (which will be defined as the near total loss of function, positive anterior drawer and talar tilt test findings, hemorrhaging, extreme point tenderness, decreased total ankle motion by > 10°, and a swelling of > 2.0 cm) [33]; a history of fracture in the same ankle during the previous year; serious disease conditions (e.g., cancer, kidney, liver, and central nervous system diseases, dementia, and blood clotting abnormalities such as hemophilia); motor or sensory disturbance due to nervous system disorders in the same leg; pregnancy or breastfeeding; medication with drugs such as nonsteroidal anti-inflammatory drugs (NSAIDs), pain relievers, or steroids for pain relief from the time of trauma to participation in the clinical trial (excluding the use of adherent inflammatory pain relievers on the day of screening); impaired hepatic (alanine aminotransferase level, ≥ 80 IU/L) or renal (creatinine level, ≥ 2 mg/dL) function; ineligibility for participation in the trial because of a history of gastrointestinal diseases that might affect the absorption of the trial medication, as judged by the principal investigator (PI); genetic conditions such as galactose intolerance, Lapp lactase deficiency, and glucose-galactose malabsorption; a history of hypersensitivity to the components of the trial medication; and participation in other clinical trials within 4 weeks of screening for the present study or concurrent participation in other clinical trials.
Ethical considerations
This study has been approved by the Institutional Review Board (IRB) of Semyung University Korean Medicine Hospital in Chungju. The purpose and potential risks of this clinical trial will be fully explained to the participants and their families. All participants will be asked to provide written informed consent before participation.
Randomization and blinding
After the acquisition of written informed consent and completion of baseline measurements, the 48 enrolled participants will be assigned serial numbers generated using a randomization tool (http://www.randomization.com) and randomly allocated to one of the two study groups (n = 24 each). The serial number codes will be inserted into opaque envelopes that will be sealed and stored in a double-locked cabinet.
Blinding will be achieved by randomized labeling and prepackaging of DS and placebo capsules, which will be identical in appearance, taste, and smell. The DS and placebo capsules provided by the pharmaceutical company will be labeled with numbers in accordance with the randomization schedule. Investigators will administer these medications in accordance with the randomization number. Randomization and uncovering will be performed by a third-party team of statisticians. Thus, participants, investigators, and outcome assessors will remain blinded to treatment allocation until study completion.
Implementation
The CRC will generate the allocation sequence, enroll participants, and assign participants to the intervention groups.
Intervention
Both groups will receive acupuncture treatment once a day for 5 days a week (excluding Saturday and Sunday) and the trial medication (DS/placebo capsule) three times a day for seven consecutive days. Treatment will be administered by Korean medicine physicians with 6 years of formal university training in Korean medicine, a licence to administer treatment, and at least 1 year of clinical experience. To ensure strict adherence to the study protocol, the physicians will receive training together and employ the same techniques.
Acupuncture treatment
All participants will receive acupuncture at the ST36, ST41, BL60, BL62, KI3, KI6, GB39, and GB40 points on the affected side [25]. Only sterile, stainless steel, disposable acupuncture needles (size, 0.25 × 30 mm; Dong Bang Acupuncture, Inc., Boryeong, Republic of Korea; product no. A84010.02) with guide tubes will be used. The depth of insertion will be 10–20 mm depending on the location of the needle [34]. After insertion, the needles will be left in position for 15 min in every session. Manual stimulation and electroacupuncture will not be applied (see Table 2).
Trial medication
The trial medications will include DS and placebo capsules, which will be identical in appearance, taste, and smell. The DS formulation composed of granular extracts of nine herbal substances, will be a gray-brown powder contained in a 0.6-g pink capsule (Fig. 2). The placebo will be composed of 300 mg of corn starch, 150 mg of lactose hydrate, 1.5 mg of magnesium stearate, and 48.5 mg of caramel colorant. Both medications will be administered at a dose of three capsules three times daily (total daily dose, nine capsules) for a period of 1 week.

Appearance of Dangguixu-san (DS) and placebo capsules used in the study
The manufacturing process for the DS capsules is as follows. First, 0.63 g of Angelicae gigantics radix, 0.42 g of Paeoniae radix rubra, 0.42 g of Linderae radix, 0.42 g of Cyperi rhizoma, 0.42 g of Sappan lignum, 0.33 g of Carthami flos, 0.29 g of Persicae semen, 0.25 g of Cinnamomum cassia, and 0.21 g of Glycyrrhizae radix are added to the extractor. The mixture is extracted with 8–10 volumes of purified water at 80–100 °C for 3–4 h. The extract is filtered (100 mesh), concentrated under reduced pressure at ≤ 60 °C, and dried to obtain 350 mg of a granular extract. Then, 45 mg of lactose hydrate, 100 mg of corn starch, and 5 mg of magnesium stearate are mixed into the granular extract, and the mixture is converted into granules, dried, crushed, and filled into capsules.
Capsules will be sourced from Kyungjin Pharmaceutical Co., Ltd. (Icheon, Gyeonggi-do, Republic of Korea (product no. 02190)). The medication will be dispensed as 0.6-g capsules packed in sealed boxes containing a 1-week dose.
Between the preintervention period and 4 weeks after treatment completion, participants will not be allowed to take the following drugs: NSAIDS, pain relievers, steroids, or adherent inflammatory pain relievers; drugs containing potassium, licorice, glycyrrhizic acid, furosemide, ethacrynic acid, or trichloromethazine; and drugs containing any of the main ingredients of DS. Participants will be asked to return their medication boxes to allow enumeration of leftover capsules and for monitoring participant adherence.
Outcome measurements
VAS, FAOS, edema, and EQ-5D-5 L scores will be assessed before, at the end of, and at 4 weeks after treatment completion. EQ-5D-5 L scores will be additionally recorded at 26 weeks after treatment completion. The number of recurrent ankle sprains will be recorded at 4, 8, 12, and 26 weeks after treatment completion.
Primary outcome
Because the objective of this study is to investigate the efficacy of DS therapy for pain relief in patients with ALAS, changes in the pain severity (measured using VAS) will be considered as the primary outcome. The VAS will be a 10-cm straight line marked at each end with the anchor labels “no pain” and “pain as bad as it could be” [35]. Patients will be asked to mark the line at a point representing their severity of pain. Scores will be recorded in millimeters, with a total score range of 0–100 mm [36].
Secondary outcomes
Secondary outcomes will include changes in FAOS, edema, EQ-5D-5 L scores, and the number of recurrent ankle sprains. The FAOS is a self-administered questionnaire specific to feet and ankles. It is designed to assess week-to-week changes in symptoms and function after foot and ankle injuries and comprises five subscales: pain (nine items), other symptoms (seven items), activities of daily living (17 items), sports and recreational activities (five items), and foot-and-ankle-related quality of life (four items). The subscales are scored separately using a Likert response format, with higher scores indicating higher levels of function [37].
Edema will be measured in centimeters using the figure-of-eight method. A measurement tape will be applied across the following landmarks in a figure-of-eight fashion: navicular tuberosity, distal tip of the lateral malleolus, distal tip of the medial malleolus, and base of the fifth metatarsal. The resulting value will be compared with the corresponding value for the healthy ankle [38].
The European Quality of Life Five-Dimension Scale (EQ-5D) is a generic instrument for assessment of health-related quality of life. It is based on a descriptive system that defines health in terms of five dimensions: mobility, self-care, usual activities, pain/discomfort, and anxiety/depression. Each dimension has three response categories: no, some, or extreme problems. The EQ-5D-5 L, which will be used in this study, is a new version of the EQ-5D that includes five levels of severity in each of the existing five EQ-5D dimensions [39].
Recurrent ankle sprain will be defined as ankle sprain occurring as a result of participation in sports or other daily activities and causing one or more of the following: discontinuation of the sports activity, inability to fully participate in the next planned sports activity, inability to go to work/school the following day, and the requirement of medical attention (ranging from onsite care administered by a general practitioner to personal care administered by a sports physician) [40].
Incidence of adverse events
AEs are undesirable and unintentional signs, symptoms, or diseases that appear during or after treatment in a clinical trial. The participants in this study will be required to voluntarily report any AEs. All AEs that occur during the trial will be documented. Adverse events that might occur in this study include skin irritation, urticaria, itching, anorexia, stomach discomfort, diarrhea, nausea, vomiting, pseudoaldosteronism, and hypokalemia. The CRC will record all AEs in detail, including the time and date of occurrence, degree of severity, any measures related to the treatment of the AE, and any potentially causal relationship between the treatment and the AE, and report all AEs to the PI and relevant IRB. In case of serious AEs (SAEs), defined as those causing severe disability or malfunction, appropriate measures will be taken and the incident will be immediately reported to the PI and relevant IRB. In case an AE occurs because of the clinical trial, participants will notify the CRC and PI and will be compensated by the “Clinical Trial Compensation.”
Quality assurance
This protocol has been reviewed and revised several times by experts on acupuncture, herbal medicine, orthopedics, statistics, and methodology. Before the trial, all researchers will be requested to attend a series of training sessions, which will ensure that the personnel involved fully understand the trial protocol and standard operating procedures (SOPs) that will be employed during the study. The Data Monitoring Committee will be composed of the PI and CRC. The clinical trial will be monitored by a clinical research associate (CRA), who will check all documents related to the clinical trial, including Case Report Forms (CRFs) and SOPs, and ensure that the clinical trial is conducted in accordance with the prescribed protocols and SOPs. Monitoring will be conducted independent of the PI. In the event that the protocol described herein is revised, the revisions will require approval from the Ministry of Food and Drug Safety and the IRB of Semyung University Korean Medicine Hospital in Chungju.
Sample size estimation
Because of the lack of adequate preliminary studies, we adopted a pilot study design with 24 participants in each group, considering the limited research funds, study period, and recruitment opportunities.
Statistical analysis
Baseline characteristics will be compared between the two groups. Repeated measures analysis of covariance (RM ANCOVA) will be performed to determine differences between groups, considering the different baseline characteristics (covariance). Continuous data will be presented as means and standard deviations and compared using the independent t test or Wilcoxon’s rank sum test, while categorical data will be presented as frequencies and percentages and compared using the chi-square or Fisher’s exact test. VAS scores for pain and the secondary outcomes (FAOS, edema, and EQ-5D-5 L scores) will be evaluated by RM ANCOVA. Dependent variables will include values measured before, at the end of, and at 4 weeks after treatment completion. Changes in the VAS score from baseline to treatment completion (visit 5) and 4 weeks after treatment completion (visit 6) will be determined by the paired t test. A repeated contrast test will be performed to account for time differences in each group. A P value of < 0.05 will be considered statistically significant. We will perform per-protocol analysis for the assessment of efficacy and a supplementary full analysis set. All statistical analyses will be performed using SPSS version 22.0 (SPSS Inc., Chicago, IL, USA).
Data for subjects who meet the dropout criteria (i.e., < 80% compliance with the protocol, incidence of SAEs, reluctance to continue the trial, incomplete data that could influence the trial, large error in protocol or significant deviation in implementation, and decision to terminate trial participation by the PI or IRB) will be excluded. Missing values will be implemented by multiple imputations. In addition, differences between subjects who complete the study and dropouts will be statistically analyzed to determine whether any particular factor is significantly associated with participation dropout. Interim analyses will not be performed.
Confidentiality and data management
Identification records of the participants will be kept confidential until publication of the results of the study. All documents related to the trial, including CRFs, will be recorded and labeled with participant identification codes and will not show the name of the participant. These serial number codes will be stored in sealed, opaque envelopes and kept in a double-locked cabinet. All participant data will be recorded in Excel files by the CRC. In additionally, raw data (CRFs) will be stored in a cabinet until the end of the study. Written informed consent for the publication of individual details and accompanying images will be obtained from the participants.
Discussion
The design of this study, including the treatment and evaluation schedules, is based on the designs of several acupuncture studies for ankle sprain [30].
There is good evidence to support the usefulness of immobilization and, occasionally, surgical correction for the management of grade III ankle sprain. However, clinical standards for the acute management of grade I and grade II ankle sprain are not well defined [4]. Therefore, we will include subjects with grade I and grade II ankle sprain in the present study and exclude those with grade III ankle sprain. We will also exclude subjects who are using analgesics that could affect the outcomes of this study and those at risk of AEs associated with DS treatment.
We will use VAS scores for pain as the primary outcome measure and FAOS, edema, EQ-5D-5 L scores, and the number of recurrences as secondary outcomes in order to evaluate the efficacy of DS for relieving pain and swelling, aiding the recovery of ankle function, improving quality of life, and decreasing the number of recurrences in patients with ALAS.
This study has some limitations. First, there is concern that the short treatment period planned for minimizing the dropout rate could affect the results of this study. Second, because preliminary data for determining the sample size is inadequate, we designed this trial as a single-center pilot study. Third, in previous studies [41,42,43,44], recurrences were followed up for 52 weeks; however, because of the short study duration, we have scheduled a follow-up period of only 26 weeks.
Nevertheless, the results of this study are expected to provide preliminary evidence regarding the usefulness and acceptability of DS treatment for the treatment of grade I and grade II ankle sprain and serve as a basis for further research.
Dissemination policy
We will report the final data to the Ministry of Health and Welfare through the Korea Health Industry Development Institute. We will also publish the results after study completion.
Trial status
This trial is ongoing. Enrollment and trial procedures are expected to be complete by the end of May 2019.
Abbreviations
- AE:
-
Adverse event
- ALAS:
-
Acute lateral ankle sprain
- BSS:
-
Blood stasis syndrome
- CAI:
-
Chronic ankle instability
- CONSORT:
-
Consolidated Standards Of Reporting Trials
- CRA:
-
Clinical research associate
- CRC:
-
Clinical research coordinator
- CRF:
-
Case Report Form
- DS:
-
Dangguixu-san
- EQ-5D-5 L:
-
European Quality of Life Five-Dimension-Five-Level scale
- FAOS:
-
Foot and ankle outcome score
- IRB:
-
Institutional Review Board
- NSAID:
-
Nonsteroidal anti-inflammatory drug
- PI:
-
Principal investigator
- SAE:
-
Serious adverse event
- SOP:
-
Standard operating procedure
- SPIRIT:
-
Standard Protocol Items: Recommendations for Interventional Trials
- TEAM:
-
Traditional East Asian medicine
- VAS:
-
Visual Analog Scale
References
Waterman BR, Owens BD, Davey S, Zaccili MA, Belmont PJ Jr. The epidemiology of ankle sprains in the United States. J Bone Joint Surg Am. 2010;92(13):2279–84.
Soboroff SH, Pappius EM, Komaroff AL. Benefits, risks, and cost of alternative approaches to the evaluation and treatment of severe ankle sprain. Clin Orthop Relat Res. 1984;183:160–8.
Bridgman SA, Clement D, Downing A, Walley G, Phair I, Maffulii N. Population based epidemiology of ankle sprains attending accident and emergency units in the West Midlands of England and a survey of UK practice for severe ankle sprains. Emerg Med J. 2003;20(6):508–10.
Brison RJ, Day AG, Pelland L, Pickett W, Johnson AP, Aiken A, Pichora DR, Brouwer B. Effect of early supervised physiotherapy on recovery from acute ankle sprain: randomized controlled trial. BMJ. 2016;355:i5650.
Pourkazemi F, Hiller C, Raymond J, Black D, Nightingale E, Refshauge K. Using balance tests to discriminate between participants with a recent index lateral ankle sprain and healthy control participants: a cross-sectional study. J Athl Train. 2016;51(3):213–22.
Hølmer P, Søndergaard L, Konradsen L, Nielsen PT, Jørgensen LN. Epidemiology of sprains in the lateral ankle and foot. Foot Ankle Int. 1994;15(2):72–4.
Birrer RB, Fani-Salek MH, Totten VY, Herman LM, Politi V. Managing ankle injuries in the emergency department. J Emerg Med. 1999;17(4):651–60.
Anandacoomarasamy A, Barnsley L. Long term outcomes of inversion ankle injuries. Br J Sports Med. 2005;39(3):e14.
Konradsen L, Bech L, Ehrenbjerg M, Nickelsen T. Seven years follow-up after ankle inversion trauma. Scand J Med Sci Sports. 2002;12(3):129–35.
Braun BL. Effects of ankle sprain in a general clinic population 6 to 18 months after medical evaluation. Arch Fam Med. 1999;8(2):143–8.
Delahunt E, Coughlan GF, Caulfield B, Nightingale EJ, Lin C, Hiller CE. Inclusion criteria when investigating insufficiencies in chronic ankle instability. Med Sci Sports Exerc. 2010;42(11):2106–21.
Harrington KD. Degenerative arthritis of the ankle secondary to long-standing lateral ligament instability. J Bone Joint Surg Am. 1979;61(3):354–61.
van den Bekerom MP, van Kimmenade R, Sierevelt IN, Eggink K, Kerkhoffs GM, van Dijk CN, Raven EE. Randomized comparison of tape versus semi-rigid and versus lace-up ankle support in the treatment of acute lateral ankle ligament injury. Knee Surg Sports Traumatol Arthrosc. 2016;24(4):978–84.
Matsumoto C, Kojima T, Ogawa K, Kamegai S, Oyama T, Shibagaki T, Fujinaga H, Takahashi K, Hikiami H, Goto H, Kiga C, Koizumi K, Sakurai H, Muramoto H, Shimada Y, Yamamoto M, Terasawa K, Takeda S, Saiki I. A proteomic approach for the diagnosis of “oketsu” (blood stasis), a pathophysiologic concept of Japanese traditional (Kampo) medicine. Evid Based Complement Alternat Med. 2008;5(4):463–74.
Li SM, Xu H, Chen KJ. The diagnostic criteria of blood-stasis syndrome: considering for standardization of pattern identification. Chin J Integr Med. 2014;20(7):483–6.
Park YJ, Yang DH, Lee JM, Park YB. Development of a valid and reliable blood stasis questionnaire and its relationship to heart rate variability. Complement Ther Med. 2013;21(6):633–40.
Chen KJ. Blood stasis syndrome and its treatment with activating blood circulation to remove blood stasis therapy. Chin J Integr Med. 2012;18(12):891–6.
Park BK, You SS, Jung JY, Lee JA, Yun KJ, Lee MS. Korean studies on blood stasis: an overview. Evid Based Complement Alternat Med. 2015;2015:316872.
Park BK, Yun KJ, Jung JY, You SS, Lee JA, Choi JA, Kang BK, Alraek T, Birch S, Lee MS. Conceptualization and utilization of blood stasis syndrome among doctors of Korean medicine: results of a web-based survey. Am J Transl Res. 2014;6(6):857–68.
Kim SG, Kim SJ, Jeong HH, Kim JH, Kim DH, Park SH, Yang SB, Kim JH, Jo EH, Kim SC, Cho NG. Comparison study for effects of pharmacopunctures of GB40 on weight bearing ratio in Grade III ankle sprain model. The Acupuncture. 2014;31(2):11–9.
Jung HK, Jeong YP, Chiang SY, Lee JE, Wi J, Im JA, Kim JH, Ryu CY, Cho MR, Yun YC. The comparative study on the acupuncture at affected limb and unaffected limb on treating ankle sprain of acute stage. J Korean Acupunct Moxibust Soc. 2008;25(3):107–13.
Koll R, Buhr M, Dieter R, Pabst H, Predel HG, Petrowicz O, Giannetti B, Klingenburg S, Staiger C. Efficacy and tolerance of a comfrey root extract (Extr. Rad. symphyti) in the treatment of ankle distorsions: results of a multicenter, randomized, placebo-controlled, double-blind study. Phytomedicine. 2004;11(6):470–7.
Qiu J. Back to the future for Chinese herbal medicines. Nat Rev Drug Discov. 2007;6(7):506–7.
Wei C. Clinical observation of Dangguixu Powder in treating 40 cases of acute pulmonary contusion. J Modern Med Health. 2013;29:1768–9.
Korea Institute of Oriental Medicine. Ankle sprain: Korean medicine clinical practice guideline, vol. 80. Seoul: Elsevier Korea LLC; 2015. p. 163–7.
Kim JH, Park SH, Kim YW, Ha JM, Bae SS, Lee GS, Cho SI, Choi BT, Shin HK. The traditional herbal medicine, Dangkwisoo-san, prevents cerebral ischemic injury through nitric oxide-dependent mechanisms. Evid Based Complement Alternat Med. 2011;2011:718302.
Seo CS, Shin HK. Simultaneous determination of nine marker compounds in the traditional Korean medicine, Dangguisu-san by high-performance liquid chromatography. Pharmacogn Mag. 2015;11(43):555–61.
Vickers AJ, Cronin AM, Maschino AC, Lewith G, MacPherson H, Foster NE, Sherman KJ, Witt C, Linde K. Acupuncture for chronic pain: individual patient data meta-analysis. Arch Intern Med. 2012;172(19):1444–53.
Diehl DL, Kaplan G, Coulter I, Glik D, Hurwitz EL. Use of acupuncture by American physicians. J Altern Complement Med. 1997;3(2):119–26.
Park JM, Hahn SK, Park JY, Park HJ, Lee HS. Acupuncture for ankle sprain; systematic review and meta-analysis. BMC Complement Altern Med. 2013;13:55–71.
Chan AW, Tetzlaff JM, Altman DG, Laupacis A, Gøtzsche PC, Krleža-Jerić K, Hróbjartsson A, Mann H, Dickersin K, Berlin JA, Doré CJ, Parulekar WR, Summerskill WS, Groves T, Schulz KF, Sox HC, Rockhold FW, Rennie D, Moher D. SPIRIT 2013 statement: defining standard protocol items for clinical trials. Ann Intern Med. 2013;158(3):200–7.
Schulz KF, Altman DG, Moher D, CONSORT Group. CONSORT 2010 statement: updated guidelines for reporting parallel group randomized trials. BMJ. 2010;340:c332.
Martin RL, Davenport TE, Paulseth S, Wukich DK, Godges JJ. Orthopaedic Section American Physical Therapy Association. Ankle stability and movement coordination impairments: ankle ligament sprains. J Orthop Sports Phys Ther. 2013;43(9):A1–40.
WHO Western Pacific Region. WHO standard acupuncture point locations in the Western Pacific region. Seoul: Elsevier Korea LLC; 2009. p. 67, 70, 133, 134, 141, 142, 195.
Huskisson EC. Measurement of pain. Lancet. 1974;2(7889):1127–31.
Stevens SS. On the theory of scales of measurement. Science. 1946;103(2684):677–80.
Roos EM, Brandsson S, Karlsson J. Validation of the foot and ankle outcome score for ankle ligament reconstruction. Foot Ankle Int. 2001;22(10):788–94.
Esterson PS. Measurement of ankle joint swelling using a figure of 8*. J Orthop Sports Phys Ther. 1979;1(1):51–2.
Herdman M, Gudex C, Lloyd A, Janssen M, Kind P, Parkin D, Bonsel G, Badia X. Development and preliminary testing of the new five-level version of EQ-5D (EQ-5D-5L). Qual Life Res. 2011;20(10):1727–36.
Janssen KW, van Mechelen W, Verhagen EA. Ankles back in randomized controlled trial (ABrCt): braces versus neuromuscular exercises for the secondary prevention of ankle sprains. Design of a randomized controlled trial. BMC Musculoskelet Disord. 2011;12:210–8.
Kemler E, van de Port I, Schmikli S, Huisstede B, Hoes A, Backx F. Effects of soft bracing or taping on a lateral ankle sprain: a non-randomised controlled trial evaluating recurrence rates and residual symptoms at one year. J Foot Ankle Res. 2015;8:13.
Van Rijn RM, van Os AG, Kleinrensink GJ, Bernsen RM, Verhaar JA, Koes BW, Bierma-Zeinstra SM. Supervised exercises for adults with acute lateral ankle sprain: a randomized controlled trial. Br J Gen Pract. 2007;57(543):793–800.
Witjes S, Gresnigt F, van den Bekerom MP, Olsman JG, van Dijk NC. The ANKLE TRIAL (ankle treatment after injuries of the ankle ligaments): what is the benefit of external support devices in the functional treatment of acute ankle sprain? A randomized controlled trial. BMC Musculoskelet Disord. 2012;13:21.
Mailuhu AK, Verhagen EA, van Ochten JM, Bindels PJ, Bierma-Zeinstra SM, van Middelkoop M. The TrAPP-study: cost-effectiveness of an unsupervised e-health supported neuromuscular training program for the treatment of acute ankle sprains in general practice: design of a randomized controlled trial. BMC Musculoskelet Disord. 2015;16:78.
Acknowledgements
The authors would like to express sincere thanks to our colleagues and staff at the College of Korean Medicine Dong-Shin University, Semyung University Korean Medicine Hospital, and DongShin University Gwangju Oriental Hospital for their support.
Funding
This study was supported by the Traditional Korean Medicine R&D program, funded by the Ministry of Health and Welfare through the Korea Health Industry Development Institute (KHIDI) (HB16C0026). The funder had no role in the study design, and will have no role in the data collection, analysis and interpretation of data, writing of the report, or the decision to submit the resulting report for publication.
Availability of data and materials
Not applicable
Author information
Authors and Affiliations
Contributions
JHK, EYL, and MRC are responsible for conceiving and designing the trial, planning data analysis, drafting the manuscript, making the final decision to terminate the trial, and approving the final manuscript. EYL and CGL will participate in data collection and are in charge of recruitment and treatment of patients. JHC is responsible for planning data analysis and analyzing the data resulting from the trial. All authors will have access to the interim results as well as the capacity to discuss, revise, and approve the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study has been approved by the Institutional Review Board (IRB) of Semyung University Korean Medicine Hospital in Chungju. The purpose and potential risks of this clinical trial will be fully explained to the participants and their families. All participants will be asked to provide written informed consent before participation. This trial has been registered with cris.nih.go.kr (registration number, KCT 0002374; 11 July 2017) and is approved by the Ministry of Food and Drug Safety (registration number, 31244).
Consent for publication
Written informed consent for the publication of their individual details and accompanying images will be obtained from the participants in the trial. The consent form is held by the authors and is available for review by the Editor-in-Chief of this journal.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1:
SPIRIT Checklist. (DOCX 367 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Kim, JH., Lee, EY., Cho, MR. et al. Effects of Dangguixu-san on acute lateral ankle sprain: study protocol for a randomized controlled trial. Trials 19, 202 (2018). https://doi.org/10.1186/s13063-018-2571-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13063-018-2571-1