
Abstract
Background
Most BRCA1 or BRCA2 mutation carriers have inherited a single (heterozygous) mutation. Transheterozygotes (TH) who have inherited deleterious mutations in both BRCA1 and BRCA2 are rare, and the consequences of transheterozygosity are poorly understood.
Methods
From 32,295 female BRCA1/2 mutation carriers, we identified 93 TH (0.3 %). “Cases” were defined as TH, and “controls” were single mutations at BRCA1 (SH1) or BRCA2 (SH2). Matched SH1 “controls” carried a BRCA1 mutation found in the TH “case”. Matched SH2 “controls” carried a BRCA2 mutation found in the TH “case”. After matching the TH carriers with SH1 or SH2, 91 TH were matched to 9316 SH1, and 89 TH were matched to 3370 SH2.
Results
The majority of TH (45.2 %) involved the three common Jewish mutations. TH were more likely than SH1 and SH2 women to have been ever diagnosed with breast cancer (BC; p = 0.002). TH were more likely to be diagnosed with ovarian cancer (OC) than SH2 (p = 0.017), but not SH1. Age at BC diagnosis was the same in TH vs. SH1 (p = 0.231), but was on average 4.5 years younger in TH than in SH2 (p < 0.001). BC in TH was more likely to be estrogen receptor (ER) positive (p = 0.010) or progesterone receptor (PR) positive (p = 0.013) than in SH1, but less likely to be ER positive (p < 0.001) or PR positive (p = 0.012) than SH2. Among 15 tumors from TH patients, there was no clear pattern of loss of heterozygosity (LOH) for BRCA1 or BRCA2 in either BC or OC.
Conclusions
Our observations suggest that clinical TH phenotypes resemble SH1. However, TH breast tumor marker characteristics are phenotypically intermediate to SH1 and SH2.
Similar content being viewed by others


Background
Women who have inherited mutations in BRCA1 or BRCA2 are at greatly increased risk of developing breast cancer (BC) and ovarian cancer (OC) [25, 38]. Identification of a mutation at these loci can lead to risk or mortality reduction if optimal surveillance, risk-reducing mastectomy (RRM), and risk-reducing salpingo-oophorectomy (RRSO) are applied [8, 29]. In addition, treatment of cancers in mutation carriers has advanced with the development of PARP inhibitors, which take advantage of the loss of BRCA1/2 function in tumors [37]. BRCA1 and BRCA2 are tumor suppressor genes, and tumors from the majority of mutation carriers have loss of heterozygosity (LOH), with loss of the normal allele, so there is no functioning protein [6, 7, 13, 31]. In early studies, including a small number of tumor samples obtained from large BC and OC families, it was suggested that greater than 85 % of BRCA1- or BRCA2-associated cancers exhibited LOH, and all showed loss of the normal allele.
The vast majority of BRCA1 and BRCA2 mutation carriers are single heterozygotes for BRCA1 (SH1) or BRCA2 (SH2). Homozygosity of missense alleles at BRCA2 (FANCD1) leads to Fanconi Anemia and increased cancer susceptibility, notably hematological malignancies [15, 22]. At least three Fanconi Anemia cases are attributable to BRCA2/FANCD1 homozygous mutations [22]. Observations of homozygosity or compound heterozygosity at BRCA1 are very rare. Domchek et al. [9] reported a female patient with short stature, microcephaly, developmental delay, significant toxicity from chemotherapy, and epithelial ovarian carcinoma diagnosed at age 28 years. This woman was a compound heterozygote at BRCA1, with mutations c.2457delC (p.Asp821Ilefs*25) and c.5207 T > C (p.Val1736Ala). Both of these mutations are likely to be deleterious variants in BRCA1-associated cancer. The only other reported case of biallelic BRCA1 mutations was in a woman with multiple congenital anomalies consistent with a Fanconi anemia-like disorder and breast cancer at age 23 [30].
Transheterozygosity (TH) is the state of heterozygosity at two different loci. Here, we define TH to be inheritance of deleterious mutations in both BRCA1 and BRCA2. Reports on several BRCA1/2 transheterozygotes (TH) have been reported in the literature, mainly without further details on tumor or patient phenotype. Ramus et al. [27] reported on one TH who had been diagnosed with both BC and OC, and was identified as having a mutation in BRCA1 c.68_69delAG (185/187delAG) and BRCA2 c.5946delT (6174delT). LOH in these tumors was not found. Additional reports identified TH for BRCA1 c.2389G > T and BRCA2 c.3068dupA [21], BRCA1 c.68_69delAG and a BRCA2 c.5946delT [36], and TH with BRCA1 c.68_69delAG and BRCA2 c.5946delT [11] in four cases. In addition, a number of reports of TH with LOH in cancer samples have been published. Randall et al. [28] reported one TH identified with a BRCA1 c.3770_3771delGA and BRCA2 c.5946delT, and being affected with both BC and OC. For the BC, only LOH at the BRCA1 locus was found (not at BRCA2), and the OC sustained LOH at both BRCA1 and BRCA2. Tesoriero et al. [35] reported a TH with BRCA1 c.3770_3771delGA and BRCA2 c.5946delT. The BC of this patient lost the wild-type BRCA2 allele. Bell et al. [1] reported on a TH with c.5266dupC BRCA1 and c.5946delT BRCA2 mutation having three independent BCs. They showed that LOH occurred in two BRCA2 and one BRCA1 tumor. A large clinic-based series of 1191 carriers from Israel [20] identified 16 TH females, 14 with the c.68_69delAG BRCA1 and c.5946delT BRCA2 mutations and two with the c.5266dupC BRCA1 and c.5946delT BRCA2 mutations. A study from Germany identified eight female TH from 8162 BC/OC families and compared the clinical characteristics of the TH to their SH relatives and to SH in the family-based study [14].
To characterize the nature of TH and clinical phenotypes of TH, we used the Consortium of Investigators of Modifiers of BRCA1/2 (CIMBA) dataset of 32,295 female BRCA1/2 mutation carriers ascertained in high-risk clinics and population-based studies. From this dataset, we investigated the occurrence of TH, we compared the characteristics and features of BC and OC in TH and single BRCA1 or BRCA2 mutations, and we examined LOH in as many cancer samples as possible.
Methods
Study sample
Details of CIMBA participating centers and data collection have been reported previously [5]. All the included mutation carriers participated in clinical and research studies at the host institutions after providing informed consent under IRB-approved protocols. Fifty-five centers and multicenter consortia (Additional file 1: Table S1) submitted data that met the CIMBA inclusion criteria [5]. Only female carriers with pathogenic BRCA1/2 mutations, concerning TH, SH1, and SH2 mutation carriers, were included in the current analysis. Pathogenicity of mutation was defined as follows: 1) generating a premature termination codon (PTC), except variants generating a PTC in exon 27 after codon 3010 of BRCA2; 2) large in-frame deletions that span one or more exons; and 3) deletion of transcription regulatory regions (promoter and/or first exon) expected to cause lack of expression of mutant allele. We also included missense variants considered pathogenic by using multifactorial likelihood approaches [4, 12]. Mutations that did not meet the above criteria but have been classified as pathogenic by Myriad Genetics, Inc. (Salt Lake City, UT, USA) were also included.
Mutations are described using the Human Genome Variation Society (HGVS) nomenclature (http://www.HGVS.org/varnomen) where the nucleotide numbering is from the A of the ATG translation initiator codon. For deletions or insertions, the most 3′ position possible was arbitrarily assigned as the altered nucleotide. The description of mutations of all types is given at the genomic level (using cDNA reference sequences NM_007294.3/BRCA1 and NM_000059/BRCA2). BIC nomenclature was also presented for common variants that are familiar to many researchers and clinicians by their BIC designation (http://research.nhgri.nih.gov/bic). For BIC nomenclature, cDNA sequences were used as reference sequence (Genbank: U14680/BRCA1 and NM_000059.1/BRCA2). The nucleotide numbering is from nucleotide 1 of the cDNA gene sequence and for deletions or insertions the most 3′ position possible was arbitrarily assigned as the altered nucleotide.
In order to compare the TH with SH1 and SH2 mutation carriers on phenotypes of interest, we created a matched case–control set, in which “cases” were defined as TH, and “controls” were SH1 and SH2 mutation carriers. Matched SH1 “controls” carried a BRCA1 mutation found in the TH “case”. Matched SH2 “controls” carried a BRCA2 mutation found in the TH “case”. SH1 and SH2 were not matched to TH for any other characteristics. Using this approach, we identified 91 TH and 9316 matched SH1 mutation carriers, and 89 TH and 3370 matched SH2 mutation carriers.
Loss of heterozygosity
From 10 TH individuals, tumor tissue was available from twelve tumors, and blood DNA from 10 TH. From one case, tumor tissue from both BC and OC was available, and from another case affected with bilateral BC, tumor samples were available from both breast tumors. Hematoxylin and eosin (H&E) slides from each tumor were examined by a specialist pathologist. Areas of >80 % tumor cells were marked for macro-dissection. DNA from two 10-micron unstained slides was extracted using the Qiagen QIAmp DNA FFPE Tissue Kit using the standard protocol but with 500 μl deparaffinization solution.
We performed micro-satellite analysis to objectively detect LOH as described previously [16]. We amplified patient tumor and blood DNA for two markers within BRCA1 (D17S855 and D17S1322) and four markers around BRCA2 (D13S290, D13S260, D13S1698, and D13S171). The heterozygosity for these markers ranged from 0.46 to 0.82 [17, 26]. Primer sequences and distance from BRCA1 or BRCA2 are given in Additional file 1 (Table S2). After polymerase chain reaction (PCR) amplification, samples were size-separated on a 96 capillary DNA analyzer (Applied Biosystems 3730xl). Data were analyzed using Genemapper Software (Applied Biosystems). For micro-satellites that were heterozygous, the ratios of allele peak heights for each tumor sample were compared to the allele peak heights for the blood DNA sample using the following formula L = (at2 X an1)/(at1 X an2), where L = the ratio; a = the height of the peak; n1 and n2 = normal allele 1 and normal allele 2; t1 and t2 = tumor allele 1 and tumor allele 2. All ten cases were informative for at least one marker in BRCA1. Where cases were informative for both markers, the LOH data were consistent for the two nearby markers. All ten cases were also informative for at least one of the four markers in BRCA2. In two cases, the data were not consistent across all markers in the 1.74 MB region and the data for the marker closest to BRCA2 was used.
To complement the information obtained from micro-satellite analysis, we also undertook DNA sequence analysis. For each individual, a small region (<200 bp) around each of their two mutations was PCR-amplified from both tumor and blood DNA. DNA from peripheral blood of a healthy control individual was also amplified for each fragment as a control for no mutation. We used 10 ng of DNA in the PCR reaction, using standard protocol and primer sequences (given in Additional file 1: Table S3). All three samples for each mutation were then treated with EXO-SAP-IT (Affymetrix) and Sanger sequenced using standard methods [32]. This sequencing was used to confirm the presence of each mutation in the blood DNA from the patient and not in the control sample. We also assessed the mutation status in the tumor to determine if LOH had occurred. Since we extracted areas of >80 % tumor cells, both alleles can be present even when LOH is present, due to contaminating normal tissue. Therefore, for each tumor we determined for each mutation if the two alleles were at an equal ratio compared to the germline sample or if there was a decrease in one of the two alleles.
Statistical Analysis
For comparison of TH and SH mutation carriers, contingency table analysis using a chi-square test was used for dichotomous variables, and a t test for continuous variables. Fisher’s exact tests were used if sample sizes in any contingency table cell were less than five. Analyses were done in STATA, v. 13.1.
Results
Characteristics of TH versus SH1 and SH2 mutation carriers
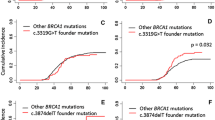
Table 1 describes the 93 female TH from 84 families identified from the CIMBA database. Among the matched TH-SH1/SH2 sets, 25 had no cancer diagnosis. The average age of these women was 39 years and the average age at diagnosis of BC was 41 years. Only 16 women were less than age 41 and 9 women were over age 41 at the time of diagnosis (mean age 49.9, range 41.4–67.9). Table 2 shows that OC age for the matched BRCA1 TH cases was 51.1 years and SH1 controls was 50.9 years (p = 0.154). For the matched BRCA2 set the average OC age for the TH cases was 54.7 years and for SH2 controls was 56.8 years (p = 0.421) (Fig. 1).

Age of breast and ovarian cancer diagnosis by mutation status
The most common TH involved inheritance of two of the three common Jewish mutations: 5 (5.4 %) women inherited BRCA1 c.5266dupC and BRCA2 c.5946delT; 31 (33.3 %) women inherited BRCA1 c.68_69delAG and BRCA2 c.5946delT. Six (6.5 %) women carried one of the three common Jewish mutations and another mutation. The majority of the remaining TH were observed only once. The majority of the TH self-identified as non-Hispanic Caucasian or Jewish. Of the 6907 women who carried one of the Jewish founder mutations, 2732 (39.6 %) self-identified as Jewish, 947 (13.7 %) were unknown, and 3225 (46.7 %) reported an ethnicity other than Jewish. We observed two TH in Hispanics and six TH in Asians (four of which were Korean). Of the 93 TH, 51 were diagnosed with BC only, 4 with OC only, 13 with both BC and OC, and 25 with no cancer diagnosis.
The matched datasets included 91 TH and 9316 SH1 for the BRCA1 matched analysis, and 89 TH and 3370 SH2 for the BRCA2 matched analysis. Two BRCA1 mutations were observed among the TH in our dataset that were not observed among SH1 (c.1390delA and c.3196G > T), and four BRCA2 mutations were observed in the TH dataset that were not observed among the SH2 (c.8633-?_8754 + ?amp, c.739_740delAT, c.5380delG, and c.2269A > T). These six carriers were not included in the analysis (denoted by asterisk in Table 1). TH were more likely to be born more recently (i.e., since 1961) than SH2 mutation carriers but not when compared to SH1s (Table 2). The TH group consisted of more individuals from Asian ancestry compared to the SH1 and SH2 groups, with an excess of women having a Jewish ancestry vs. the SH1 group. TH were more likely to have ever been diagnosed with BC than SH1 or SH2 individuals (68.1 % vs. 52.0 %; p = 0.002, and 67.4 % vs. 50.4 %; p = 0.002), and TH were more likely to be diagnosed with OC than SH2 women (16.9 % vs. 9.3 %; p = 0.017), which was not observed in TH vs. SH1 women, perhaps due to the lower incidence of OC in BRCA2 vs. BRCA1. Age at BC diagnosis was significantly different for TH vs. SH2 (40.5 years vs. 45.0 years; p < 0.001), but there was no difference between TH and SH1.
There were 64 TH cases with BC. Of these, 62 TH were matched to 4846 SH1s and 60 TH were matched to 1699 SH2 (Table 3). TH were more likely to have estrogen receptor (ER)- and progesterone receptor (PR)-positive BC than SH1s (ER: 42.9 % vs. 24.0 %; p = 0.010; PR: 40.6 % vs. 20.0 %; p = 0.013). In contrast, the BCs of TH were less likely ER- and PR-positive than in SH2s (ER: 42.9 % vs. 76.5 %; p < 0.0001; PR: 40.6 % vs. 62.8 %; p = 0.012). The proportion of ER- and PR-positive BCs in TH was intermediate to that of SH1 and SH2. No difference was seen regarding the HER2 status between the BCs of TH and SH1s and SH2s, respectively, although the available numbers were small. No differences in other BC characteristics (morphology, grade, stage) were observed.
Only 17 TH were diagnosed with OC, and thus we had limited data on features of OC to make inferences regarding differences in TH compared with SH1 or SH2. No statistically significant differences were observed for OC traits between TH and SHs (Table 4). Surprisingly, four borderline tumors were reported in both the SH1 and SH2 groups.
Loss of heterozygosity
Due to the frequent LOH in SH individuals, we examined the hypothesis that either BRCA1 or BRCA2 would be lost in each of the TH individuals due to LOH, and that whichever gene was lost could have an impact on their tumor characteristics. Of the 68 TH individuals with cancer, LOH analysis of three tumors from two cases had previously been published by our group using the same methods as the newly identified cases [27]. In the context of the current study, 12 additional tumor samples from 10 patients were analyzed (Table 5). We first used micro-satellite markers and an objective ratio of peak heights to determine if there was loss of one of the alleles when an individual was heterozygous [3] (Additional file 1: Tables S4 and S5). LOH analysis with micro-satellite markers normally includes linkage or segregation data to determine if the normal allele is lost. Since we did not have samples from other family members, we performed Sanger sequencing at the position of the mutations in both germline and tumor samples to determine which allele was lost. One sample failed for the sequencing so it was not possible to determine whether the normal or mutated allele was lost. Some samples showed loss of the mutant allele, which would suggest random loss. Tumors that exhibited LOH by micro-satellite analysis but did not indicate a decrease of the normal allele by sequencing were not considered to exhibit classic LOH. Following both sets of analyses and including our previously published data, one breast tumor (case 8) and one OC (case 2) showed LOH for BRCA1, two breast tumors (cases 9 and 11) showed LOH of BRCA2, and the remaining tumors provided no evidence for LOH at either BRCA1 or BRCA2 (Table 5).
Discussion
This study describes the characteristics of TH compared with SH1 and SH2 mutation carriers and supplements the existing literature regarding LOH in TH. Previously, 35 female TH individuals have been reported in the literature in a series of papers [1, 11, 14, 20, 21, 27, 28, 35, 36]. Only three relatively small studies have so far compared the characteristics of TH to SH women. Lavie et al. [20] reported a non-significant difference in BC occurrence; seven of the 16 TH women (46.7 %) had a personal history of breast carcinoma compared with 372 of 926 (40.2 %) carriers of a single mutation (odds ratio (OR) = 1.3, 95 % confidence interval (CI) 0.4–4.0) [20]. The mean age at diagnosis in TH was 44.6 years, compared with 48.1 in SH. In contrast, Heidemann et al. [14] based on a study of 8 TH individuals suggested that TH develop BC at an earlier age and have more severe disease than those with single heterozygous BRCA mutation [14]. Zuradelli et al. [39] reported TH, and provided the possible association between TH and gastric cancer. Similar to the results from the study by Lavie et al. on 16 Ashkenazi Jewish female TH [20], we report that TH were more likely than both SH1 or SH2 to be diagnosed with BC, which was also observed in our series. In addition to prior reports, we observed that TH were more likely to be diagnosed with OC compared with SH2s, but not compared with with SH1s. TH breast tumors were more likely to be ER-/PR-positive than in SH1, but less likely than in SH2 patients, without other different tumor or disease characteristics.
A number of TH had not been diagnosed with cancer by the time this analysis was completed. Twenty-five TH in our cohort had no BC or OC diagnosis at the time of counseling or genotyping. The average age of these TH individuals was 39.1 years (range 20–68). Of these, 16 (64 %) were less than 41 years old at the time of study, which is the average age of BC diagnosis, and 23 (92 %) were younger than the average age of OC diagnosis (54 years) in the CIMBA data. Of these 25 unaffected TH women, 7 (28 %) reported a RRSO compared to 2751 (22.6 %) who underwent RRSO among the total set of SH controls without BC or OC (12,154). Two (8.0 %) cancer-free TH underwent bilateral risk-reducing mastectomy compared to 1076 (8.9 %) SH. In addition, we had missing data for a number of relevant variables that could have impacted some inferences. For example, of the 62 breast cancers in the TH groups, only 21 (34 %) reported stage information.
Although this is the largest series of TH women reported to date, the study is still limited in a number of ways. TH were more likely to be born more recently (i.e., since 1961) than SH2, but not SH1. Since there is evidence that birth cohort may have an important effect on cancer risk [18], the risk associations reported here may require additional evaluation in the future. The higher incidence of BC in the TH group versus both SH1 and SH2 groups, and of OC in the TH vs. the SH2 cohort could be explained by non-random inclusion of TH in the sample, leading to potential biases in associations, and this may limit generalizability of the dataset. Our analyses also do not account for potentially important confounders and the longitudinal nature of the data to follow cancer cases from time of testing to either cancer diagnosis or censoring after risk-reducing surgery. Furthermore, the great majority of missing data on cancer features avoids that certain questions may be appropriately addressed from this type of dataset. Additional future studies are required to completely evaluate these clinically important unresolved issues, and hopefully with the ongoing multinational collaboration within consortia like CIMBA this will be possible in time.
Differences in breast tumor hormone receptor status suggest that TH cases developing BC have an intermediate cancer phenotype between BRCA1 and BRCA2, which would be consistent with the tumors being driven by loss of either BRCA1 or BRCA2. We attempted to determine the frequency of loss of each gene in a subset of cases where tumor material was available. Previously published data suggest a high rate of LOH with loss of the normal allele in the majority of BRCA1 and BRCA2 cases with strong family history at approximately 80 and 70 years, respectively [24]. However, we did not find loss of either BRCA1 or BRCA2 in the majority of tumors. The low frequency of LOH was consistent with the results from a previously published case (case 12) where we did not find LOH of either gene in either the breast or ovarian tumor [27]. Three other papers on TH showed LOH with loss of the normal allele [1, 28, 35]. One potential reason for the low frequency of LOH in this study could be that seven of the breast tumor samples were areas of ductal carcinoma in situ (DCIS) with micro-invasion rather than a region of the invasive breast tumor. However, we identified two tumors with LOH in these types of samples so this explanation is unlikely to be the major cause of the low rate of LOH.
The observed ages at diagnosis of BC in TH, SH1, and SH2, and the distributions of tumor characteristics may also reveal the interactions of BRCA1 and BRCA2 mutations, which may have implications for modeling the cancer susceptibility in TH. The observed age distributions rule out a multiplicative model for the interactions of BRCA1 and BRCA2 mutations on BC risk. Given the well-established BC risks for BRCA1 and BRCA2 mutations, a multiplicative model would imply very high cancer risk at young ages. However, the present study suggests that ages at BC diagnosis in TH are not significantly different from those in BRCA1 mutation carriers. Therefore, a multiplicative model of cancer risk for BRCA1 and BRCA2 is inconsistent with the current observations. This observation, combined with the fact that the tumor characteristics are intermediate to SH1 and SH2, suggests that an additive model for the joint effects of BRCA1 and BRCA2 mutations is more plausible. These results could be used for modeling the cancer risks for TH carriers and could be incorporated into risk prediction models.
Micro-satellite analysis alone did show a decrease in one of the two alleles in more of the tumors (6 out of 12 BRCA1 and 5 out of 12 BRCA2); however, the sequencing data suggested that the mutant allele rather than the normal allele was lost in many of the tumors. Although the early publications in high-risk families showed very high rates of LOH, exclusively with loss of the normal allele, more recently there have been many publications showing larger numbers of cases with no LOH [19, 23, 24] and an increasing number of tumors with loss of the mutant allele [19, 24]. The second hit in these tumors could be due to a somatic mutation of the normal chromosome or due to promoter methylation, rather than LOH. Unfortunately, the amount of material from these tumors was very limited, and it was not possible to preform additional experiments to investigate alternative mechanisms. Methylation of BRCA1 has been shown to be a mechanism of decreased BRCA1 expression in sporadic BC [2, 34], although this is less frequent in BRCA1 carriers [10, 33]. Why the mechanism of LOH with loss of the normal allele in TH might be different compared with SH is unclear. Tumor material was only available in a small proportion of the cases with cancer. Therefore, it is difficult to interpret the results of the tumor study more broadly. Despite the small numbers, we did not find evidence to support the hypothesis that the tumors would either have LOH of BRCA1 or BRCA2. The TH breast tumor characteristics, however, do appear to be intermediate in phenotype to SH1 and SH2, suggesting some cancers are being driven by inactivation of BRCA1 and some by inactivation of BRCA2. Additional studies that explore other causes of inactivation (e.g., methylation or somatic mutation) are warranted.
Conclusions
We report evidence that the BRCA1 mutation in TH may drive these clinical TH phenotypes based on elevated OC risk in TH vs. SH2 but not SH1, and earlier age of BC diagnosis in TH vs. SH2 but not SH1. Therefore, TH may be managed more like BRCA1 mutation carriers than BRCA2 mutation carriers. In contrast, TH breast tumor characteristics (e.g., ER/PR status) are intermediate in phenotype to SH1 and SH2. Future studies are warranted to understand whether TH should be managed differently to SH1 or SH2 carriers, and, if so, to enable individualized counseling and clinical management appropriate for TH mutation carriers.
Abbreviations
- BC:
-
Breast cancer
- CIMBA:
-
Consortium of Investigators of Modifiers of BRCA1/2
- DCIS:
-
Ductal carcinoma in situ
- ER:
-
Estrogen receptor
- LOH:
-
Loss of heterozygosity
- OC:
-
Ovarian cancer
- PTC:
-
Premature termination codon
- PR:
-
Progesterone receptor
- RRSO:
-
Risk-reducing salpingo-oophorectomy
- SH1:
-
Single mutation at BRCA1
- SH2:
-
Single mutation at BRCA2
- TH:
-
Transheterozygosity, transheterozygote
References
Bell DW, Erban J, Sgroi DC, Haber DA. Selective loss of heterozygosity in multiple breast cancers from a carrier of mutations in both BRCA1 and BRCA2. Cancer Res. 2002;62(10):2741–3.
Birgisdottir V, Stefansson OA, Bodvarsdottir SK, Hilmarsdottir H, Jonasson JG, Eyfjord JE. Epigenetic silencing and deletion of the BRCA1 gene in sporadic breast cancer. Breast Cancer Res. 2006;8(4):R38.
Canzian F, Salovaara R, Hemminki A, Kristo P, Chadwick RB, Aaltonen LA, de la Chapelle A. Semiautomated assessment of loss of heterozygosity and replication error in tumors. Cancer Res. 1996;56(14):3331–7.
Chenevix-Trench G, Healey S, Lakhani S, Waring P, Cummings M, Brinkworth R, Deffenbaugh AM, Burbidge LA, Pruss D, Judkins T, et al. Genetic and histopathologic evaluation of BRCA1 and BRCA2 DNA sequence variants of unknown clinical significance. Cancer Res. 2006;66(4):2019–27.
Chenevix-Trench G, Milne RL, Antoniou AC, Couch FJ, Easton DF, Goldgar DE. An international initiative to identify genetic modifiers of cancer risk in BRCA1 and BRCA2 mutation carriers: the Consortium of Investigators of Modifiers of BRCA1 and BRCA2 (CIMBA). Breast Cancer Res. 2007a;9(2):104
Collins N, McManus R, Wooster R, Mangion J, Seal S, Lakhani SR, Ormiston W, Daly PA, Ford D, Easton DF. Consistent loss of the wild type allele in breast cancers from a family linked to the BRCA2 gene on chromosome 13q12-13. Oncogene. 1995;10(8):1673–5.
Cornelis RS, Neuhausen SL, Johansson O, Arason A, Kelsell D, Ponder BA, Tonin P, Hamann U, Lindblom A, Lalle P. High allele loss rates at 17q12-q21 in breast and ovarian tumors from BRCAl-linked families. The Breast Cancer Linkage Consortium. Genes Chromosomes Cancer. 1995;13(3):203–10.
Domchek SM, Friebel TM, Singer CF, Evans DG, Lynch HT, Isaacs C, Garber JE, Neuhausen SL, Matloff E, Eeles R, et al. Association of risk-reducing surgery in BRCA1 or BRCA2 mutation carriers with cancer risk and mortality. JAMA. 2010;304(9):967–75.
Domchek SM, Tang JB, Stopfer J, Lilli DR, Hamel N, Tischkowitz M, Monteiro AN, Messick TE, Powers J, Yonker A, et al. Biallelic Deleterious BRCA1 mutations in a woman with early-onset ovarian cancer. Cancer Discov. 2013;3(4):399–405.
Dworkin AM, Spearman AD, Tseng SY, Sweet K, Toland AE. Methylation not a frequent “second hit” in tumors with germline BRCA mutations. Fam Cancer. 2009;8(4):339–46.
Friedman E, Bar-Sade Bruchim R, Kruglikova A, Risel S, Levy-Lahad E, Halle D, Bar-On E, Gershoni-Baruch R, Dagan E, Kepten I, et al. Double heterozygotes for the Ashkenazi founder mutations in BRCA1 and BRCA2 genes. Am J Hum Genet. 1998;63(4):1224–7.
Goldgar DE, Easton DF, Deffenbaugh AM, Monteiro AN, Tavtigian SV, Couch FJ. Integrated evaluation of DNA sequence variants of unknown clinical significance: application to BRCA1 and BRCA2. Am J Hum Genet. 2004;75(4):535–44.
Gudmundsson J, Johannesdottir G, Bergthorsson JT, Arason A, Ingvarsson S, Egilsson V, Barkardottir RB. Different tumor types from BRCA2 carriers show wild-type chromosome deletions on 13q12-q13. Cancer Res. 1995;55(21):4830–2.
Heidemann S, Fischer C, Engel C, Fischer B, Harder L, Schlegelberger B, Niederacher D, Goecke TO, Doelken SC, Dikow N, et al. Double heterozygosity for mutations in BRCA1 and BRCA2 in German breast cancer patients: implications on test strategies and clinical management. Breast Cancer Res Treat. 2012;134(3):1229–39.
Howlett NG, Taniguchi T, Olson S, Cox B, Waisfisz Q, De Die-Smulders C, Persky N, Grompe M, Joenje H, Pals G, et al. Biallelic inactivation of BRCA2 in Fanconi anemia. Science. 2002;297(5581):606–9.
Khalique L, Ayhan A, Weale ME, Jacobs IJ, Ramus SJ, Gayther SA. Genetic intra-tumour heterogeneity in epithelial ovarian cancer and its implications for molecular diagnosis of tumours. J Pathol. 2007;211(3):286–95.
Khoo US, Chan KY, Cheung AN, Xue WC, Shen DH, Fung KY, Ngan HY, Choy KW, Pang CP, Poon CS, et al. Recurrent BRCA1 and BRCA2 germline mutations in ovarian cancer: a founder mutation of BRCA1 identified in the Chinese population. Hum Mutat. 2002;19(3):307–8.
King MC, Marks JH, Mandell JB. Breast and ovarian cancer risks due to inherited mutations in BRCA1 and BRCA2. Science. 2003;302(5645):643–6.
King TA, Li W, Brogi E, Yee CJ, Gemignani ML, Olvera N, Levine DA, Norton L, Robson ME, Offit K, et al. Heterogenic loss of the wild-type BRCA allele in human breast tumorigenesis. Ann Surg Oncol. 2007;14(9):2510–8.
Lavie O, Narod S, Lejbkowicz F, Dishon S, Goldberg Y, Gemer O, Rennert G. Double heterozygosity in the BRCA1 and BRCA2 genes in the Jewish population. Ann Oncol. 2011;22(4):964–6.
Liede A, Rehal P, Vesprini D, Jack E, Abrahamson J, Narod SA. A breast cancer patient of Scottish descent with germ-line mutations in BRCA1 and BRCA2. Am J Hum Genet. 1998;62(6):1543–4.
Mathew CG. Fanconi anaemia genes and susceptibility to cancer. Oncogene. 2006;25(43):5875–84.
Merajver SD, Frank TS, Xu J, Pham TM, Calzone KA, Bennett-Baker P, Chamberlain J, Boyd J, Garber JE, Collins FS. Germline BRCA1 mutations and loss of the wild-type allele in tumors from families with early onset breast and ovarian cancer. Clin Cancer Res. 1995;1(5):539–44.
Meric-Bernstam F. Heterogenic loss of BRCA in breast cancer: the “two-hit” hypothesis takes a hit. Ann Surg Oncol. 2007;14(9):2428–9.
Miki Y, Swensen J, Shattuck-Eidens D, Futreal PA, Harshman K, Tavtigian S, Liu Q, Cochran C, Bennett LM, Ding W. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science. 1994;266(5182):66–71.
Miolo G, Puppa LD, Santarosa M, De Giacomi C, Veronesi A, Bidoli E, Tibiletti MG, Viel A, Dolcetti R. Phenotypic features and genetic characterization of male breast cancer families: identification of two recurrent BRCA2 mutations in north-east of Italy. BMC Cancer. 2006;6:156.
Ramus SJ, Friedman LS, Gayther SA, Ponder BA, Bobrow L, van der Looji M, Papp J, Olah E. A breast/ovarian cancer patient with germline mutations in both BRCA1 and BRCA2. Nat Genet. 1997;15(1):14–5.
Randall TC, Bell KA, Rebane BA, Rubin SC, Boyd J. Germline mutations of the BRCA1 and BRCA2 genes in a breast and ovarian cancer patient. Gynecol Oncol. 1998;70(3):432–4.
Saslow D, Boetes C, Burke W, Harms S, Leach MO, Lehman CD, Morris E, Pisano E, Schnall M, Sener S, et al. American Cancer Society guidelines for breast screening with MRI as an adjunct to mammography. CA Cancer J Clin. 2007;57(2):75–89.
Sawyer SL, Tian L, Kähkönen M, Schwartzentruber J, Kircher M, Majewski J, Dyment DA, Innes AM, Boycott KM, Moreau LA, et al. Biallelic mutations in BRCA1 cause a new Fanconi anemia subtype. Cancer Discov. 2015;5(2):135–42.
Smith SA, Easton DF, Evans DG, Ponder BA. Allele losses in the region 17q12-21 in familial breast and ovarian cancer involve the wild-type chromosome. Nat Genet. 1992;2(2):128–31.
Soegaard M, Kjaer SK, Cox M, Wozniak E, Høgdall E, Høgdall C, Blaakaer J, Jacobs IJ, Gayther SA, Ramus SJ. BRCA1 and BRCA2 mutation prevalence and clinical characteristics of a population-based series of ovarian cancer cases from Denmark. Clin Cancer Res. 2008;14(12):3761–7.
Suijkerbuijk KP, Fackler MJ, Sukumar S, van Gils CH, van Laar T, van der Wall E, Vooijs M, van Diest PJ. Methylation is less abundant in BRCA1-associated compared with sporadic breast cancer. Ann Oncol. 2008;19(11):1870–4.
Tapia T, Smalley SV, Kohen P, Muñoz A, Solis LM, Corvalan A, Faundez P, Devoto L, Camus M, Alvarez M, et al. Promoter hypermethylation of BRCA1 correlates with absence of expression in hereditary breast cancer tumors. Epigenetics. 2008;3(3):157–63.
Tesoriero A, Andersen C, Southey M, Somers G, McKay M, Armes J, McCredie M, Giles G, Hopper JL, Venter D. De novo BRCA1 mutation in a patient with breast cancer and an inherited BRCA2 mutation. Am J Hum Genet. 1999;65(2):567–9.
Tsongalis GJ, Linfert DR, Johnson RC, Ackroyd R, Berman MM, Ricci A. Double heterozygosity for mutations in the BRCA1 and BRCA2 genes in a breast cancer patient. Arch Pathol Lab Med. 1998;122(6):548–50.
Tutt A, Robson M, Garber JE, Domchek SM, Audeh MW, Weitzel JN, Friedlander M, Arun B, Loman N, Schmutzler RK, et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: a proof-of-concept trial. Lancet. 2010;376(9737):235–44.
Wooster R, Neuhausen SL, Mangion J, Quirk Y, Ford D, Collins N, Nguyen K, Seal S, Tran T, Averill D. Localization of a breast cancer susceptibility gene, BRCA2, to chromosome 13q12-13. Science. 1994;265(5181):2088–90.
Zuradelli M, Peissel B, Manoukian S, Zaffaroni D, Barile M, Pensotti V, Cavallari U, Masci G, Mariette F, Benski AC, et al. Four new cases of double heterozygosity for BRCA1 and BRCA2 gene mutations: clinical, pathological, and family characteristics. Breast Cancer Res Treat. 2010;124(1):251–8.
Acknowledgments
ACA and the CIMBA data management are funded by Cancer Research UK (C12292/A20861 and C12292/A11174). TRR was supported by R01-CA083855, R01-CA102776, and P50-CA083638. KLN, TMF, and SMD are supported by the Basser Research Center at the University of Pennsylvania. BP is supported by R01-CA112520. Cancer Research UK provided financial support for this work. ACA is a Senior Cancer Research UK Cancer Research Fellow. DFE is Cancer Research UK Principal Research Fellow. Tumor analysis was funded by STOP CANCER (to SJR). Study-specific acknowledgements are as follows:
Study | Funding | Acknowledgements |
BCFR - all | This work was supported by grant UM1 CA164920 from the National Cancer Institute. The content of this manuscript does not necessarily reflect the views or policies of the National Cancer Institute or any of the collaborating centers in the Breast Cancer Family Registry (BCFR), nor does mention of trade names, commercial products, or organizations imply endorsement by the US Government or the BCFR. | |
BCFR-AU | Maggie Angelakos, Judi Maskiell, Gillian Dite, Helen Tsimiklis. | |
BCFR-NY | We wish to thank members and participants in the New York site of the Breast Cancer Family Registry for their contributions to the study. | |
BCFR-ON | We wish to thank members and participants in the Ontario Familial Breast Cancer Registry for their contributions to the study. | |
BFBOCC-LT | BFBOCC is partly supported by: Lithuania (BFBOCC-LT): Research Council of Lithuania grant SEN-18/2015 | BFBOCC-LT acknowledge Vilius Rudaitis and Laimonas Griškevičius. BFBOCC-LV acknowledge Drs Janis Eglitis, Anna Krilova and Aivars Stengrevics. |
BIDMC | BIDMC is supported by the Breast Cancer Research Foundation | |
BMBSA | BRCA gene mutations and breast cancer in South African women (BMBSA) was supported by grants from the Cancer Association of South Africa (CANSA) to Elizabeth J. van Rensburg | BMBSA wish to thank the families who contribute to the BMBSA study |
BRICOH | SLN was partially supported by the Morris and Horowitz Families Endowed Professorship. | We wish to thank Yuan Chun Ding and Linda Steele for their work in participant enrollment and biospecimen and data management. |
CBCS | We thank Bent Ejlertsen Ejlertsen and Anne-Marie Gerdes for the recruitment and genetic counseling of participants | |
CNIO | This work was partially supported by Spanish Association against Cancer (AECC08), RTICC 06/0020/1060, FISPI08/1120, Mutua Madrileña Foundation (FMMA) and SAF2010-20493 | We thank Alicia Barroso, Rosario Alonso and Guillermo Pita for their assistance. |
COH-CCGCRN | City of Hope Clinical Cancer Genetics Community Network and the Hereditary Cancer Research Registry, supported in part by Award Number RC4CA153828 (PI: J. Weitzel) from the National Cancer Institute and the Office of the Director, National Institutes of Health. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health | |
CONSIT TEAM | Funds from Italian citizens who allocated the 5×1000 share of their tax payment in support of the Fondazione IRCCS Istituto Nazionale Tumori, according to Italian laws (INT-Institutional strategic projects ‘5×1000’) to SM. | Bernard Peissel, Jacopo Azzollini and Daniela Zaffaroni of the Fondazione IRCCS Istituto Nazionale dei Tumori, Milan, Italy; Bernardo Bonanni, Monica Barile and Irene Feroce of the Istituto Europeo di Oncologia, Milan, Italy; Alessandra Viel and Riccardo Dolcetti of the CRO Aviano National Cancer Institute, Aviano (PN), Italy; Barbara Pasini and Francesca Vignolo-Lutati of the University of Turin, Turin, Italy; Laura Papi and Gabriele Capone of the University of Florence, Florence, Italy; Laura Ottini and Giuseppe Giannini of the “Sapienza” University, Rome, Italy; Liliana Varesco of the IRCCS AOU San Martino – IST, Istituto Nazionale per la Ricerca sul Cancro, Genoa, Italy; Maria Grazia Tibiletti of the Ospedale di Circolo-Università dell’Insubria, Varese, Italy; Antonella Savarese and Aline Martayan of the Istituto Nazionale Tumori Regina Elena, Rome, Italy; Stefania Tommasi of the Istituto Nazionale Tumori “Giovanni Paolo II” - Bari, Italy. |
CORE | The CIMBA data management and data analysis were supported by Cancer Research UK grants C12292/A20861, C12292/A11174. ACA is a Cancer Research UK Senior Cancer Research Fellow. GCT is an NHMRC Senior Principal Research Fellow. | UK grants C12292/A20861. We wish to thank Sue Healey for her many contributions to CIMBA. |
DEMOKRITOS | This research has been co-financed by the European Union (European Social Fund – ESF) and Greek national funds through the Operational Program “Education and Lifelong Learning” of the National Strategic Reference Framework (NSRF) - Research Funding Program of the General Secretariat for Research & Technology: SYN11_10_19 NBCA. Investing in knowledge society through the European Social Fund. | |
DKFZ | The DKFZ study was supported by the DKFZ. | |
EMBRACE | EMBRACE is supported by Cancer Research UK Grants C1287/A10118 and C1287/A11990. D. Gareth Evans and Fiona Lalloo are supported by an NIHR grant to the Biomedical Research Centre, Manchester. The Investigators at The Institute of Cancer Research and The Royal Marsden NHS Foundation Trust are supported by an NIHR grant to the Biomedical Research Centre at The Institute of Cancer Research and The Royal Marsden NHS Foundation Trust. Ros Eeles and Elizabeth Bancroft are supported by Cancer Research UK Grant C5047/A8385. Ros Eeles is also supported by NIHR support to the Biomedical Research Centre at The Institute of Cancer Research and The Royal Marsden NHS Foundation Trust | RE is supported by NIHR support to the Biomedical Research Centre at The Institute of Cancer Research and The Royal Marsden NHS Foundation Trust |
FCCC | The authors acknowledge support from The University of Kansas Cancer Center (P30 CA168524) and the Kansas Bioscience Authority Eminent Scholar Program. A.K.G. was funded by 5U01CA113916, R01CA140323, and by the Chancellors Distinguished Chair in Biomedical Sciences Professorship. | We thank Ms. JoEllen Weaver and Dr. Betsy Bove for their technical support. |
GC-HBOC | The German Consortium of Hereditary Breast and Ovarian Cancer (GC-HBOC) is supported by the German Cancer Aid (grant no 110837), Rita K. Schmutzler) and by the Center for Molecular Medicine Cologne (CMMC) | |
GEMO | The study was supported by the Ligue Nationale Contre le Cancer; the Association “Le cancer du sein, parlons-en!” Award; the Canadian Institutes of Health Research for the “CIHR Team in Familial Risks of Breast Cancer” program and the French National Institute of Cancer (INCa). | Genetic Modifiers of Cancer Risk in BRCA1/2 Mutation Carriers (GEMO) study : National Cancer Genetics Network «UNICANCER Genetic Group», France. We wish to pay a tribute to Olga M. Sinilnikova, who with Dominique Stoppa-Lyonnet initiated and coordinated GEMO until she sadly passed away on the 30th June 2014, and to thank all the GEMO collaborating groups for their contribution to this study. GEMO Collaborating Centers are: Coordinating Centres, Unité Mixte de Génétique Constitutionnelle des Cancers Fréquents, Hospices Civils de Lyon - Centre Léon Bérard, & Equipe «Génétique du cancer du sein», Centre de Recherche en Cancérologie de Lyon: Olga Sinilnikova†, Sylvie Mazoyer, Francesca Damiola, Laure Barjhoux, Carole Verny-Pierre, Mélanie Léone, Nadia Boutry-Kryza, Alain Calender, Sophie Giraud; and Service de Génétique Oncologique, Institut Curie, Paris: Dominique Stoppa-Lyonnet, Marion Gauthier-Villars, Bruno Buecher, Claude Houdayer, Etienne Rouleau, Lisa Golmard, Agnès Collet, Virginie Moncoutier, Muriel Belotti, Antoine de Pauw, Camille Elan, Catherine Nogues, Emmanuelle Fourme, Anne-Marie Birot. Institut Gustave Roussy, Villejuif: Brigitte Bressac-de-Paillerets, Olivier Caron, Marine Guillaud-Bataille. Centre Jean Perrin, Clermont–Ferrand: Yves-Jean Bignon, Nancy Uhrhammer. Centre Léon Bérard, Lyon: Christine Lasset, Valérie Bonadona, Sandrine Handallou. Centre François Baclesse, Caen: Agnès Hardouin, Pascaline Berthet, Dominique Vaur, Laurent Castera. Institut Paoli Calmettes, Marseille: Hagay Sobol, Violaine Bourdon, Tetsuro Noguchi, Audrey Remenieras, François Eisinger. CHU Arnaud-de-Villeneuve, Montpellier: Isabelle Coupier, Pascal Pujol. Centre Oscar Lambret, Lille: Jean-Philippe Peyrat, Joëlle Fournier, Françoise Révillion, Philippe Vennin†, Claude Adenis. Centre Paul Strauss, Strasbourg: Danièle Muller, Jean-Pierre Fricker. Institut Bergonié, Bordeaux: Emmanuelle Barouk-Simonet, Françoise Bonnet, Virginie Bubien, Nicolas Sevenet, Michel Longy. Institut Claudius Regaud, Toulouse: Christine Toulas, Rosine Guimbaud, Laurence Gladieff, Viviane Feillel. CHU Grenoble: Dominique Leroux, Hélène Dreyfus, Christine Rebischung, Magalie Peysselon. CHU Dijon: Fanny Coron, Laurence Faivre. CHU St-Etienne: Fabienne Prieur, Marine Lebrun, Caroline Kientz. Hôtel Dieu Centre Hospitalier, Chambéry: Sandra Fert Ferrer. Centre Antoine Lacassagne, Nice: Marc Frénay. CHU Limoges: Laurence Vénat-Bouvet. CHU Nantes: Capucine Delnatte. CHU Bretonneau, Tours: Isabelle Mortemousque. Groupe Hospitalier Pitié-Salpétrière, Paris: Florence Coulet, Chrystelle Colas, Florent Soubrier, Mathilde Warcoin. CHU Vandoeuvre-les-Nancy : Johanna Sokolowska, Myriam Bronner. CHU Besançon: Marie-Agnès Collonge-Rame, Alexandre Damette. Creighton University, Omaha, USA: Henry T. Lynch, Carrie L. Snyder. |
GEORGETOWN | CI received support from the Non-Therapeutic Subject Registry Shared Resource at Georgetown University (NIH/NCI grant P30-CA051008), the Fisher Center for Familial Cancer Research, and Swing Fore the Cure. | |
HCSC | Was supported by a grant RD12/0036/0006 and 15/00059 from ISCIII (Spain), partially supported by European Regional Development FEDER funds | We acknowledge Alicia Tosar and Paula Diaque for their technical assistance |
HEBCS | The HEBCS was financially supported by the Helsinki University Hospital Research Fund, Academy of Finland (266528), the Finnish Cancer Society and the Sigrid Juselius Foundation. | HEBCS would like to thank Dr. Kristiina Aittomäki, Taru A. Muranen, Drs. Carl Blomqvist and Kirsimari Aaltonen and RNs Irja Erkkilä and Virpi Palola for their help with the HEBCS data and samples. |
HEBON | The HEBON study is supported by the Dutch Cancer Society grants NKI1998-1854, NKI2004-3088, NKI2007-3756, the Netherlands Organization of Scientific Research grant NWO 91109024, the Pink Ribbon grant 110005 and the BBMRI grant NWO 184.021.007/CP46. HEBON thanks the registration teams of the Comprehensive Cancer Centre Netherlands and Comprehensive Centre South (together the Netherlands Cancer Registry) and PALGA (Dutch Pathology Registry) for part of the data collection. | The Hereditary Breast and Ovarian Cancer Research Group Netherlands (HEBON) consists of the following Collaborating Centers: Coordinating center: Netherlands Cancer Institute, Amsterdam, NL: M.A. Rookus, F.B.L. Hogervorst, F.E. van Leeuwen, S. Verhoef, M.K. Schmidt, N.S. Russell, J.L. de Lange, R. Wijnands; Erasmus Medical Center, Rotterdam, NL: J.M. Collée, A.M.W. van den Ouweland, M.J. Hooning, C. Seynaeve, C.H.M. van Deurzen, I.M. Obdeijn; Leiden University Medical Center, NL: C.J. van Asperen, J.T. Wijnen, R.A.E.M. Tollenaar, P. Devilee, T.C.T.E.F. van Cronenburg; Radboud University Nijmegen Medical Center, NL: C.M. Kets, A.R. Mensenkamp; University Medical Center Utrecht, NL: M.G.E.M. Ausems, R.B. van der Luijt, C.C. van der Pol; Amsterdam Medical Center, NL: C.M. Aalfs, T.A.M. van Os; VU University Medical Center, Amsterdam, NL: J.J.P. Gille, Q. Waisfisz, H.E.J. Meijers-Heijboer; University Hospital Maastricht, NL: E.B. Gómez-Garcia, M.J. Blok; University Medical Center Groningen, NL: J.C. Oosterwijk, A.H. van der Hout, M.J. Mourits, G.H. de Bock; The Netherlands Foundation for the detection of hereditary tumours, Leiden, NL: H.F. Vasen; The Netherlands Comprehensive Cancer Organization (IKNL): S. Siesling, J.Verloop; The Dutch Pathology Registry (PALGA): L.I.H. Overbeek. The HEBON study is supported by the Dutch Cancer Society grants NKI1998-1854, NKI2004-3088, NKI2007-3756, the Netherlands Organization of Scientific Research grant NWO 91109024, the Pink Ribbon grants 110005 and 2014-187.WO76, the BBMRI grant NWO 184.021.007/CP46 and the Transcan grant JTC 2012 Cancer 12-054. HEBON thanks the registration teams of IKNL and PALGA for part of the data collection. |
HRBCP | HRBCP is supported by The Hong Kong Hereditary Breast Cancer Family Registry and the Dr. Ellen Li Charitable Foundation, Hong Kong | We wish to thank Hong Kong Sanatorium and Hospital for their continued support |
HUNBOCS | Hungarian Breast and Ovarian Cancer Study was supported by Hungarian Research Grants KTIA-OTKA CK-80745, OTKA K-112228 and the Norwegian EEA Financial Mechanism Hu0115/NA/2008-3/OP-9 | We wish to thank the Hungarian Breast and Ovarian Cancer Study Group members (Janos Papp, Tibor Vaszko, Aniko Bozsik, Timea Pocza, Judit Franko, Maria Balogh, Gabriella Domokos, Judit Ferenczi, Department of Molecular Genetics, National Institute of Oncology, Budapest, Hungary) and the clinicians and patients for their contributions to this study. |
HVH | We wish to thank the Oncogenetics Group (VHIO) and the High Risk and Cancer Prevention Unit of the University Hospital Vall d’Hebron. Acknowledgements to the Cellex Foundation for providing research facilities and equipment. | |
ICO | ICO: Contract grant sponsor: Asociación Española Contra el Cáncer, Spanish Health Research Fund; Carlos III Health Institute; Catalan Health Institute and Autonomous Government of Catalonia. Contract grant numbers: ISCIIIRETIC RD06/0020/1051, RD12/0036/008, PI10/01422, PI10/00748, PI13/00285, PIE13/00022, 2009SGR290 and 2014SGR364. | We wish to thank the ICO Hereditary Cancer Program team led by Dr. Gabriel Capella. |
IHCC | The IHCC was supported by Grant PBZ_KBN_122/P05/2004 | |
INHERIT | This work was supported by the Canadian Institutes of Health Research for the “CIHR Team in Familial Risks of Breast Cancer” program, the Canadian Breast Cancer Research Alliance-grant #019511 and the Ministry of Economic Development, Innovation and Export Trade – grant # PSR-SIIRI-701. | We would like to thank Dr Martine Dumont, Martine Tranchant for sample management and skillful technical assistance. J.S. is Chairholder of the Canada Research Chair in Oncogenetics. J.S. and P.S. were part of the QC and Genotyping coordinating group of iCOGS (BCAC and CIMBA). |
IOCHBOCS | IOCHBOCS is supported by Ministero della Salute and “5×1000” Istituto Oncologico Veneto grant. | |
IPOBCS | This study was in part supported by Liga Portuguesa Contra o Cancro. | We wish to thank Drs. Ana Peixoto, Catarina Santos, Patrícia Rocha and Pedro Pinto for their skillful contribution to the study. |
KCONFAB | kConFab is supported by a grant from the National Breast Cancer Foundation, and previously by the National Health and Medical Research Council (NHMRC), the Queensland Cancer Fund, the Cancer Councils of New South Wales, Victoria, Tasmania and South Australia, and the Cancer Foundation of Western Australia; | We wish to thank Heather Thorne, Eveline Niedermayr, all the kConFab research nurses and staff, the heads and staff of the Family Cancer Clinics, and the Clinical Follow Up Study (which has received funding from the NHMRC, the National Breast Cancer Foundation, Cancer Australia, and the National Institute of Health (USA)) for their contributions to this resource, and the many families who contribute to kConFab. |
KOHBRA | KOHBRA is supported by a grant from the National R&D Program for Cancer Control, Ministry for Health, Welfare and Family Affairs, Republic of Korea (1020350). | |
MAYO | MAYO is supported by NIH grants CA116167, CA128978 and CA176785, an NCI Specialized Program of Research Excellence (SPORE) in Breast Cancer (CA116201), a grant from the Breast Cancer Research Foundation, and a generous gift from the David F. and Margaret T. Grohne Family Foundation. | |
MCGILL | Jewish General Hospital Weekend to End Breast Cancer, Quebec Ministry of Economic Development, Innovation and Export Trade | |
MODSQUAD | MODSQUAD was supported by MH CZ - DRO (MMCI, 00209805) and by the European Regional Development Fund and the State Budget of the Czech Republic (RECAMO, CZ.1.05/2.1.00/03.0101) to LF, and by Charles University in Prague project UNCE204024 (MZ). | Modifier Study of Quantitative Effects on Disease (MODSQUAD): MODSQUAD acknowledges ModSQuaD members Csilla Szabo (National Human Genome Research Institute, National Institutes of Health, Bethesda, MD, USA); Lenka Foretova and Eva Machackova (Department of Cancer Epidemiology and Genetics, Masaryk Memorial Cancer Institute and MF MU, Brno, Czech Republic); and Michal Zikan, Petr Pohlreich and Zdenek Kleibl (Oncogynecologic Center and Department of Biochemistry and Experimental Oncology, First Faculty of Medicine, Charles University, Prague, Czech Republic). |
MSKCC | MSKCC is supported by grants from the Breast Cancer Research Foundation, the Robert and Kate Niehaus Clinical Cancer Genetics Initiative, and the Andrew Sabin Research Fund. | Anne Lincoln, Lauren Jacobs |
NCI | The research of Drs. MH Greene and PL Mai was supported by the Intramural Research Program of the US National Cancer Institute, NIH, and by support services contracts NO2-CP-11019-50 and N02-CP-65504 with Westat, Inc, Rockville, MD. For CIMBA PRS paper: The research of Drs. MH Greene and JT Loud was supported by the Intramural Research Program of the US National Cancer Institute, NIH, and by support services contracts NO2-CP-11019-50 and N02-CP-65504 with Westat, Inc, Rockville, MD. | |
NNPIO | This work has been supported by the Russian Federation for Basic Research (grants 14-04-93959 and 15-04-01744). | |
NRG Oncology | This study was supported by NRG Oncology Operations grant number U10 CA180868 as well as NRG SDMC grant U10 CA180822, Gynecologic Oncology Group (GOG) Administrative Office and the GOG Tissue Bank (CA 27469) and the GOG Statistical and Data Center (CA 37517). Drs. Greene, Mai and Savage were supported by funding from the Intramural Research Program, NCI. | We thank the investigators of the Australia New Zealand NRG Oncology group |
OCGN | We wish to thank members and participants in the Ontario Cancer Genetics Network for their contributions to the study. | |
OSU CCG | OSUCCG is supported by the Ohio State University Comprehensive Cancer Center. | Leigha Senter, Kevin Sweet, Caroline Craven, Julia Cooper, and Michelle O’Conor were instrumental in accrual of study participants, ascertainment of medical records and database management. |
PBCS | This work was supported by the ITT (Istituto Toscano Tumori) grants 2011-2013. | |
SEABASS | Ministry of Science, Technology and Innovation, Ministry of Higher Education (UM.C/HlR/MOHE/06) and Cancer Research Initiatives Foundation | We would like to thank Yip Cheng Har, Nur Aishah Mohd Taib, Phuah Sze Yee, Norhashimah Hassan and all the research nurses, research assistants and doctors involved in the MyBrCa Study for assistance in patient recruitment, data collection and sample preparation. In addition, we thank Philip Iau, Sng Jen-Hwei and Sharifah Nor Akmal for contributing samples from the Singapore Breast Cancer Study and the HUKM-HKL Study respectively. The Malaysian Breast Cancer Genetic Study is funded by research grants from the Malaysian Ministry of Science, Technology and Innovation, Ministry of Higher Education (UM.C/HIR/MOHE/06) and charitable funding from Cancer Research Initiatives Foundation. |
SMC | This project was partially funded through a grant by the Israel cancer association and the funding for the Israeli Inherited breast cancer consortium | SMC team wishes to acknowledge the assistance of the Meirav Comprehensive breast cancer center team at the Sheba Medical Center for assistance in this study. |
SWE-BRCA | SWE-BRCA collaborators are supported by the Swedish Cancer Society | Swedish scientists participating as SWE-BRCA collaborators are: from Lund University and University Hospital: Åke Borg, Håkan Olsson, Helena Jernström, Karin Henriksson, Katja Harbst, Maria Soller, Ulf Kristoffersson; from Gothenburg Sahlgrenska University Hospital: Anna Öfverholm, Margareta Nordling, Per Karlsson, Zakaria Einbeigi; from Stockholm and Karolinska University Hospital: Anna von Wachenfeldt, Annelie Liljegren, Annika Lindblom, Brita Arver, Gisela Barbany Bustinza, Johanna Rantala; from Umeå University Hospital: Beatrice Melin, Christina Edwinsdotter Ardnor, Monica Emanuelsson; from Uppsala University: Hans Ehrencrona, Maritta Hellström Pigg, Richard Rosenquist; from Linköping University Hospital: Marie Stenmark-Askmalm, Sigrun Liedgren |
UCHICAGO | UCHICAGO is supported by NCI Specialized Program of Research Excellence (SPORE) in Breast Cancer (CA125183), R01 CA142996, 1U01CA161032 and by the Ralph and Marion Falk Medical Research Trust, the Entertainment Industry Fund National Women’s Cancer Research Alliance and the Breast Cancer research Foundation. OIO is an ACS Clinical Research Professor. | We wish to thank Cecilia Zvocec, Qun Niu, physicians, genetic counselors, research nurses and staff of the Cancer Risk Clinic for their contributions to this resource, and the many families who contribute to our program. |
UCLA | Jonsson Comprehensive Cancer Center Foundation; Breast Cancer Research Foundation | We thank Joyce Seldon MSGC and Lorna Kwan, MPH for assembling the data for this study. |
UCSF | UCSF Cancer Risk Program and Helen Diller Family Comprehensive Cancer Center | We would like to thank Dr. Robert Nussbaum and the following genetic counselors for participant recruitment: Beth Crawford, Kate Loranger, Julie Mak, Nicola Stewart, Robin Lee, Amie Blanco and Peggy Conrad. And thanks to Ms. Salina Chan for her data management. |
UKFOCR | UKFOCR was supported by a project grant from CRUK to Paul Pharoah. | We thank Simon Gayther, Carole Pye, Patricia Harrington and Eva Wozniak for their contributions towards the UKFOCR. |
UPENN | National Institutes of Health (NIH) (R01-CA102776 and R01-CA083855; Breast Cancer Research Foundation; Susan G. Komen Foundation for the cure, Basser Research Center for BRCA | |
UPITT/MWH | Frieda G. and Saul F. Shapira BRCA-Associated Cancer Research Program;Hackers for Hope Pittsburgh | |
VFCTG | Victorian Cancer Agency, Cancer Australia, National Breast Cancer Foundation | Geoffrey Lindeman, Marion Harris, Martin Delatycki of the Victorian Familial Cancer Trials Group. We thank Sarah Sawyer and Rebecca Driessen for assembling this data and Ella Thompson for performing all DNA amplification. |
WCP | Dr Karlan is funded by the American Cancer Society Early Detection Professorship (SIOP-06-258-01-COUN) and the National Center for Advancing Translational Sciences (NCATS), Grant UL1TR000124 | |
Funding for the iCOGS infrastructure came from: the European Community’s Seventh Framework Programme under grant agreement n° 223175 (HEALTH-F2-2009-223175) (COGS), Cancer Research UK (C1287/A10118, C1287/A 10710, C12292/A11174, C1281/A12014, C5047/A8384, C5047/A15007, C5047/A10692, C8197/A16565), the National Institutes of Health (CA128978) and Post-Cancer GWAS initiative (1U19 CA148537, 1U19 CA148065 and 1U19 CA148112 - the GAME-ON initiative), the Department of Defence (W81XWH-10-1-0341), the Canadian Institutes of Health Research (CIHR) for the CIHR Team in Familial Risks of Breast Cancer, Komen Foundation for the Cure, the Breast Cancer Research Foundation, and the Ovarian Cancer Research Fund . |
Funding
Described above.
Authors’ contributions
The following authors made substantial contributions to conception and design, or acquisition of data, or analysis and interpretation of data, and were involved in drafting the manuscript or revising it critically for important intellectual content: TRR, TMF, NM, FW, SC, ILA, PA, NA, BKA, DB, JB, RB, PB, AB, SSB, TC, JCa, JCh, KBMC, FJC, CC, MBD, MdlH, OD, SMD, KLN, KD, SE, EMBRACE, DGE, LF, EF, DF, PAG, JGa, GG, AKG, MHG, JGr, EHah, EHal, UH, TVOH, HEBON, ENI, CI, AJ, RJ, KJ-B, EMJ, BYK, BK, KConFab Investigators, AK, YL, CLas, CLaz, JLe, NL, JLu, SM, GM, MM, SLN, HN, DN, RLN, KO, EO, OIO, SKP, MP, PR, CR-F, MAR, CS, JS, CFS, PS, MS, DS-L, GS, CIS, MTa, MRT, S-HT, MBT, MTh, LT, MTi, AET, AT-G, NT, EJvR, DV, SW-G, BW, JNW, JZ, KKZ, LM, DE, GC-T, ACA, and SJR. The following authors were involved in the statistical analysis and oversaw the writing of the manuscript: TRR, TMF, NM, FW, ACA, and SJR. The following author was responsible for the laboratory data generation and analysis: SJR. All authors have given final approval of the version to be published and have participated sufficiently in the work to take public responsibility for appropriate portions of the content. All authors have agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Competing interests
The authors declare that they have no competing interests.
Ethics approval and consent to participate
Ethics approval was obtained at each participating center for the collection of the data described in this report. Informed consent was obtained from each participant for inclusion in this research.
Author information
Authors and Affiliations
Consortia
Corresponding author
Additional files
Additional file 1: Table S1.
Ethics committees that granted approval for the access and use of the data for this study. Table S2. Participant counts by center and mutation. Table S3. Primers used for PCR and Sanger sequencing. Table S4. Primers used in micro-satellite analysis for loss of heterozygosity. Table S5. Micro-satellite loss of heterozygosity and sequencing analysis results. (DOC 177 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Rebbeck, T.R., Friebel, T.M., Mitra, N. et al. Inheritance of deleterious mutations at both BRCA1 and BRCA2 in an international sample of 32,295 women. Breast Cancer Res 18, 112 (2016). https://doi.org/10.1186/s13058-016-0768-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13058-016-0768-3