
Abstract
Background
Platelet-derived growth factor receptor α (PDGFRα) expression is increased in activated hepatic stellate cells (HSCs) in cirrhotic liver, while normal hepatocytes express PDGFRα at a negligible level. However, cancerous hepatocytes may show upregulation of PDGFRα, and hepatocellular carcinoma is preceded by chronic liver injury. The role of PDGFRα in non-cancerous hepatocytes and liver fibrosis is unclear. We hypothesized that upon liver injury, PDGFRα in insulted hepatocytes contributes to liver fibrosis by facilitating intercellular crosstalk between hepatocytes and HSCs.
Methods
Hepatocytes were isolated from normal and thioacetamide (TAA)-induced cirrhotic livers for assessment of PDGFRα expression. Conditional knock-out (KO) C57BL/6 mice, in which PDGFRα was selectively deleted in hepatocytes, were generated. Liver fibrosis was induced by injecting TAA for 8 weeks. Hep3B cells were transfected with a small interfering RNA (siRNA) (PDGFRα or control) and co-cultured with LX2 cells.
Results
PDGFRα expression was increased in hepatocytes from fibrotic livers compared to normal livers. Conditional PDGFRα KO mice had attenuated TAA-induced liver fibrosis with decreased HSC activation and proliferation. Immunoblot analyses revealed decreased expression of phospho-p44/42 MAPK in TAA-treated KO mice; these mice also showed almost complete suppression of the upregulation of mouse double minute 2. Although KO mice exhibited increased expression of transforming growth factor (TGF)-β and Smad2/3, this was compensated for by increased expression of inhibitory Smad7. LX2 cells co-cultured with PDGFRα siRNA-infected Hep3B cells showed decreased PDGFRα, α smooth muscle actin, collagen α1(I), TGFβ, and Smad2/3 expression. LX2/PDGFRα-deleted hepatocyte co-culture medium showed decreased PDGF-BB and PDGF-CC levels.
Conclusions
Deletion of PDGFRα in hepatocytes attenuated the upregulation of PDGFRα in HSCs after TAA treatment, resulting in decreased liver fibrosis and HSC activation. This suggests that in the event of chronic liver injury, PDGFRα in hepatocytes plays an important role in liver fibrosis by affecting PDGFRα expression in HSCs.
Similar content being viewed by others


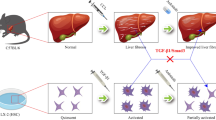
Background
Liver fibrosis is a multicellular response to liver injury in which hepatic stellate cells (HSCs) are responsible for 90% of extracellular matrix (ECM) production [1,2,3,4,5]. Liver fibrosis upon acute liver injury may be beneficial by providing mechanical stability and facilitating the restoration of hepatic architecture and function. However, repetitive chronic liver insults result in liver fibrosis, which may progress to cirrhosis [3, 6, 7], the major contributing factor to development of hepatocellular carcinoma (HCC) [8].
Upon liver injury, HSCs are activated by cytokines, including platelet-derived growth factors (PDGFs), which are potent mitogens [9, 10]. PDGFs exist as five isoforms: PDGF-AA, PDGF-AB, PDGF-BB, PDGF-CC, and PDGF-DD. The cellular effects of PDGFs require the function of cell surface receptor tyrosine kinases comprising platelet-derived growth factor receptor (PDGFR)α and PDGFRβ subunits [11, 12]. Elevated PDGFR expression is observed in human cardiac fibrosis after myocardial infarct, pulmonary fibrosis, kidney fibrosis, and liver fibrosis [13,14,15,16].
PDGFR expression is very low in the normal adult liver but is significantly upregulated in HSCs in cirrhotic liver [17]. PDGFRα participates in liver fibrosis, as evidenced by increased PDGFRα expression in cirrhotic human livers, human cell lines, and a mouse model of liver fibrosis [18, 19]. Stimulation of PDGFRα expression in HSCs induces their activation and proliferation [16, 20,21,22,23], and blocking of PDGFRα expression suppresses HSC proliferation [24]. In addition, stimulation of PDGFRα expression by excessive production of PDGF-CC induces liver cirrhosis and HCC in PDGF-C transgenic (Tg) mice [25, 26]. PDGFRα expression in the liver is markedly increased in PDGF-C Tg mice, although the responsible cell type is unclear.
Normal adult hepatocytes show negligible PDGFRα expression. However, hepatocytes in embryos may have enhanced PDGFRα expression, which later is markedly attenuated [27]. Therefore, the role of PDGFRα expression in adult hepatocytes in liver fibrosis is unknown. However, the increased PDGFRα expression in HCC subsets in the absence of background liver cirrhosis suggests that abnormal hepatocytes overexpress PDGFRα [28].
Our preliminary study demonstrated that PDGFRα expression was increased in not only stimulated HSCs but also injured hepatocytes. We hypothesized that PDGFRα in hepatocytes in the setting of chronic liver injury plays an important role in liver fibrosis by facilitating intercellular crosstalk between hepatocytes and HSCs.
We report for the first time that conditional abolition of PDGFRα expression in hepatocytes attenuates chemically induced liver fibrosis by suppressing the upregulation of PDGFRα and TGFβ in HSCs, reducing their activation and proliferation.
Methods
Generation of PDGFRα conditional KO mice
Homozygous PDGFRαloxp/Loxp mice with targeted deletion of exons 1 to 4 and albumin (alb)-Cre mice, both on the C57BL/6 background, were obtained from Jackson Laboratories (Bar Harbor, ME). PDGFRαloxp/Loxp mice were crossbred with Alb-Cre mice to obtain offspring carrying PDGFRαloxp/wt;Alb-Cre. Next, PDGFRαloxp/wt;Alb-Cre were bred to PDGFRαloxp/Loxp mice, resulting in PDGFRαloxp/Loxp;Alb-Cre+/− or KO mice. Mice of all other genotypes, including PDGFRαloxp/Loxp;Alb-Cre−/− and PDGFRαloxp/wt;Alb-Cre−/−, are referred to as wild-type (WT) controls.
Animals and thioactetamide-induced liver fibrosis
All experiments on mice were approved by the Institutional Animal Use and Care Committee (IACUC) at Gangnam Severance Hospital, Yonsei University College of Medicine (permit number 0031) in accordance with the recommendations and restrictions of the IACUC, which follows the guidelines of the United States National Institutes of Health.
Male 8-week old C57BL/6 (WT) and PDGFRα conditional KO mice (PDGFRα KO) were used. The animals were housed with a 12 h light:dark cycle and fed standard chow. For the fibrosis groups, thioacetamide (TAA) (300 mg/kg body weight) was injected intraperitoneally into male WT and PDGFRα KO mice during the light cycle twice weekly for 8 weeks. One week after the final TAA treatment, the mice were fasted for 14–16 h with free access to water and subsequently euthanized. Untreated age-matched WT or PDGFRα KO male mice were euthanized after the fast.
The animal experiment groups were as follows: WT-control (n = 5), untreated WT mice; KO-control (n = 5), untreated PDGFRα conditional KO mice; WT-TAA (n = 5), WT mice treated with TAA for 8 weeks; and KO-TAA (n = 5), PDGFRα conditional KO mice treated with TAA for 8 weeks.
Hepatocyte and HSC isolation
Hepatocytes and HSCs were isolated from WT and WT-TAA mice by an in situ collagenase perfusion method as described previously with modifications [29]. Protein and RNA were extracted from the isolated hepatocytes for analyses.
HCC tissue
Freshly frozen liver-tissue specimens from livers resected due to HCC or metastatic liver cancer were used. The liver specimens were archived at the Tissue Bank of Yonsei University College of Medicine, Gangnam Severance Hospital with the patients’ consent. The non-tumor sites of liver specimens with hepatitis but not liver cirrhosis as determined via pathologic evaluation (n = 7) were compared to liver specimens from metastatic liver cancer patients (n = 7) that appeared normal on pathologic assessment. This study was approved by the Institutional Review Board of Gangnam Severance Hospital, Yonsei University College of Medicine, Seoul, Republic of Korea (3–2015–0045).
Immunohistochemical analyses and terminal dUTP nick end-labeling assay
Liver-tissue specimens were stained with Masson’s trichrome and the fibrosis ratio was calculated using an image analysis system as described previously with modifications [30]. The total area was calculated as the sum of the area of the microscopic fields, including parenchyma and fibrosis. For each slide, the area of fibrosis was evaluated in 20 consecutive fields at a magnification of × 200, and averaged.
Sections of mouse liver tissue specimens were immunostained using a mouse anti-human alpha-smooth muscle actin (αSMA) primary antibody (DAKO, Carpinteria, CA). Detection of the primary antibody was carried out by the immunoperoxidase technique using an ABC Kit (Vector Laboratories) as a measure of HSC activation. Peroxidase activity was determined by reaction with diaminobenzidine tetrahydrochloride (DAB). Data are total numbers of αSMA-positive cells present in 10 high-power fields (× 400) per sample.
Apoptosis was evaluated by terminal dUTP nick end-labeling (TUNEL) assay. The TUNEL assay detects fragmentation of nuclear DNA during apoptotic cell death in situ, and was performed using an ApopTag Peroxidate In Situ Apoptosis Detection Kit (Millipore, USA). For each slide, the area of immunostained apoptotic nuclei by 3’-OH-end labeling of fragmented DNA was evaluated in 20 consecutive fields at a magnification of × 200, and averaged.
Sections of human liver-tissue specimens were immunostained with a mouse anti-PDGFRα antibody (Santa Cruz Biotechnology, Santa Cruz, CA) and assessed at a magnification of × 400.
siRNA transfection
Transient knockdown assays were performed using DharrmaFECT 1 (Dharmacon, Lafayette, CO), according to the manufacturer’s instructions. PDGFRα (#5156) and control (D-001206-13) siRNAs were purchased from Dharmacon.
Cell lines and in vitro experiments
Hep3B (KCLB #88064) and LX2 (cat #SCC064) cells were purchased from the Korean Cell Line Bank (Seoul, Republic of Korea), and EMD Millipore (Temecula, CA), respectively, and maintained as described previously [31, 32].
Cells were cultured using hanging cell-culture inserts (1 μm pore size, Falcon) to separate cell populations. Wells and inserts with medium were allowed to stabilize for 24 h at 37 °C according to the manufacturer’s recommendations. Hep3B cells were seeded in the insert (3 × 103 cells/cm2) and allowed to attach overnight in Dulbecco’s modified Eagle’s medium (DMEM) with 10% fetal bovine serum (FBS). The following day, siRNA (PDGFRα or control) was transfected into Hep3B cells using DharmaFECT 1 (Dharmacon, Lafayette, CO), according to the manufacturer’s instructions. Briefly, LX2 cells (3 × 105/cm2) were seeded on a six-well plate and incubated for 24 h. Hep3B cells were plated separately on a culture insert and transfected with the PDGFRα or control siRNA. After a 6 h stabilizing period, the insert containing treated Hep3B cells was placed above the plate with LX cells and incubated for a further 24 h. LX2 Cells and supernatants were harvested and recovered for further analyses.
Cell viability assay
The 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay was used to evaluate the proliferation of LX cells. Following co-culture for 24 h, 5 mg/mL MTT was added to the culture medium (Sigma-Aldrich, St. Louis, MO) and incubated for 4 h at 37 °C. The supernatant was removed, the cells were treated with 150 μL/well dimethyl sulfoxide, and the absorption at 470 nm was measured. The assay was carried out in triplicate and each experiment was repeated at least three times. Data are percentages of surviving cells relative to the control.
RNA isolation and real-time polymerase chain reaction
Total RNA was extracted from frozen whole livers or isolated cells using TRIzol reagent (Invitrogen, Carlsbad, CA) or Qiagen mini columns (Qiagen Inc. Valencia, CA) according to the manufacturer’s protocol. RNA concentrations were quantified by spectrophotometry. RNA integrity was assessed by agarose gel electrophoresis and ethidium bromide staining. The RNA samples were diluted in RNase-free water and stored at − 70 °C until use. Five micrograms of RNA were reverse-transcribed using the RNA PCR Kit, version 1.2 (TaKaRa Bio Inc., Japan) according to the manufacturer’s recommendations. Oligonucleotide primers and a TaqMan probe for PDGFRα; TGF-β; Smad 2, 3, and 7; collagen α1(I) (Col1α(I)); αSMA; bax; bcl-2; and MDM2 were used, with 18S as the internal control. The probes were obtained from Applied Biosystems (Perkin-Elmer/PE Applied Biosystems, Foster City, CA). The TaqMan probe was labeled at the 5′-end with the reporter dye FAM and at the 3′-end with a minor groove binder (MGB) nonfluorescent quencher. Quantitative polymerase chain reaction (qPCR) was performed in triplicate for each sample on a Step One Plus Real Time System (Applied Biosystems). Each 20 μL reaction contained 10 μL TaqMan Fast Universal Master Mix (Applied Biosystems, Darmstadt, Germany), 1 μL Gene Expression Mix, and 2 μL cDNA diluted in 7 μL RNase-free water. The thermal cycler conditions were 20 s at 95 °C, followed by 40 cycles of 5 s at 95 °C and 20 s at 60 °C. Fold changes in the expression of target genes relative to the endogenous 18S control were calculated as described previously [33].
Protein extraction and immunoblotting
Whole-liver lysates were prepared in Triton-X 100 lysis buffer containing protease inhibitors and protein concentrations were quantified using the Bradford method with bovine serum albumin (BSA) as the standard. Resolution by SDS-PAGE was followed by immunoblotting using the following antibodies: rabbit anti-PDGFRα (#3164), rabbit anti-phospho-p44/42 MAPK (#9101), rabbit anti-p44/42 MAPK (#9102), rabbit anti-β actin (#4(a967), all from Cell Signaling (Danvers, MA) and anti-phospho-Smad3 (#ab52903) from Abcam (Cambridge, MA). Epitope-primary antibody complexes were detected using species-specific secondary antibodies conjugated to horseradish peroxidase (HRP) followed by enhanced chemiluminescence (ECL) (Thermo Fisher Scientific Pierce, IL).
Determination of PDGF-CC and -BB levels in culture medium
The levels of PDGF-CC and PDGF-BB in culture medium were quantified using a Quantikine enzyme-linked immunosorbent assay (ELISA) Kit (R&D Systems, Minneapolis, MN) according to the manufacturer’s instructions.
Statistical analyses
Results are shown as means ± standard errors of the mean (SEMs). Data were subjected to nonparametric analyses (Kruskal–Wallis or Mann–Whitney test) or one-way analysis of variance (ANOVA) with Tukey’s post hoc test. A value of P < 0.05 was considered indicative of statistical significance. All calculations were performed in IBM SPSS Statistics version 23 for Windows.
Results
PDGFRα deletion in hepatocytes attenuated the TAA-induced increase in PDGFRα expression in the whole liver
The level of PDGFRα expression is increased in fibrotic liver due to its overexpression in HSCs [19, 34]. Non-insulted hepatocytes scarcely express PDGFRα [19].
Hepatocytes from TAA-treated livers showed increased PDGFRα expression compared to those from normal livers (Fig. 1a). To address the role of hepatocyte PDGFRα in liver fibrosis, we generated hepatocyte-specific conditional PDGFRα KO mice (Fig. 1b). The non-insulted livers (control) of WT and conditional KO mice showed no significant differences in PDGFRα expression (Fig. 1c). However, the livers of TAA-treated WT mice, but not those of TAA-treated KO mice, had enhanced PDGFRα expression (Fig. 1c).

Attenuation of PDGFRα expression in TAA-treated livers after conditional deletion of PDGFRα in hepatocytes. a PDGFRα in hepatocytes. b Identification of hepatocyte PDGFRα-deleted knockout (KO) mice by genotyping PCR (lane 2). c PDGFRα expression in the liver of wild-type (WT) and hepatocyte PDGFRα-deleted KO mice (d) Western blotting analyses; densitometry values normalized to that of β-actin are shown. *P < 0.05, compared to that of TAA-untreated WT mice. **P < 0.05, compared to WT mice under identical conditions
Immunoblot analyses of the whole liver showed an increased PDGFRα protein level in TAA-treated WT mice, and a significantly reduced PDGFRα protein level in TAA-treated KO mice (Fig. 1d).
PDGFRα deletion in hepatocytes alleviated TAA-induced HSC activation and liver fibrosis
The fibrotic area in the liver was increased by TAA in both WT and KO mice (Fig. 2a). However, PDGFRα conditional KO mice showed significantly attenuated liver fibrosis compared to WT mice (Fig. 2a). Quantitative analyses of col1α(I) expression yielded a result comparable with the histologic findings (Fig. 2b).

Suppression of TAA-induced liver fibrosis by hepatocyte platelet-derived growth factor receptor (PDGFR) α deletion. a Area of fibrosis. b Expression of (b) Col1α(I) and c αSMA. d Number of αSMA-positive cells. e Western blotting analyses; densitometry values are shown. *P < 0.05, compared to TAA-untreated WT mice. **P < 0.05, compared to WT mice under identical conditions
The TAA-induced increase in the expression of αSMA, a marker of HSC activation, was significantly abolished by deletion of PDGFRα (Fig. 2c). The number of αSMA-positive cells was significantly increased and decreased by TAA in the liver of WT and KO mice, respectively (Fig. 2d).
Deletion of PDGFRα in hepatocytes attenuates ERK activation
ERK is a downstream signaling factor involved in PDGF-induced activation of PDGFR [35]. Immunoblot analyses of phospho-p44/42 MAPK (ERK1/2) revealed increased phosphorylation of ERK1/2 in the livers of WT and KO TAA-treated mice compared to the controls. However, KO mice showed significantly attenuated phosphorylation of ERK1/2 compared to WT TAA-treated mice (Fig. 2e).
Deletion of PDGFRα did not influence hepatocyte apoptosis
TAA treatment increased the number of cells displaying nuclear fragmentation compared to the control. Deletion of PDGFRα in hepatocytes did not affect apoptosis irrespective of TAA treatment, similar to the WT control (Fig. 3a, b). Bcl-2 inhibits apoptosis whereas Bax promotes it [36], and the Bcl-2/Bax ratio is a marker of survival after apoptotic stimuli [37]. The liver Bcl-2/Bax ratio did not differ significantly between WT and KO mice after TAA treatment (Fig. 3c).

Effects of conditional PDGFRα deletion on hepatocyte morphology and apoptosis. a Hematoxylin and eosin (HE) staining (× 400), Masson’s trichrome staining (× 200), and TUNEL staining (× 400) of the liver. b Area of immunostained liver. c Liver Bcl-2: Bax expression ratios. *P < 0.05, compared to TAA-untreated WT mice. **P < 0.05, compared to WT mice under identical conditions
Effect of hepatocyte PDGFRα deletion on TGFβ signaling
Activation of PDGFR by excessive PDGF ligands results in upregulation of the TGFβ/Smad signaling pathway to facilitate HSC proliferation and collagen production [38]. In this study, deletion of PDGFRα in hepatocytes resulted in increased TGFβ expression in normal and TAA-treated livers compared to the WT (Fig. 4a). Downstream signaling of TGFβ and Smad2/3 was also upregulated by deletion of PDGFRα in hepatocytes in both normal and TAA-induced fibrotic livers compared to the WT (Fig. 4b, c). The protein level of Smad3 showed a similar trend to the mRNA level (Fig. 4d). Smad7 was significantly upregulated in the liver of conditional PDGFRα-deleted mice; this likely compensated for the effects of enhanced TGF-β signaling (Fig. 4d).

Effect of hepatocyte PDGFRα deletion on TGF-β signaling in TAA-treated mouse liver. Expression of (a) TGFβ (b), Smad2 (c), and Smad3. d Western blotting analyses of Smads; densitometry values normalized to that of β-actin are shown. e Expression of inhibitory Smad7. *P < 0.05, compared to TAA-untreated WT mice. **P < 0.05, compared to WT mice under identical conditions
Deletion of PDGFRα in hepatocytes blocked upregulation of MDM2
MDM2 is a negative regulator of p53 and is considered an oncogene [39, 40]. Activation of PDGRα independently of PDGF results in activation of MDM2 and suppression of p53 [41, 42]. TAA-induced liver injury was accompanied by upregulation of MDM2 in the liver; this was blocked by deletion of PDGFRα in hepatocytes (Fig. 5a).

Effect of hepatocyte PDGFRα deletion on MDM2 upregulation after TAA-induced liver injury. a MDM2 expression in the liver normalized to that of the 18S rRNA gene. *P < 0.05, compared to TAA-untreated WT mice. **P < 0.05, compared to WT mice under identical conditions. b Expression of MDM2 in hepatocytes and HSCs. *P < 0.05, compared to normal liver cells
To speculate on the cellular components responsible for the TAA-induced increase in MDM2 expression, hepatocytes and HSCs were isolated from TAA-treated livers and MDM2 expression was evaluated. Although both hepatocytes and HSCs from injured livers showed increased MDM2 expression, the magnitude of the upregulation was significantly greater in hepatocytes than in HSCs (Fig. 5b).
Co-culture with PDGFRα-deleted Hep3B cells decreased PDGFRα expression in, and suppressed the activation of, LX2 cells
Hep3B cells have elevated PDGFRα expression [19, 43]. Culture of LX2 cells with PDGFRα-silenced Hep3B cells attenuated PDGFRα expression in the former (Fig. 6a). They had decreased αSMA and col1α(I) expression compared to that on LX2 cells, co-cultured with Hep3B cells with normally expression PDGFRα (Fig. 6b, c).

Changes in LX2 cells co-cultured with Hep3B cells with or without PDGFRα deletion. a Expression of (a) PDGFRα, b col1α(I) and c αSMA in LX2 cells co-cultured with Hep3B cells with or without PDGFRα silencing. d MTT assay results. e Expression of TGFβ, Smad2, Smad3, and Smad7 in LX2 cells. *P < 0.05, compared to LX2 cells co-cultured with PDGFRα-preserved Hep3B cells
LX2 cells were co-cultured with PDGFRα-silenced or control Hep3B cells for 24 h. The proliferation of LX2 cells was reduced by co-culture with PDGFRα-silenced Hep3B cells (Fig. 6d).
TGF β signaling in LX2 cells co-cultured with PDGFRα-silenced Hep3B cells
LX2 cells cultured with PDGFRα-silenced Hep3B cells exhibited attenuated expression of TGFβ, and Smad2/3 (Fig. 6e). However, expression of Smad7 was not downregulated by co-culture with PDGFRα-silenced Hep3B cells (Fig. 6e).
PDGF ligands in PDGFRα-silenced Hep3B cell co-culture medium
The levels of PDGF-BB and PDGF-CC were significantly reduced in the co-culture medium of LX2 cells with PDGFRα-silenced Hep3B cells, and the reduction in the PDGF-BB level was of greater magnitude than that in the PDGF-CC level (Fig. 7).

Levels of PDGF-BB and -CC in Hep3B and LX2 co-culture medium. *P < 0.05, compared to LX2 cells co-cultured with PDGFRα-preserved Hep3B cells
PDGFRα expression in human liver with hepatitis without advanced fibrosis
Expression of PDGFRα was examined in normal human liver and human liver with hepatitis but not advanced fibrosis. Immunohistochemistry showed expression of PDGFRα at the hepatocyte membrane and in nonparenchymal cells in the liver with hepatitis, whereas the normal liver exhibited no PDGFRα staining (Fig. 8a).

Assessment of PDGFRα expression in the human liver with hepatitis without liver fibrosis. a Immunohistochemistry of liver specimens with hepatitis showed the presence of PDGFRα at the hepatocyte membrane (arrows) and in nonparenchymal cells (arrowheads), whereas normal-liver specimens showed no PDGFRα staining. Original magnification, × 400. b Whole-cell lysates from pathologically proven hepatitis tissue without significant fibrosis (n = 7) were examined for col1α(I) and PDGFRα mRNA levels compared to those in normal liver tissue (n = 7)
Whole-cell lysates from livers with pathologically proven hepatitis but not advanced fibrosis (n = 7) were examined for col1α(I) and PDGFRα expression and compared to that in normal liver tissue (n = 7). Livers with hepatitis but not significant liver fibrosis showed elevated PDGFRα expression compared to the normal livers, but col1α(I) expression was not significantly different (Fig. 8b).
Discussion
PDGFRα expression is markedly elevated in chronic liver injury and enhanced PDGFRα activation contributes to liver fibrosis [19, 34]. The proliferation of PDGFRα-positive HSCs during liver fibrosis can be suppressed by blocking of PDGFRα in HSCs [24]. While only PDGFRα in HSCs has been thought to play a role in liver fibrosis, we report that PDGFRα is upregulated in injured hepatocytes, which contributes to HSC proliferation, resulting in liver fibrosis. Deletion of PDGFRα in hepatocytes significantly attenuated TAA-induced liver fibrosis. PDGFRα in hepatocytes plays an important role in liver fibrosis by inducing HSC activation and proliferation; in vitro, HSCs co-cultured with PDGFRα-deleted hepatocytes exhibited attenuated activation and decreased collagen production.
Normal adult hepatocytes express a low level of PDGFRα, the role of which in hepatocytes in liver fibrosis was unclear. However, PDGFRα is reportedly expressed in cancerous hepatocytes, and facilitates the proliferation and migration of HCC cells, which are related to invasion and metastasis [44, 45]. Hepatocytes expressing dominant-negative PDGFRα revealed decreased TGF-β-induced migration and tumor formation [44]. Therefore, our data suggest that PDGFRα in injured hepatocytes also contributes to liver fibrosis, the most important risk factor for HCC.
siRNA KO of PDGFRα in hepatocytes resulted in decreased PDGF-BB and PDGF-CC levels in co-culture medium compared to co-culture of non-KO hepatocytes. Because activated HSCs are possible sources of PDGF ligands [46], and HSC activation was attenuated by PDGFRα deletion in hepatocytes, the reduced PDGF-BB and PDGF-CC levels were likely due to decreased production by HSCs. In addition, the decrement in the PDGF-BB level was of greater magnitude than that in the PDGF-CC level. PDGF-BB is reportedly the most potent mitogen for HSC activation, and some animal models of fibrosis, such as the bile-duct ligation model, show upregulation of PDGF-BB and PDGF-CC, but the latter to a lesser extent [47]. The mechanism by which PDGFRs on hepatocytes are activated is unclear. Increased ERK activation was detected in the livers of TAA-treated WT and KO mice, suggesting that PDGF ligands mediate PDGFR activation upon liver insult; the PDGFRα KO mice exhibited attenuated ERK activation. However, upregulation of MDM2, which is activated at the time of PDGF-independent PDGFR activation, was almost completely blocked by PDGFRα deletion in hepatocytes when WT mice liver showed upregulation of MDM2 after TAA treatment. Although both hepatocytes and HSCs from TAA-treated livers had elevated MDM2 expression, the upregulation in hepatocytes was about one thousand-fold higher than that in HSCs. Therefore, PDGFRα in hepatocytes might be predominantly activated by non-PDGF mediated mechanisms; this warrants further research.
Elevated levels of TGFβ are seen in liver fibrosis and TGFβ overexpression results in liver fibrosis [48, 49]. TGFβ binds to cell surface receptors to initiate intracellular signal transduction pathways, including activation of Smad proteins [50,51,52]. Smad2 and Smad3 form complexes with Smad4 to activate gene expression. Smad7, an inhibitory Smad, disrupts receptor activation of Smad2/3, which inhibits TGFβ signaling in a negative feedback loop [53,54,55]. Activation of PDGFRα ligands by excess PDGF-C upregulates TGFβ/Smad3 signaling to facilitate liver fibrosis and blocking of Smad3 signaling attenuates PDGF-C–induced liver fibrosis [38]. Deletion of PDGFRα in hepatocytes results in upregulation of TGFβ and Smad2/3 expression in the whole liver. However, this increase in TGFβ and Smad2/3 expression is offset by overexpression of Smad7, resulting in attenuation of TAA-induced liver fibrosis. The cellular source of elevated TGFβ, Smad2/3, and Smad7 in the liver after hepatocyte PDGFRα deletion is unclear. However, co-culture of PDGFRα-deleted hepatocytes and normal HSCs resulted in significant upregulation of TGFβ and Smad2/3 expression in HSCs and maintenance of Smad7 expression.
Particles of injured hepatocytes, as apoptotic bodies, promote secretion of proinflammatory and fibrogenic cytokines from neighboring inflammatory cells such as macrophages [56]. In addition, damage-associated molecular patterns (DAMPs) have been suggested to be released from injured hepatocytes; this may induce liver fibrosis [57]. It can be speculated that PDGFRα in injured hepatocytes induces liver fibrosis by promoting the release of DAMPs and activating HSCs. Although PDGRα-deleted HCC cells exhibit enhanced apoptosis [43], this was not so for non-cancerous hepatocytes in this study.
Conclusions
Our findings suggest that although PDGFRα expression in normal hepatocytes is negligible, it is upregulated by liver insult and plays a vital role in liver fibrosis by facilitating the activation and proliferation of HSCs.
Abbreviations
- Alb:
-
Albumin
- Col1α(I):
-
Collagen 1α(I)
- DAMP:
-
Damage-associated molecular pattern
- ECM:
-
Extracellular matrix
- HCC:
-
Hepatocellular carcinoma
- HSC:
-
Hepatic stellate cell
- KO:
-
Knockout
- MDM2:
-
Mouse double minute 2
- MTT:
-
Methyl thiazolyl tetrazolium
- PDGF:
-
Platelet-derived growth factor
- PDGFR:
-
Platelet-derived growth factor receptor
- TAA:
-
Thioacetamide
- Tg:
-
Transgenic
- TGFβ:
-
Transforming growth factor beta
- TUNEL:
-
Terminal dUTP nick end-labeling
- WT:
-
Wild-type
- αSMA:
-
Alpha-smooth muscle actin
References
Friedman SL. Mechanisms of hepatic fibrogenesis. Gastroenterology. 2008;134:1655–69.
Kisseleva T, Brenner DA. Role of hepatic stellate cells in fibrogenesis and the reversal of fibrosis. J Gastroenterol Hepatol. 2007;22(Suppl 1):S73–8.
Pellicoro A, Ramachandran P, Iredale JP. Reversibility of liver fibrosis. Fibrogenesis Tissue Repair. 2012;5(Suppl 1):S26.
Wells RG. Cellular sources of extracellular matrix in hepatic fibrosis. Clin Liver Dis. 2008;12:759–68 viii.
Mederacke I. Liver fibrosis: mouse models and relevance in human liver diseases. Z Gastroenterol. 2013;51:55–62.
Lim YS, Kim WR. The global impact of hepatic fibrosis and end-stage liver disease. Clin Liver Dis. 2008;12:733–46 vii.
Pinzani M, Vizzutti F. Fibrosis and cirrhosis reversibility: clinical features and implications. Clin Liver Dis. 2008;12:901–13 x.
Severi T, van Malenstein H, Verslype C, van Pelt JF. Tumor initiation and progression in hepatocellular carcinoma: risk factors, classification, and therapeutic targets. Acta Pharmacol Sin. 2010;31:1409–20.
Bataller R, Brenner DA. Liver fibrosis. J Clin Invest. 2005;115:209–18.
Hernandez-Gea V, Friedman SL. Pathogenesis of liver fibrosis. Annu Rev Pathol. 2011;6:425–56.
Oseini AM, Roberts LR. PDGFRalpha: a new therapeutic target in the treatment of hepatocellular carcinoma? Expert Opin Ther Targets. 2009;13:443–54.
Heldin CH. Targeting the PDGF signaling pathway in tumor treatment. Cell Commun Signal. 2013;11:97.
Zymek P, Bujak M, Chatila K, Cieslak A, Thakker G, Entman ML, Frangogiannis NG. The role of platelet-derived growth factor signaling in healing myocardial infarcts. J Am Coll Cardiol. 2006;48:2315–23.
Abdollahi A, Li M, Ping G, Plathow C, Domhan S, Kiessling F, Lee LB, McMahon G, Grone HJ, Lipson KE, et al. Inhibition of platelet-derived growth factor signaling attenuates pulmonary fibrosis. J Exp Med. 2005;201:925–35.
Chen YT, Chang FC, Wu CF, Chou YH, Hsu HL, Chiang WC, Shen J, Chen YM, Wu KD, Tsai TJ, et al. Platelet-derived growth factor receptor signaling activates pericyte-myofibroblast transition in obstructive and post-ischemic kidney fibrosis. Kidney Int. 2011;80:1170–81.
Kocabayoglu P, Lade A, Lee YA, Dragomir AC, Sun X, Fiel MI, Thung S, Aloman C, Soriano P, Hoshida Y, et al. Beta-PDGF receptor expressed by hepatic stellate cells regulates fibrosis in murine liver injury, but not carcinogenesis. J Hepatol. 2015;63:141–7.
Breitkopf K, Roeyen C, Sawitza I, Wickert L, Floege J, Gressner AM. Expression patterns of PDGF-A, -B, -C and -D and the PDGF-receptors alpha and beta in activated rat hepatic stellate cells (HSC). Cytokine. 2005;31:349–57.
Iwayama T, Olson LE. Involvement of PDGF in fibrosis and scleroderma: recent insights from animal models and potential therapeutic opportunities. Curr Rheumatol Rep. 2013;15:304.
Hayes BJ, Riehle KJ, Shimizu-Albergine M, Bauer RL, Hudkins KL, Johansson F, Yeh MM, Mahoney WM Jr, Yeung RS, Campbell JS. Activation of platelet-derived growth factor receptor alpha contributes to liver fibrosis. PLoS One. 2014;9:e92925.
Pinzani M. Platelet-derived growth factor receptor expression in hepatic stellate cells: how too much of a good thing can be bad. Hepatology. 1995;22:997–9.
Wong L, Yamasaki G, Johnson RJ, Friedman SL. Induction of beta-platelet-derived growth factor receptor in rat hepatic lipocytes during cellular activation in vivo and in culture. J Clin Invest. 1994;94:1563–9.
Thieringer F, Maass T, Czochra P, Klopcic B, Conrad I, Friebe D, Schirmacher P, Lohse AW, Blessing M, Galle PR, et al. Spontaneous hepatic fibrosis in transgenic mice overexpressing PDGF-A. Gene. 2008;423:23–8.
Czochra P, Klopcic B, Meyer E, Herkel J, Garcia-Lazaro JF, Thieringer F, Schirmacher P, Biesterfeld S, Galle PR, Lohse AW, et al. Liver fibrosis induced by hepatic overexpression of PDGF-B in transgenic mice. J Hepatol. 2006;45:419–28.
Kikuchi A, Pradhan-Sundd T, Singh S, Nagarajan S, Loizos N, Monga S. Platelet-derived growth factor receptor alpha contributes to human hepatic stellate cell proliferation and migration. Am J Pathol. 2017;187:2273–87.
Campbell JS, Hughes SD, Gilbertson DG, Palmer TE, Holdren MS, Haran AC, Odell MM, Bauer RL, Ren HP, Haugen HS, et al. Platelet-derived growth factor C induces liver fibrosis, steatosis, and hepatocellular carcinoma. Proc Natl Acad Sci U S A. 2005;102:3389–94.
Campbell JS, Johnson MM, Bauer RL, Hudkins KL, Gilbertson DG, Riehle KJ, Yeh MM, Alpers CE, Fausto N. Targeting stromal cells for the treatment of platelet-derived growth factor C-induced hepatocellular carcinogenesis. Differentiation. 2007;75:843–52.
Awuah PK, Nejak-Bowen KN, Monga SPS. Role and regulation of PDGFR alpha signaling in liver development and regeneration. Am J Pathol. 2013;182:1648–58.
Yu JH, Kim JM, Kim JK, Choi SJ, Lee KS, Lee JW, Chang HY, Lee JI. Platelet-derived growth factor receptor alpha in hepatocellular carcinoma is a prognostic marker independent of underlying liver cirrhosis. Oncotarget. 2017;8:39534–46.
Seglen PO. Preparation of isolated rat liver cells. Methods Cell Biol. 1976;13:29–83.
O’Brien MJ, Keating NM, Elderiny S, Cerda S, Keaveny AP, Afdhal NH, Nunes DP. An assessment of digital image analysis to measure fibrosis in liver biopsy specimens of patients with chronic hepatitis C. Am J Clin Pathol. 2000;114:712–8.
Friedman SL, Rockey DC, McGuire RF, Maher JJ, Boyles JK, Yamasaki G. Isolated hepatic lipocytes and Kupffer cells from normal human liver: morphological and functional characteristics in primary culture. Hepatology. 1992;15:234–43.
Hiron M, Daveau M, Arnaud P, Bauer J, Lebreton JP. The human hepatoma Hep3B cell line as an experimental model in the study of the long-term regulation of acute-phase proteins by cytokines. Biochem J. 1992;287(Pt 1):255–9.
Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(T)(−Delta Delta C) method. Methods. 2001;25:402–8.
Borkham-Kamphorst E, Kovalenko E, van Roeyen CR, Gassler N, Bomble M, Ostendorf T, Floege J, Gressner AM, Weiskirchen R. Platelet-derived growth factor isoform expression in carbon tetrachloride-induced chronic liver injury. Lab Investig. 2008;88:1090–100.
Kazlauskas A. PDGFs and their receptors. Gene. 2017;614:1–7.
Ikeguchi M, Hirooka Y, Kaibara N. Quantitative analysis of apoptosis-related gene expression in hepatocellular carcinoma. Cancer. 2002;95:1938–45.
Oltvai ZN, Milliman CL, Korsmeyer SJ. Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that accelerates programmed cell death. Cell. 1993;74:609–19.
Lee JI, Wright JH, Johnson MM, Bauer RL, Sorg K, Yuen S, Hayes BJ, Nguyen L, Riehle KJ, Campbell JS. Role of Smad3 in platelet-derived growth factor-C-induced liver fibrosis. Am J Physiol Cell Physiol. 2016;310:C436–45.
Martin K, Trouche D, Hagemeier C, Sorensen TS, La Thangue NB, Kouzarides T. Stimulation of E2F1/DP1 transcriptional activity by MDM2 oncoprotein. Nature. 1995;375:691–4.
Momand J, Zambetti GP. Mdm-2: “big brother” of p53. J Cell Biochem. 1997;64:343–52.
Lei H, Velez G, Kazlauskas A. Pathological signaling via platelet-derived growth factor receptor {alpha} involves chronic activation of Akt and suppression of p53. Mol Cell Biol. 2011;31:1788–99.
Pennock S, Haddock LJ, Eliott D, Mukai S, Kazlauskas A. Is neutralizing vitreal growth factors a viable strategy to prevent proliferative vitreoretinopathy? Prog Retin Eye Res. 2014;40:16–34.
Stock P, Monga D, Tan X, Micsenyi A, Loizos N, Monga SP. Platelet-derived growth factor receptor-alpha: a novel therapeutic target in human hepatocellular cancer. Mol Cancer Ther. 2007;6:1932–41.
Gotzmann J, Fischer AN, Zojer M, Mikula M, Proell V, Huber H, Jechlinger M, Waerner T, Weith A, Beug H, et al. A crucial function of PDGF in TGF-beta-mediated cancer progression of hepatocytes. Oncogene. 2006;25:3170–85.
Fischer AN, Fuchs E, Mikula M, Huber H, Beug H, Mikulits W. PDGF essentially links TGF-beta signaling to nuclear beta-catenin accumulation in hepatocellular carcinoma progression. Oncogene. 2007;26:3395–405.
Greuter T, Shah VH. Hepatic sinusoids in liver injury, inflammation, and fibrosis: new pathophysiological insights. J Gastroenterol. 2016;51:511–9.
Borkham-Kamphorst E, Weiskirchen R. The PDGF system and its antagonists in liver fibrosis. Cytokine Growth Factor Rev. 2016;28:53–61.
Sanderson N, Factor V, Nagy P, Kopp J, Kondaiah P, Wakefield L, Roberts AB, Sporn MB, Thorgeirsson SS. Hepatic expression of mature transforming growth factor beta 1 in transgenic mice results in multiple tissue lesions. Proc Natl Acad Sci U S A. 1995;92:2572–6.
Kanzler S, Lohse AW, Keil A, Henninger J, Dienes HP, Schirmacher P, Rose-John S, zum Buschenfelde KH, Blessing M. TGF-beta1 in liver fibrosis: an inducible transgenic mouse model to study liver fibrogenesis. Am J Phys. 1999;276:G1059–68.
Dooley S, ten Dijke P. TGF-beta in progression of liver disease. Cell Tissue Res. 2012;347:245–56.
Inagaki Y, Okazaki I. Emerging insights into transforming growth factor beta Smad signal in hepatic fibrogenesis. Gut. 2007;56:284–92.
Wells RG. Fibrogenesis. V. TGF-beta signaling pathways. Am J Physiol Gastrointest Liver Physiol. 2000;279:G845–50.
Lonn P, Moren A, Raja E, Dahl M, Moustakas A. Regulating the stability of TGFbeta receptors and Smads. Cell Res. 2009;19:21–35.
Moustakas A, Heldin CH. The regulation of TGFbeta signal transduction. Development. 2009;136:3699–714.
Moustakas A, Souchelnytskyi S, Heldin CH. Smad regulation in TGF-beta signal transduction. J Cell Sci. 2001;114:4359–69.
Seki E, Schwabe RF. Hepatic inflammation and fibrosis: functional links and key pathways. Hepatology. 2015;61:1066–79.
Luedde T, Kaplowitz N, Schwabe RF. Cell death and cell death responses in liver disease: mechanisms and clinical relevance. Gastroenterology. 2014;147:765–83 e764.
Acknowledgments
Not applicable.
Funding
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT & Future Planning (NRF-2016R1A2B4015192), and the Next-Generation 21 Program (PJ01323001).
Availability of data and materials
All data generated or analyzed during this study are included in this article.
Author information
Authors and Affiliations
Contributions
JIL conceived and designed experiments. BJL, WKL, HWL, and KSL contributed to experimental planning and performed the experiments. HYC performed the experiments. HWL, JKK, and HYC analyzed the data. BJL and WKL drafted the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All experiments involving mice were approved by the Institutional Animal Use and Care Committee (IACUC) at Gangnam Severance Hospital, Yonsei University College of Medicine (permit number 0031) in accordance with the recommendations and restrictions of the IACUC, which follows the National Institutes of Health guidelines. Use of human specimens was approved by the Institutional Review Board of Gangnam Severance Hospital, Yonsei University College of Medicine, Seoul, Republic of Korea (3–2015–0045). The liver specimens were archived at the Tissue Bank of Yonsei University College of Medicine, Gangnam Severance Hospital after acquiring patient consent.
Consent for publication
This work does not contain individual patient’s personal details or information.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Lim, B.J., Lee, WK., Lee, H.W. et al. Selective deletion of hepatocyte platelet-derived growth factor receptor α and development of liver fibrosis in mice. Cell Commun Signal 16, 93 (2018). https://doi.org/10.1186/s12964-018-0306-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12964-018-0306-2