
Abstract
Background
Several problems exist with current methods used to align DNA sequences for comparative sequence analysis. Most dynamic programming algorithms assume that conserved sequence elements are collinear. This assumption appears valid when comparing orthologous protein coding sequences. Functional constraints on proteins provide strong selective pressure against sequence inversions, and minimize sequence duplications and feature shuffling. For non-coding sequences this collinearity assumption is often invalid. For example, enhancers contain clusters of transcription factor binding sites that change in number, orientation, and spacing during evolution yet the enhancer retains its activity. Dot plot analysis is often used to estimate non-coding sequence relatedness. Yet dot plots do not actually align sequences and thus cannot account well for base insertions or deletions. Moreover, they lack an adequate statistical framework for comparing sequence relatedness and are limited to pairwise comparisons. Lastly, dot plots and dynamic programming text outputs fail to provide an intuitive means for visualizing DNA alignments.
Results
To address some of these issues, we created a stand alone, platform independent, graphic alignment tool for comparative sequence analysis (GATA http://gata.sourceforge.net/). GATA uses the NCBI-BLASTN program and extensive post-processing to identify all small sub-alignments above a low cut-off score. These are graphed as two shaded boxes, one for each sequence, connected by a line using the coordinate system of their parent sequence. Shading and colour are used to indicate score and orientation. A variety of options exist for querying, modifying and retrieving conserved sequence elements. Extensive gene annotation can be added to both sequences using a standardized General Feature Format (GFF) file.
Conclusions
GATA uses the NCBI-BLASTN program in conjunction with post-processing to exhaustively align two DNA sequences. It provides researchers with a fine-grained alignment and visualization tool aptly suited for non-coding, 0–200 kb, pairwise, sequence analysis. It functions independent of sequence feature ordering or orientation, and readily visualizes both large and small sequence inversions, duplications, and segment shuffling. Since the alignment is visual and does not contain gaps, gene annotation can be added to both sequences to create a thoroughly descriptive picture of DNA conservation that is well suited for comparative sequence analysis.
Similar content being viewed by others
Background
The most widely used methods for aligning DNA sequences rely on dynamic programming algorithms initially developed by Smith-Waterman and Needleman-Wunsch [1, 2]. These algorithms create the mathematically best possible alignment of two sequences by inserting gaps in either sequence to maximize the score of base pair matches and minimize penalties for base pair mismatches and sequence gaps. Although these methods have proven invaluable in understanding sequence conservation and gene relatedness, they make several assumptions. One of their assumptions in generating the "best" alignment is that sequence features are collinear. For example, segments X, Y, Z in sequence one are also ordered as X, Y, and Z in sequence two. Another assumption is that short segments, like Y, have not become inverted or duplicated (e.g. X, Y, Y', Z). These rearrangement events are prone to be gapped out in dynamic programming and thus described as unrelated. Local alignment algorithms can be used to identify these rearrangements provided an exhaustive search is performed, but typically, only the highest scoring local alignments are considered valid and other, lower scoring local alignments are assumed to be spurious matches between unrelated sequences.
When aligning protein coding sequences, dynamic programming works quite well. Evolution exerts significant functional constraint on protein coding sequences. When an inversion, duplication or segment-shuffling event occurs, the protein is often compromised by truncation due to the introduction of frame shifts and stop codons. These deleterious mutations are typically lost and not observed in the surviving population. When aligning this type of constrained sequence element, dynamic programming works quite well.
Functional non-coding sequences do not appear to be as constrained in the ordering of elements as protein coding sequences [3–6]. Compact cis-regulatory modules, for example, enhance or suppress eukaryotic gene expression in response to external stimuli and play key roles in development and differentiation. One of the best characterized eukaryotic enhancers is the even-skipped stripe 2 element in Drosophila that controls transcription of the second transverse stripe of even-skipped mRNA during embryogenesis. Functional and comparative sequence analysis of stripe 2 clearly demonstrate that the enhancer maintains its specific activity across species yet displays significant small-scale insertions, deletions, and rearrangements of transcription factor binding sites within the module [7, 8]. Tracing the evolutionary path of such non-coding elements is proving difficult with current alignment tools and may be assisted by a visual alignment program like GATA.
Implementation
GATA employs a two tiered architecture in aligning DNA sequences. GATAligner executes and processed BLASTN output. GATAPlotter displays the processed alignments and annotation from GATAligner.
GATAligner
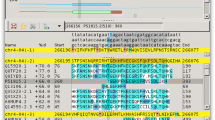
The GATAligner application (figure 1) uses the NCBI bl2seq and BLASTN programs [9, 10] to generate all possible local alignments between two input DNA sequences that score above a very low cut off (see Table 1). To avoid problems associated with visualizing both large and small local alignments, see Results/ Discussion, a sliding window is advanced at one base intervals across each local alignment. Windowed sequences scoring above a defined score are saved. To reduce the number of windowed sequences, each is compared to its neighbours and joined if they are of the same score and orientation. The score is not changed. These "sub-alignment" objects contain several features: a score, an orientation, a reference to the parental local alignment, the aligned sequences, and start and stop coordinates for each sequence. The sub-alignments are then saved to disk. This alignment and post-processing takes less than a minute for two 50 kb sequences using a window size of 24 and a score cut off of 25 bits on an 800 MHz PowerPC laptop computer.

Screen capture of the GATAligner program.
Our initial goal was to create a high resolution sequence alignment and visualization tool to use in identifying small sequence rearrangements, like those associated with evolving non-coding regulatory DNA. We initially divided the first sequence into overlapping windows offset by one base pair. A Smith-Waterman dynamic programming algorithm was then used to align each window against the entire second sequence. Windows were scored, merged, and saved as above. Although this method is more rigorous than using BLASTN, it took 20–50 times as long, and did not produce significantly different results (data not shown). It should be noted that BLASTN requires seven consecutive identical bases to align two sequences. Thus in rare cases, some windows will be missed, for example, GGGGGGcTTTTTTaCCCCCCgAAAAAA versus GGGGGGaTTTTTTgCCCCCCtAAAAAA.
GATAPlotter
The GATAPlotter application (figure 2) takes sub-alignment objects created by GATAligner and displays them graphically. Two boxes connected by a line are used to represent each sub-alignment. The boxes are plotted against horizontal representations of the input sequences with the reference sequence on top. The size of each box is determined by the start and stop positions in the sub-alignment. The shading of the boxes and connector line are scaled according to the sub-alignment score where solid black represents the highest score obtained, light grey the lowest. Lastly the colour of the connecting line is used to indicate the sub-alignment orientation, black for +/+, red for +/-. Where windows overlap, those with the highest score are displayed on top. Single clicking on overlapping windows retrieves all of the underlying windowed sequence alignment information. Double clicking fetches all of the associated local alignment information as parsed from BLAST.

Screen capture of the GATAPlotter program. An alignment between D. melanogaster and D. pseudobscura surrounding gene CG1877.
GATAPlotter also has the capability to display extensive gene annotation for one or both input sequences. The principle component of gene annotation rendering by GATA is the "GeneGroup" (figure 3). Each GeneGroup is drawn independent of other GeneGroups and is allowed to float within the panel to avoid overlap. A typical GeneGroup contains one DNA sequence from which one or more "TransGroups" are derived. Each TransGroup contains exons, an RNA transcript and possibly a protein translation. Each of these features are described using the Berkeley Drosophila Genome Project GFF format [11]. Coding and non-coding DNA sub features are only created in the presence of translation features and represent the most conservative estimation of what is protein coding sequence. If any translation predicts a larger coding region than the others, this is adopted for the entire GeneGroup. A point of confusion by many is that exons encode protein peptides. This is not necessarily true (i.e. 5' and 3' UTRs) and has lead to a variety of annotation rendering errors. When parsing a GFF file, GATAPlotter looks for the following GFF features: exon, translation, transcript, *gene*, *rna*, *transpos*, *misc*, where an * represents one or more wild cards. These wild card "genes" are interpreted as the closing feature on the GeneGroup from which all the proceeding TransGroups are derived. Annotation for both strands is drawn together; arrows are used to indicate orientation. Features not recognized by the parser are interpreted as novel user defined elements and rendered in their own tracks. GFF annotation examples, templates, and extensive descriptions are provided under the GATAPlotter "Documentation" menu. See table 2 and 3 for a complete listing of program options.

Rendered gene annotation in GATA. A typical protein coding gene is visualized as a GeneGroup comprised of multiple TransGroups containing Exons, Introns, and a Protein transcript. Arrows designate orientation. The DNA glyph is rendered as both Non-Coding and Coding elements.
Results and discussion
To illustrate the types of rearrangements GATA can distinguish, examine figures 4 and 5. Both contain alignments between Drosophila melanogaster and D. pseudoobscura. Figure 4 contains three highly similar genes. Figure 5, inversions of putative enhancers. Annotation for each was obtained from whole_genome_annotation_dmel_RELEASE3-1.gff [11]. Orthologous sequences were isolated using the FlyCatcher program [12]. In cases where alignment windows overlap, the lowest scoring windows are drawn first and higher scoring windows placed on top. Both, connecting lines and their associated boxes are shaded according to score.

Example: gene triplication. An example of a gene triplication in D. melanogaster and D. pseudobscura surrounding gene CG14745

Example: sequence inversion. An example of a sequence inversion event between D. melanogaster and D. pseudobscura surrounding gene CG8930.
Several related alignment and visualization tools have proven useful in comparative sequence analysis. Dot plot analysis can be used to identify duplications and inversions. Programs such as Dotter, JDotter, Dotlet, and Family Relations [13–16] generate graphical representations of sequence conservation by scoring identity between two perpendicular sequence representations. Although, mapping annotation to dot plots containing duplications and inversions is rather difficult and counter intuitive. Programs such as Artemis/ACT, LALNVIEW and to some extent, PLALIGN [17–20], utilize alignment information generated from dynamic programming algorithms to create box-line-box representations of each local alignment. These are similar to GATA but do not divide local alignments into window scored sub-alignments. This is unfortunate since window scoring enables a more detailed view of the actual sequence similarity within a large local alignment. Moreover, meaningful visualization using these browsers requires setting a high cut off score for the visualized local alignments. This effectively eliminates smaller, lower scoring local alignments that may provide alternative or even better inverted local alignments. GATA's windowed post-processing overcomes these associated problems.
One program that is proving quite useful in avoiding the collinearity problem while still using a dynamic programming algorithm is Shuffle LAGAN [21]. Alignments generated by Shuffle LAGAN are combine with alignment annotation viewers such as VISTA [22, 23] to align entire genomes. K-BROWSER/ MAVID and Mauve are two additional genome browser/ aligners that look equally promising [24–26]. Although, it should be noted, these programs are designed to provide genome wide alignments and identify large-scale rearrangements, GATA is best suited at interrogating non-coding DNA sequences between 0–200 kb in size for both large and small rearrangements.
One of the major challenges facing bioinformaticians is the development of alignment and visualization tools for multi-species comparative sequence analysis. Within the fly community alone, 12 divergent species of diptera and hymenoptera will be sequenced within 3 years. A variety of higher eukaryotes including human, mouse, rat, dog, chimp, cow, chicken, opossum, and platypus have or are in the process of being completely sequenced. How can one visualize the alignment and species-specific annotation for 12 orthologs of a particular gene or a genomic segment? The GATA alignment paradigm is well suited to this challenge and will play a prominent role in GATA's development.
Conclusions
As comparative sequence analysis accelerates, scientists need more sophisticated alignment and visualization tools to define the evolutionary relationships and functional significance between particular orthologous sequences. This is especially true for regulatory, non-coding DNA that can show significant small-scale rearrangements. These new tools must incorporate detailed annotation alongside views of sequence conservation while providing easy access to the underlying sequence information. GATA provides one such solution.
Availability and requirements
Project name:
GATA, graphic alignment tool for comparative sequence analysis.
Project home pages:
http://gata.sourceforge.net/ and http://rana.lbl.gov/GATA
Operating system(s):
Platform independent
Programming language:
Java
Other requirements:
Java 1.4 or higher
License:
GNU GPL
Any restrictions to use by non-academics:
None
References
Smith TF, Waterman MS: Identification of common molecular subsequences. J Mol Biol 1981, 147(1):195–197. 10.1016/0022-2836(81)90087-5
Needleman SB, Wunsch CD: A general method applicable to the search for similarities in the amino acid sequences of two proteins. J Mol Biol 1970, 48: 443–453.
LudWig MZ: Functional evolution of noncoding DNA. Curr Opin Genet Dev 2002, 12: 634–639. 10.1016/S0959-437X(02)00355-6
Markstein M, Levine M: Decoding cis-regulatory DNAs in the Drosophila genome. Curr Opin Genet Dev 2002, 12(5):601–606. 10.1016/S0959-437X(02)00345-3
Wray GA, Hahn MW, Abouheif E, Balhoff JP, Pizer M, Rockman MV, Romano LA: The evolution of transcriptional regulation in eukaryotes. Mol Biol Evol 2003, 20(9):1377–1419. 10.1093/molbev/msg140
McGregor AP, Shaw PJ, Hancock JM, Bopp D, Hediger M, Wratten NS, Dover GA: Rapid restructuring of bicoid-dependent hunchback promoters within and between Dipteran species: implications for molecular coevolution. Evol Dev 2001, 3(6):397–407. 10.1046/j.1525-142X.2001.01043.x
Ludwig MZ, Patel NH, Kreitman M: Functional analysis of eve stripe 2 enhancer evolution in Drosophila: rules governing conservation and change. Development 1998, 125(5):949–958.
Ludwig MZ, Bergman C, Patel NH, Kreitman M: Evidence for stabilizing selection in a eukaryotic enhancer element. Nature 2000, 403(6769):564–567. 10.1038/35000615
Tatusova TA, Madden TL: BLAST 2 Sequences, a new tool for comparing protein and nucleotide sequences. FEMS Microbiol Lett 1999, 174(2):247–250. 10.1016/S0378-1097(99)00149-4
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ: Basic local alignment search tool. J Mol Biol 1990, 215(3):403–410. 10.1006/jmbi.1990.9999
Misra S, Crosby MA, Mungall CJ, Matthews BB, Campbell KS, Hradecky P, Huang Y, Kaminker JS, Millburn GH, Prochnik SE, Smith CD, Tupy JL, Whitfied EJ, Bayraktaroglu L, Berman BP, Bettencourt BR, Celniker SE, de Grey AD, Drysdale RA, Harris NL, Richter J, Russo S, Schroeder AJ, Shu SQ, Stapleton M, Yamada C, Ashburner M, Gelbart WM, Rubin GM, Lewis SE: Annotation of the Drosophila melanogaster euchromatic genome: a systematic review. Genome Biol 2002, 3(12):research0083.1–0083.22. 10.1186/gb-2002-3-12-research0083
Nix DA: FlyCatcher.[http://rana.lbl.gov/FlyCatcher/]
Sonnhammer EL, Durbin R: A dot-matrix program with dynamic threshold control suited for genomic DNA and protein sequence analysis. Comput Appl Biosci 1996, 12(6):507–510.
Brodie R, Roper RL, Upton C: JDotter: a Java interface to multiple dot plots generated by dotter. Bioinformatics 2004, 20(2):279–281. 10.1093/bioinformatics/btg406
Pagni M, Junier T: Dotlet.[http://www.isrec.isb-sib.ch/java/dotlet/Dotlet.html]
Brown CT, Rust AG, Clarke PJ, Pan Z, Schilstra MJ, De Buysscher T, Griffin G, Wold BJ, Cameron RA, Davidson EH, Bolouri H: New computational approaches for analysis of cis-regulatory networks. Dev Biol 2002, 246(1):86–102. 10.1006/dbio.2002.0619
Rutherford K, Parkhill J, Crook J, Horsnell T, Rice P, Rajandream M-A, Barrell B: Artemis: sequence visualisation and annotation. Bioinformatics 2000, 16(10):944–945. 10.1093/bioinformatics/16.10.944
ACT: Artemis Comparison Tool[http://www.sanger.ac.uk/Software/ACT/]
Duret L, Gasteiger E, Perriere G: LALNVIEW: a graphical viewer for pairwise sequence alignments. Comput Appl Biosci 1996, 12(6):507–510.
Pearson WR: PLALIGN.[http://fasta.bioch.virginia.edu/fasta_www/plalign.htm]
Brudno M, Malde S, Poliakov A, Do CB, Couronne O, Dubchak I, Batzoglou S: Glocal alignment: finding rearrangements during alignment. Bioinformatics 2003, 19(Suppl 1):i54–62. 10.1093/bioinformatics/btg1005
Brudno M, Poliakov A, Salamov A, Cooper GM, Sidow A, Rubin EM, Solovyev V, Batzoglou S, Dubchak I: Automated whole-genome multiple alignment of rat, mouse, and human. Genome Res 2004, 14(4):685–92. 10.1101/gr.2067704
Shah N, Couronne O, Pennacchio LA, Brudno M, Batzoglou S, Bethel EW, Rubin EM, Hamann B, Dubchak I: Phylo-VISTA: interactive visualization of multiple DNA sequence alignments. Bioinformatics 2004, 20(5):636–643. 10.1093/bioinformatics/btg459
Chakrabarti K, Pachter L: Visualization of multiple genome annotations and alignments with the K-BROWSER. Genome Res 2004, 14(4):716–720. 10.1101/gr.1957004
Bray N, Pachter L: MAVID: Constrained Ancestral Alignment of Multiple Sequences. Genome Res 2004, 14(4):693–699. 10.1101/gr.1960404
Darling AC, Mau B, Blattner FR, Perna NT: Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res 2004, 14(7):1394–1403. 10.1101/gr.2289704
Acknowledgements
DAN received postdoctoral support from the American Cancer Society and would like to thank Lisa Simerinko for her assistance with Java and GUIs.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
DAN designed and constructed the GATA programs with advice and supervision from MBE.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Nix, D.A., Eisen, M.B. GATA: a graphic alignment tool for comparative sequence analysis. BMC Bioinformatics 6, 9 (2005). https://doi.org/10.1186/1471-2105-6-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2105-6-9