
Abstract
In the last decades, optical tweezers have progressively emerged as a unique tool to investigate the biophysical world, allowing to manipulate and control forces and movements of one molecule at a time with unprecedented resolution. In this review, we present the use of optical tweezers to perform single-molecule force spectroscopy investigations from an experimental perspective. After a comparison with other single-molecule force spectroscopy techniques, we illustrate at an introductory level the physical principles underlying optical trapping and the main experimental configurations employed nowadays in single-molecule experiments. We conclude with a brief summary of some remarkable results achieved with this approach in different biological systems, with the aim to highlight the great variety of experimental possibilities offered by optical tweezers to scientists interested in this research field.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Since ancient times, humans have being fascinated by light, which is intimately involved with our existence and strongly affects our daily lives. Already in the Ancient Greece, mathematicians and philosophers were interested in light phenomena, just think of Euclid, who summarized the knowledge at his time on light reflection and refraction in his work Optica, where he proposed for the first time mathematical formulas to describe such effects. For a long time, the studies on light were strictly connected to astronomy and it was only in the 17–18th centuries that scientists began wondering about the physical nature of light. The understanding that light is a form of electromagnetic wave marked a turning point in physics and opened the way to a new driving force in scientific investigations: the creation and control of light at will and the possibility to exploit it to make progress also in research fields other than physics. The potentiality of using light to probe the structure of matter has been then boosted by the discovery of photons. In this scenario, surely the invention of optical tweezers more than three decades ago represents a milestone.
The first optical trapping of microscopic particles was reported in 1970 by Arthur Ashkin [1,2,3] at Bell Laboratories, who was awarded by the Nobel prize in 2018 for this discovery. Although the underlying physical principle of Optical Tweezers (OT), the so-called radiation pressure, was known since long time, only thanks to the technological developments achieved in the ’60s, it was possible to overcome the limitations that seemed to make unfeasible till then any practical use of this manipulation method. Ashkin’s work pioneered the field of optical trapping and since then the interest of the scientific community has been progressively increasing, so much that nowadays hundreds of research teams worldwide have an optical tweezers instrument in their laboratories. Besides the possibility to better understand the interaction between light and matter, optical tweezers are also (and above all) used as a powerful tool to manipulate micro-sized objects, thanks to the forces exerted by highly focused laser beams, which find application in different research areas, from molecular biology to statistical mechanics or microrheological studies [4,5,6,7]. Of course, other micro-manipulation techniques based on light have been developed apart from optical tweezers, as optoelectronic tweezers [8] and photovoltaic ones [9,10,11]. In those cases, the trapping force is not directly provided by photons themselves, but rather by electrical effects mediated by light. The main advantages are here represented by the use of less powerful light sources required for the experiments and the possibility to manipulate large ensembles of particles in parallel. However, these light-induced tweezers are two-dimensional and cannot be exploited to actuate particles in three dimensions. Moreover, their response times are typically slow compared with standard OT systems, due to the physical properties of the intermediate light-sensitive material, and they are not very efficient to manipulate sub-micrometric objects.
In the last years, the new technologies have certainly contributed to spread the use of optical tweezers in various research areas, expanding the potentialities offered by optical trapping and manipulation [12]. Among the others, one of the main achievements is represented by holographic optical tweezers (HOTs), where a single laser beam is split into many optical traps by using a computer-controlled diffractive optical element, like a commercial Spatial Light Modulator(SLM). Besides the simultaneous creation of multiple traps, the main exciting feature of HOTs is represented by the possibility to individually move in three-dimensions the trapped objects [4, 13, 14]. This makes holographic tweezers highly suitable to investigate the hydrodynamic behaviour of colloidal systems [15,16,17] and to control optically trapped particles for studying statistical physics phenomena and ensembles out of equilibrium [18, 19]. HOTs also provide new insights into the operation of micromachines [20, 21] and represent a powerful manipulation tool in optofluidic lab-on-a-chip devices [13], for sorting and sensing applications on particles, droplet and biomolecules [22,23,24]. Another significant success granted by optical tweezers is represented by its unprecedented access to the nanoscopic world. As a matter of fact, recently new approaches have been developed to overcome the difficulties related to scale down to nanometres the optical tweezing techniques [25]. In the last years, Nanotweezers have been successfully applied to a wide variety of nanostructures, such as plasmonic nanoparticles [26, 27], nano-semiconductors [28,29,30,31], quantum dots [32] and graphene [33, 34]. Moreover, nanotweezers have also been combined with spectroscopy techniques, so that the physical and chemical properties of a nanostructure can be characterized by investigating its light-induced vibrational, electronic and plasmonic response [35,36,37,38]. Besides the fascinating applications mentioned previously, it has to be recognized to optical tweezers the great merit to have bridged the gap between physics and biology in the last thirty years. For a long time, these two sciences diverged and awkwardly looked at each other, but in the second part of the 20th century, they started to overcome their reciprocal diffidence. A famous example of this collaboration is represented by the discovery of the double helix structure of DNA made in 50s by means of X-ray diffraction experiments, a technique typical of physical studies on matter and nowadays widely exploited also in chemistry, mineralogy, material science and biology [39]. Over the years, biologists have taken keen interest in the experimental techniques and analysis methods used in physics, recognizing their potentialities for understanding the biological world, whereas physicists were progressively attracted by the marvellous variety and complexity of the biological forms that were also used to test new physical theories. Most important, physicists became aware that force is a fundamental factor in biological systems, where it acts at all length scales, from cells to single molecules [40]. In this scenario, optical tweezers play a key role, since they are able to apply and control forces in the same range experienced by molecular systems in their native environment (that is from a few tens of femtoNewtons to almost 100 picoNewtons) and they are nearly non-destructive for biological specimens. A large variety of biophysical investigations have been successfully carried out by exploiting single-beam, dual-beam and holographic optical tweezers [41,42,43,44], and the ongoing technological developments have extended the range of applicability of these optical manipulation strategies.
In this work, we will review different methods and experimental approaches based on OT to apply and measure forces on single-molecule (SM) systems. At present, there is a wide variety of optical trap designs and an exhaustive discussion of all those available for single-molecule investigations is beyond the scope of this review. Rather, we would like to present some aspects and experimental results which could highlight the potentialities offered by these manipulation methods, first by comparing OT with other used SM techniques, then by presenting the main physical phenomena underlying the light-induced manipulation mechanisms and, finally, by discussing some remarkable applications of optical tweezing strategies applied to single molecules of DNA, proteins and molecular motors.
2 Techniques for single-molecule force spectroscopy
Among the experimental potentialities offered by optical tweezers technique, the possibility to manipulate biological systems at the single-molecule level is surely one of the most amazing. Single-molecule (SM) experiments were made possible not only by the advent of nanotechnology and molecular biology, but also by the advances in materials science that allowed optical tweezers and other techniques to reach unprecedented force and spatial resolutions.
In the last decades, single-molecule experiments have emerged in the biophysical research area, thanks to their capability to provide a complementary and different perspective with respect to traditional ensemble-based methods [45, 46]. As a matter of fact, the capability to follow individual molecules allows investigating aspects of molecular thermodynamics and kinetics that are sometimes difficult to be obtained via bulk experiments. First, single-molecule methods yield both averages and distributions of the investigated biophysical properties, highlighting that each molecule is unique and presents random and stochastics dynamics. In contrast, bulk methods provide results that are obtained over an ensemble made by a huge number of molecules (also in the case of a small volume of diluted solutions we are speaking of at least \(10^{9}\) molecules), so that rare events could not be revealed due to the fact that the measured signals are unsynchronized. An explanatory example is represented by proteins and their unfolding–folding mechanisms (as it will be explained in details in Sect. 4.2). A protein can exist in its folded (or misfolded) state or the unfolded one and a full characterization of the transition between these configurations can be provided, for instance, by calorimetry bulk experiments. However, it is well-known that during the folding/unfolding process, some proteins visit one or more intermediate states, which are usually characterized by very short lifetimes. Due to this, the fraction of molecules that are in the intermediate state can be small enough to preclude their detection in bulk calorimetry investigations, being the corresponding signal masked by those that are in most populated conformational ensembles. On the contrary, in SM measurements the protein conformation transition is followed in real time and, by using the proper time resolution, it is possible to reveal each transition pathways and the corresponding kinetics. Moreover, SM experiments grant the investigation of the molecular heterogeneity, a key parameter for those molecules (like some molecular motors) which can exist in multiple spatial conformations, each of them having a characteristic energy. The mean conformation revealed by bulk experiments surely represents the most stable among the possible ones, but in such experiments crucial information on the other existing states could be missing, even if these are related to precise biological functions and essential to obtain a complete picture of the molecule behaviour. For these reasons, single-molecule experiments are nowadays considered a source of invaluable information to better and more deeply understand several biomolecular processes.

Simplified representations of the most used techniques for single-molecule experiments. a Atomic Force Spectroscopy (AFM) consists of a flexible cantilever with a sharp tip, which is used as force-transducer. Tip movements are usually monitored by an optical method, where a laser beam is focused on the back of the cantilever and is reflected to a position-sensitive detector (PSD): any deflection of the cantilever results in a displacement of the beam on the PSD, whose electric signal is converted into a force value. The molecules lie on a surface supported by a piezoelectric stage, which is moved with respect to the cantilever to investigate the morphology of the biological sample. AFM is often used to characterize protein aggregation, or to perform force spectroscopy studies on the unfolding/refolding mechanisms of single proteins, where the molecule is tethered between the tip and the underlying surface. b Magnetic tweezers (MT) employ magnets to create a magnetic field gradient which results in a force acting on a paramagnetic bead and, consequently, to the single molecule attached to it. The second end of the molecule is linked to one coverslip of the fluidic chamber, where the experiments are carried out. A motorised holder moves the magnets in the vertical direction, thus tuning the force and inducing a molecule stretch; a magnets rotation can be also realised, which causes a torsion of the bead and the molecule. An optical system equipped with a lens objective and a CCD camera is used for real-time detection of the bead position. c Optical tweezers (OT) exploit an highly focused laser beam to trap micro- and nanosized objects at its focal point. A typical geometry in single-molecule experiments performed with OT consists of a single-molecule tethered between two beads, which are trapped by two focused laser beams: when one bead is moved with respect to the other, a mechanical stimulus is applied to the investigated biological system
Of course, the capability to handle an individual molecule requires the proper combination of instrumentation and biological assays. As a matter of fact, current single-molecule manipulation methods span several orders of magnitude both in force (from \(10^{-14}\) to \(10^{-8}\) N) and length (from \(10^{-10}\) to \(10^{-4}\) m) and often have complementary forces and loading rate regimes. Over the past two decades, several single-molecule methods have been developed to apply and measure forces on one molecule at a time. Among these, atomic force microscopy (AFM), magnetic tweezers (MT) and optical tweezers (OT) are the most commonly employed [47] and in the following they will be briefly discussed (more exhaustive reviews can be found in [47, 48]).
AFM is a well-established technique to study the surface morphology of a sample, by monitoring distance-dependent interaction forces between a sharp probe and the substrate. The probe is represented by the tip of a flexible cantilever (see Fig. 1a), commonly made of silicon or silicon nitride and with pyramidal or conical shape. The sample is moved with respect to the probe by means of piezoelectric actuators, so the tip interacts with the substrate by scanning its surface in a raster way. The probe-surface interaction force is derived by monitoring the deflection of the cantilever tip, whose amplitude is measured exploiting an optical lever method, where a laser beam is focused on the back of the cantilever and its reflected component is detected by a position-sensitive four-quadrant photodiode. The most exploited feature of AFM is the imaging, which has been widely used to resolve heterogeneous conformational states of various biomolecular samples [49], such as protein aggregates. However, AFM is also employed to manipulate and exert mechanical force on single molecules. In single-molecule experiments with AFM, usually the substrate supported by the piezoelectric stage is covered with the molecules to be analysed, and by approaching the tip to the surface a link with the free end of one molecule can be created. As a result, the molecule is tethered between the cantilever tip and the underlying surface (see Fig. 1a), and it can be stretched with sub-nanometric resolution by retracting away the tip and exploiting the piezo-electric actuators. The forces which can be exerted with AFM methods lie in a wide range, between 20 pN and 10 nN, with stiffness values of 10–10000 pN/nm and a remarkable spatial resolution (\({\sim }\) 0.2nm) [48]. These properties make AFM an ideal tool to investigate strong covalent bonds, to probe stiff intermolecular and intramolecular interactions and to image single molecules on a flat surface, like for protein aggregation. Moreover, in the last years several technological developments have permitted to improve the time resolution of this technique down to the millisecond range, meaning that now it is possible to watch single biomolecules in action, like the case of myosin V [50]. Despite this, the relative low force resolution (\({\sim }\) 10pN) clearly highlights that AFM has some limitations on studying the mechano-response of single molecules in the lower piconewton regime, as well as to investigate the dynamics of molecular motors in the sub-millisecond range. Moreover, specificity represents another potential drawback of this technique: indeed, sometimes it is difficult to discriminate between interactions of the tip with the desired molecule from other non-specific interactions, as well as to avoid that the AFM tip establishes the link in a position on the molecule other than the desired one.
Concerning the magnetic tweezers [45], they use a super-paramagnetic micro-bead as force sensor to apply controlled mechano-stresses to single molecules. In standard configurations, the investigated molecule is tethered via chemical bonds between the super-paramagnetic bead and one of the coverslips used to assemble the fluidic chamber, where the experiments are carried out (see Fig. 1b). The fluidic cell is exposed to a magnetic field created by means of a magnets array (or electromagnets) suspended in its proximity. The sample is illuminated through the gap between the magnets with a light emitting diode and the interference pattern created by the unscattered light and that scattered by the bead is recorded by a CCD camera and exploited to determine the sphere position. The magnetic field exerts an upward (or downward) force on the bead, which results in a mechanical stimulus applied to the biological system; by moving the translation stage which supports the magnets it is possible to vary the magnetic force and perform stretching experiments on the molecule. Typical values of spatial resolution in magnetic tweezers are between 2 and 10 nm, the temporal resolution is in the range \(10^{-2}\) and \(10^{-4}\) sec and the magnetic trap stiffness is very low, around \(10^{-4}\) pN/nm. Therefore, in contrast to AFM technique, the magnetic tweezers are very sensitive to low forces, which can be varied from 100 pN down to 0.01pN. Moreover, MT have the remarkable advantage to be able to really exert a constant force to the molecule when they operate in passive mode, since the spatial fluctuation of the bead during the measurements is small enough for the magnetic field gradient to be considered constant. On the contrary, in AFM and OT instruments the constant-force mode can be achieved only by feedback-control mechanisms that usually do not allow monitoring dynamic phenomena in the sub-millisecond scale. Furthermore, the magnetic field only acts on the paramagnetic bead used as probe and this assures the possibility to perform noninvasive force measurements also in a heterogeneous environment, like the interior of a cell. Finally, MT are widely and mainly exploited to twist single molecules and detect the consequent structural transitions, as in the studies on the chiral nature of DNA reported in [51, 52].
Optical tweezers exploit focused laser beams to trap objects ranging in size from a few tens of nanometres to several microns. However, in single-molecule experiments, the typical configuration consists of a microsized bead used as force sensor to which the molecule under investigation is attached (see Fig. 1c), whereas the other end of the molecule can be linked to a fixed or moving surface. As it will be explained more in detail in Sect. 3, the bead is trapped in the focus of the laser beam and the scattering and gradient forces exerted by light allow moving it in three dimensions with a high spatial resolution, down to 0.1–0.2 nm. OT are able to exert forces in the range 0.1–100 pN, depending on the size of the bead and the power of the laser, and the values of trap stiffness (\(\sim \) 0.01–1 pN/nm) and force resolution (\(\sim \) 0.05 pN) are about 10 and 100–1000 times smaller than AFM methods, respectively. Moreover, OT have usually better spatiotemporal resolution compared with magnetic tweezers, since the light-induced displacement of the bead has a sub-nanometric accuracy with a detection system characterized by a sub-millisecond resolution. Single-molecule experiments with optical tweezers have been performed on very different biological systems, from nucleic acids to proteins, molecular motors and cells [53], and the wide existing literature confirms the notable versatility of this technique.
However, also OT suffer from some limitations and drawbacks. The first one is that OT lack of selectively and any object in proximity of the beam focus can be easily trapped. It is then important to keep as low as possible the number of uncontrolled particles flowing close to the experimental area, in order to avoid an object from being trapped, while the experiments is being carried out with an other one. A further limitation is represented by the possible damages that can be induced by laser beams with high intensity that also restricts the use of conventional OT for in vivo experiments; in this case, it is suggested to use spheres with larger diameters (more than 2 microns) or silica beads to keep the local heating below 1 \(^\circ \)C, as well as light wavelengths in the near infrared. Moreover, optical tweezers suffer for any instability of the beam pointing or laser power, which result in undesired motions of the optical trap and fluctuations in the applied force, respectively. These drawbacks particularly invalidate the experiments where a high reproducibility as well as quantitative measurements are needed. A more stable optical trap can be obtained by using a high-quality laser source, which can be quite expensive but significantly enhances the performance of the setup. Another source of instabilities is represented by air flows, which can introduce local fluctuations of the refractive index, thus deviating the optical path of the laser beam. Thus, often the main optical components of the setup are enclosed in tubes or boxes, which, however, complicate the periodic alignment of the whole optical path. For the same reason, high-resolution measurements with OT are often limited to homogeneous solution and purified samples, to reduce as much as possible optical perturbations. Mechanical vibrations could also prevent from performing measurements with high spatial and force resolutions. As a consequence, the stabilization of both the optical and detection components (lasers, microscope, light detectors, etc), as well as the fluidic chamber where the experiments are carried out, plays a crucial role in determining the quality of the experimental data. In this case, an optical table with passive or active vibration-insulating system should be used to reduce instrumental noise; moreover, for more challenging experiments, it could be also necessary to locate the experimental setup in a room located in the ground floor of the building and acoustically insulated [4]. In the last years, several strategies have been implemented to overcome this last limitation, like feedback systems to achieve an active stabilization of the instrument or to control light intensity noise, damped suspending boxes and closed fluidic chambers filled with gas; with these shrewdnesses, a spatial resolution at the angstrom scale was achieved in single-molecule manipulation with optical tweezers [53]. Since the stiffness of the optical trap depends on the gradient of the light distribution, besides the quality of the shape of the laser, also the objectives used to focus the light beam are crucial elements. Indeed, objectives with a numerical aperture (NA) as higher as possible have to be used, in order to generate a tight focus and thus obtain a stiff and stable optical trap. This requires the use of oil or water immersion objectives, which present some limits. The firsts allow realizing stiffer optical traps (therefore higher force values), but they suffer for aberrations and the working distance is mainly limited to few microns below the coverslip of the fluidic chamber; on contrary, the water-immersion objectives permit to optically manipulate objects deeper in solution, but the trap strength is reduced, due to the lower NA of these objectives compared to those using oil as immersion medium. Furthermore, since optical forces scale down approximately with the particle volume, in conventional OT the size of the trapped objects is restricted by the diffraction-limit, which makes challenging the use of this technique to the nanoscopic world, preventing the direct manipulation of minuscule molecules as DNA/RNA or proteins. Several new technologies and approaches have been developed to manipulate nanoscopic objects, as metal or plasmonic nanoparticles and nanotubes [4, 54], and biomolecules [55], but further steps have still to be made in order to optimize the manipulation accuracy, enlarge the number of nano-objects that can be simultaneously controlled, and, more in general, make nano-tweezers as versatile as conventional OT have demonstrated to be in manipulating micro-sized objects.
3 Optical trapping: principles and methods
The ability of light to exert a force is known since long time. Already in the seventeenth century Kepler in his opera De Cometis ascribed to a mysterious force originated from solar light the experimental observation that comet tails always point away from the Sun. It was necessary to wait till the late nineteenth century to give a theoretical explanation to this phenomenon, thanks to the physicists James Clerk Maxwell and Adolfo Bartoli, who first introduced the concept of radiation pressure. Some years later, the first experimental demonstration of the existence of the radiation pressure was carried out independently in Russia and USA, by Lebedev and by Nichols and Hull, respectively, by using a torsion balance in vacuum. However, it was immediately clear that the effects induced by the radiation pressure were extremely small and negligible in the daily life. Till the advent of the technological progresses achieved in the 60s–70s, specifically the invention of the laser and the possibility to realise structures at the microscopic scale. From one side, the lasers allowed obtaining light sources significantly more intense than before, above all if the light intensity is concentrated in a tiny spot by focusing the beam; on the other side, the availability of microscopic objects enabled to scale down the optical manipulation experiments to sizes where optical forces become relevant. Arthur Ashkin had the great merit of having understood the potentialities offered by these technologies and taken advantage of them to create the first optical tweezers prototye.
3.1 Physical principle
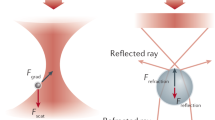
The radiation pressure is defined as the pression exerted by the electromagnetic radiation impinging on a surface and it derives from different phenomena, as absorption and scattering of light. In the case of an optically trapped spherical particle, this interaction can be described by the electromagnetic theory and the Maxwell stress tensor, but this approach can result in complex and computationally intensive calculations. Therefore, often suitable approximations are introduced to simplify the calculations and obtain a quantitative understanding of the phenomenon in the frame of different regimes. However, regardless of the simplifications applied, all these regimes rely on the same physical principle, that is the momentum conservation law. Indeed, also light carries momentum, which is altered when the trapping beam is scattered by the illuminated dielectric particle. As a consequence of the momentum conservation principle, an equal and opposite momentum change is imparted to the particle, which results in a net force acting on it. There are three main theoretical approaches to describe the physics of optical trapping, depending on the ratio between the wavelength \(\lambda \) of the light and the diameter d of the particle. In the ray optics regime, the particle, that in typical optical tweezing experiments is around one micrometre, is larger then the light wavelength (\(d>>\lambda \)), whereas in the Rayleigh regime the opposite situation is verified (\(d<<\lambda \)). Nevertheless, most of single-molecule experiments done with optical tweezers fall in an intermediate regime, where the light wavelength is comparable with the dimension of the trapped object, which could also be non-spherical and non-homogeneous. In this case a full wave-optical modelling of the particle-light interaction is required, making the calculations not trivial. However, the first two regimes represent often a valuable source of physical insights to describe optical trapping phenomena, therefore in the following the ray optics approximation in the case of a spherical particle will be presented to obtain a qualitative understanding of the phenomenon. For an exhaustive explanation of the other two regimes, the reader is referred to [4].

Qualitative pictures on the origin of the optical forces acting on a spherical particle illuminated by a laser beam. In the ray optics approximation, the beam is decomposed in rays, each of them carrying a linear momentum p which is modified after the interaction with the particle; for the momentum conservation law, this results in a force acting on the sphere. The momentum carried by the incoming light is labelled as \({\varvec{p}}_{i}\), whereas the ray reflected at the first surface and the one transmitted (after two refraction events) are indicated as \({\varvec{p}}_{r}\) and \({\varvec{p}}_{t}\), respectively. For sake of clarity, the reflected rays due to multiple scattering events are not represented. a Homogeneous beam. Considering a homogeneous light beam, the vertical components of the forces exerted by two opposite rays on either side of the sphere cancel out. Therefore, there is a net force which acts on the sphere and it is oriented along the beam propagation direction. This axial component is called scattering force. b Gaussian beam. For a laser beam, the light intensity has a Gaussian profile, which is more intense in correspondence of the optical axis. In this case, a full compensation of the vertical components of the forces occurs only if the particle lies at the centre of the beam. On the contrary, if the sphere slightly moves towards one of the beam borders, the momentum carried by ray 1, \({\varvec{p}}_{i,1}\), is higher than that of its symmetric ray 2, \({\varvec{p}}_{i,2}\), since this latter lies in the less intense part of the beam. Therefore, the vertical components of the forces do not cancel out, and the net force has a vertical contribution which pushes the particle back to the beam centre if the refractive index of the sphere is higher than that of the surrounding medium. The force component perpendicular to the beam propagation direction is defined as gradient force. c Focused Gaussian beam. If the laser beam is highly focused the gradient force significantly exceeds the scattering one and presents a further component along the beam propagation direction which allows trapping the particle in the beam focus
In the ray optics approximation, a monochromatic light beam is decomposed into individual rays that follow the laws of geometrical optics. Each ray of light has a momentum \({\varvec{p}} = \, n_\lambda \; (h/\lambda ) \; {\varvec{e}}_\lambda \) which depends on the intensity of the ray (i.e. the number of photons which compose the ray, \(n_\lambda \)), the light wavelength \(\lambda \), the Planck constant h and the propagation direction \({\varvec{e}}_\lambda \). Therefore, any variation in the intensity of the light or in its propagation direction results in a variation of the linear momentum of the ray. Giving one ray of light i, when it hits a micro-sphere a small fraction of it is reflected (r), whereas most of its intensity is refracted (t). Depending on the reflection and refraction coefficients, the linear momentum relative to the incident light (\({\varvec{p}}_i\)) is divided between the two outcoming rays, where \({\varvec{p}}_r\) and \({\varvec{p}}_t\) are the linear momentum carried by the reflected and the transmitted rays, respectively. The transmitted ray t propagates inside the sphere till reaching the opposite surface, where it undergoes multiple refraction and reflection events. Since the momentum of the system (bead and light) must be conserved, the change of momentum experienced by the light is transferred to the illuminated sphere. The balance between all these contributions is used to derive the momentum transferred to the bead \({\varvec{p}}_\mathrm{b}\), as well as the force exerted to the bead, obtained as \({\varvec{F}}_\mathrm{b} = d{\varvec{p}}_\mathrm{b}/\text {d}t\). The exact solutions for \({\varvec{F}}_\mathrm{b}\) were reported by Ashkin in [56] by considering all the scattered rays. However, usually for a transparent (no-absorbing) bead the reflected components are significantly weaker than the transmitted one, so the main contribution to the light momentum is carried by the ray which exits the bead (see Fig. 2a), thus the momentum transferred to the bead can be defined as the difference between the momenta of the incoming and transmitted rays \({\varvec{p}}_\mathrm{b} = {\varvec{p}}_i - {\varvec{p}}_t\) . When the micro-particle is illuminated with a wave plane, the sum of the forces exerted by all the rays leads to a net force oriented along the propagation direction of the light, since the orthogonal components cancel out each other. This net force is usually referred as scattering force. However, in trapping experiments laser beams are generally employed, which have a Gaussian intensity profile (Fig. 2b), meaning that light rays coming from the centre of the beam are more intense than those at the edges. Thus, the rays with higher intensity apply a greater force to the bead with respect to the light at the beam edges and so the resulting force has also a component perpendicular to the light propagation direction, which is known as gradient force. When a bead of refractive index \(n_\mathrm{b}\) immersed in a medium of refractive index \(n_m\) is exactly at the centre of a Gaussian beam, it experiences only the scattering force, as in the case of a wave plane; however, when the particle slightly moves from the beam optical axis a gradient force arises, which brings back the sphere if \(n_\mathrm{b} > n_m\) or pushes it away from the region of higher intensity if \(n_\mathrm{b} < n_m\). Clearly, the ray optics approach can be also exploited to study optical trapping in particles with shapes more complex than the spherical one, where other interesting effects can occur, as optical torque or lift effect [4].
As anticipated before, the Rayleigh regime represents the lower size limit, that is, when the particle is smaller than the light wavelength and the conditions for Rayleigh scattering are satisfied. Few optical trapping experiments are done in biophysics under this condition, anyway this approach is convenient to obtain simple expressions for the scattering force and the gradient one, as well as to clarify their physical origin. In the Rayleigh regime the particle is approximated as an induced dipole immersed in an electromagnetic field, which can be considered homogeneous inside the object. From the interaction between the particle and the inducing time-varying electromagnetic field an optical force arises, which acts on the trapped object and enables its trapping. Besides a spin-dependent force term due to non-homogeneous light polarization which is usually negligible in optical trapping experiments [4], two other components contribute to the light-induced force, which represent the gradient and scattering forces discussed before. The scattering force \({\varvec{F}}_\mathrm{scat}\) derives from the absorption and scattering of the incident light by the particle and it is expressed by the formula:
where c is the speed of light in vacuum, \(\left\langle {\varvec{S}} \right\rangle \) is the time-averaged Poynting vector and \(\sigma \) is the extinction cross section, which depends on both scattering and absorption. Since \(\left\langle {\varvec{S}} \right\rangle \) is oriented along the direction of light propagation and it is proportional to the light intensity \(I({\varvec{r}})\), it means that the scattering force is higher for more intense light beam, as it was easily guessed also in the ray optics approach. Concerning the gradient force \({\varvec{F}}_\mathrm{grad}\), it is given by [57]:
where \(\epsilon _m\) is the dielectric permittivity of the medium, \(\alpha \) is the complex polarisability of the particle with respect to the surrounding medium and I(r) is the light intensity profile of the beam. When the real part of the polarisability \(\textit{Re}(\alpha )\) is positive (i.e. \(n_\mathrm{b} > n_m\)), the particle is attracted towards the region of the beam where the intensity is higher, whereas if Re(\(\alpha \)) is negative (\(n_\mathrm{b} < n_m\)) then the particle is repelled by the high intensity areas. This component of the optical force is the most relevant and it is the responsible for the final trapping of the particle. Moreover, the Eq. 2 clearly shows that the gradient of the light intensity \(\nabla I({\varvec{r}})\) plays a key role in determining the relevance of the gradient force with respect to the scattering one. This effect was already noticed by Ashkin in his first experiments, when he used Gaussian laser beams to actuate micrometric spheres suspended in water and he observed that besides the scattering force, there was also a gradient force along the intensity gradient. Even if in that configuration, the particle can still move along the beam propagation direction, the discovery of the optical gradient force represented a turning point in the path towards the development of the first optical trap. Indeed, Ashkin understood that the possible presence of a gradient force component also along the optical axis of the beam could be exploited to control and compensate the scattering contribution. Thanks to his intuition, in 1986 Ashkin and his collaborators presented the first optical tweezers instrument [3], based on the use of tightly focused laser beams. As a matter of fact, if the light beam is sufficiently focused, the intensity gradient around the focus becomes steep, leading to the appearance of a strong gradient force also in the direction parallel to the scattering one. This gradient force points toward the beam focal point and significantly surpasses the scattering component, so that an efficient and stable particle trapping at the focus can be finally achieved (see Fig. 2c). Experimental results and theoretical analyses demonstrated that the optical trap behaves like an elastic spring for small displacements (\({\sim }\) 150 nm) of the bead centre with respect to the centre of the focal point (\(x_\mathrm{bead}\)). The force F acting on the particle can be expressed as \(F=-\,x_\mathrm{bead}\, \cdot \,k_\mathrm{trap}\), where the proportionality factor \(k_{trap}\) represents the stiffness of the optical trap and depends on both the light intensity and the focusing geometry employed. The minus sign indicates that the force is opposite to the particle displacement and brings it back to the beam focus. Usually, the bead displacement \(x_\mathrm{bead}\) is derived by monitoring the outcoming beam, which is projected to a position-sensitive detector. The light beam is deflected when the trapped particle moves away from its centre and a proper calibration of the detectors allows converting the voltage signal relative to the beam centroid position to the corresponding bead displacement and force values. In this way, an optical force-transduction device is obtained, which can be applied to a great variety of single-molecule biophysical investigations.
3.2 Experimental methods
In most of single-molecule experiments, the biological system is not directly manipulated by the focused laser beam, both because the optical forces exerted on it are too small and to avoid possible light-induced damage effects. On the contrary, the molecules are attached to a surface, usually a polystyrene or silica micro-sized bead, which is optically trapped and used as a handle to manipulate single molecules bound to it. In this way, the particle works as force-transducer which simultaneously applies a mechanical stimulus to the molecule and monitor its response in real time.
The core elements of optical tweezers are the laser source and the objective lens used to focus it. Thus, optical traps are usually realized by modifying an inverting microscope, where the laser beam is introduced in the optical path before the objective. In this way, the microscope can be exploited to provide all the functions relative to the light beam focusing, the movement of the microfluidic chamber where the experiments are carried out and the imaging of the sample. Moreover, accurate and precise measurements with optical tweezers can be achieved only if careful attention is paid to the whole opto-mechanical design, as well as to the calibration protocols. As a matter of fact, the determination of bead displacements and applied forces requires the previous calibration of the position detectors and the measurement of the optical trap stiffness \(k_\mathrm{trap}\) (a detailed description of this aspect can be found in [4, 41, 58]). Finally, the potentialities offered by the optical tweezers instruments can be significantly enhanced by implementing dynamic systems for controlling the position of the optical trap in the specimen plane and that of the trapping chamber: piezoelectric or acousto-optic actuators are usually employed to this aim. The integration of such systems in a feedback loop allows realizing position-clamp measurements, in which the position of the trapped bead is kept fixed by varying the force, and force-clamp measurements, where the force applied to the trapped object is maintained constant by controlling the trap position [53]: these experimental methods have been successfully exploited for investigating molecular motors and biopolymers unfolding mechanisms, as it will be presented in the next sections.
The proper match of the OT instrument with the biological system to be studied is another key parameter to consider when starting a single-molecule experiment. The remarkable versatility of optical tweezers in this field is doubtless evidenced by the wide variety of approaches and experimental geometries that have been developed in the last years to perform force spectroscopy measurements on single molecules. In the following, we will briefly focus on the configurations most employed to manipulate biological specimens at the single-molecule level, whose application to bio-polymers and molecular motors have been growing rapidly in the new century.

Simplified pictures of optical trapping geometries for DNA single-molecule experiments. a Single-trap configuration. A single optical trap is created by focusing a laser beam; in the focus a microscopic bead is trapped, which works like force-tranducer. The investigated single molecule is tethered between the bead in the optical trap and a glass coverslip, via biochemical bonds. b Dual-trap configuration. Two focused laser beams are employed to create two independent optical traps, which actuate two beads trapped at the focal points. The distance between the beads can be varied at will, thus exerting a mechanical force to the molecule tethered between them. c Single-trap/micropipette configuration. The molecule is attached between two beads, one of which is kept fixed at the tip of a glass micropipette. A focused laser beam is used to trap the second beam, which can be used both as handle to actuate the molecule and as force-tranducer
Typically, in single-molecule experiments with optical tweezers, one end of the molecule is attached to a surface, while the other end is linked to the optically trapped bead, which works as force-transduction element and is used to apply a mechanical stimulus to the molecule. The usual attachment scheme for DNA/RNA molecules involves ligand–receptor pairs as binding factors, thus realizing efficient and specific attachments between the molecule and the surfaces. Often, one end of the biological system is labelled with biotin, while the surface is covered with the complementary molecules, avidin or streptavidin; in this way, the DNA under investigation can be linked to the surface through the specific binding biotin-streptavidin. On the contrary, the second end of the single molecule is labelled with a different substance, to avoid that the two ends attach to the same surface. For instance, a digoxigenin-labelling of the molecule can be used, while the bead is coated with the corresponding antibody, the anti-digoxigenin, so that a strong antibody-antigene bound is created between the molecule and the optically trapped particle. In the case of proteins, often reactive cystein residues are exploited, which can be naturally present or specifically introduced in the amino acid sequence; double-stranded DNA sequences (dsDNA) are attached to the target protein, via disulfide bond linkage with the cystein residues, and they are used as molecular-handles, which work as spacers to avoid undesired bead–bead or bead–protein interactions.
The most widely used optical tweezers geometry is that depicted in Fig. 3a, which is based on a single optical trap. In this configuration, the molecule is tethered between a fixed surface (chamber coverslip) and a bead trapped by the laser beam. In static measurements, the optical trap is kept fixed and the bead displacement with respect to the beam centre is monitored, so that the time evolution of the force applied to the biological system is derived. However, this setup has been also extensively used to perform active force-clamp measurements, where a feedback system controls the trap movement in order to apply a constant force to the molecule. This single-beam configuration is particularly suitable to investigate processive molecular motors, as for instance kinesin proteins walking on fixed microtubules [59] or polimerase enzymes [60], providing information on their conformational transitions and energy landscapes. The second geometry illustrated in Fig. 3b is the so-called dual-trap optical tweezers, or two-bead assays, where two beads are held in separate optical traps and the molecule under investigation is bound between them. In this configuration, a higher spatial resolution is achieved, since the noise associated with surface-coupled assays is removed; moreover, the direction and amplitude of the applied force is better defined. Thanks to this, the dual-trap geometry has been successfully employed to measure the step size of RNA polymerase down to one base-pair (0.34 nm) resolution [61, 62], as well as to characterize the mechanical properties of DNA molecules and proteins. The two-bead setup is also exploited to perform constant-velocity measurements, where the molecule is pulled at constant rate and its kinetic and thermodynamic parameters are determined [63].

Optical mini-tweezers instrument described by Smith et al. [64]. This setup employs two counter-propagating beams, which are focused into the same spatial position: in this way the scattering forces of the two beams compensate each other, thus leading to a more stable trap as compared with other single-trap instruments. a Schematic representation of the setup, where the two beams follow an identical optical path. Each laser is coupled with an optical fibre, whose free end is anchored into a wiggler system. The wiggler gently moves the head of the fibre by means of piezoelectric actuators, thus varying the final position of the optical trap. Almost 4% of the light is directed by a pellicle beam splitter (BS) to a position-sensitive detector (PSD), which monitors the trap displacement. The remaining light passes through a collimating lens, a polarizing beam-splitter (PBS) and a quarter-wave plate (\(\lambda \)/4) before entering into a high numerical aperture objective, which focuses the beam inside a microfluidic chamber. The scattered light is collected by a second identical objective and directed to a PSD and a photodiode, which converts any beam displacement into a force signal. A motorised stage supports the fluidic chamber where the experiments are carried out, which is held between the two objectives. Finally, a LED light source and a CCD camera are exploited to image the experimental area. b Detail of the two focusing objectives and the fluidic chamber mounted between them. c Image of the area inside the fluidic chamber where the experiments are carried out. An anti-digoxigenin coated bead (AD) is trapped by the laser beams, whereas the streptavidin coated bead (SA) is kept fixed at the tip of the glass micropipette. A single-molecule (illustrated as red line) is tethered between the two beads, and by varying the position of the AD bead with respect to the SA, it is possible to apply a force to the molecule. The molecule is invisible due to its nanometric size. The laser beams are not visible too, since a filter is used to avoid CCD saturation. The two photos have been taken at the Physics and Astronomy department of the University of Padua, where a setup similar to that of [64] is present
This kind of experiments has been successfully carried out also with the configuration shown in Fig. 3c, where the molecule is tethered between two beads: one is optically trapped, whereas the other is kept fixed at the tip of a glass micropipette by air suction. This geometry recalls the one shown in Fig. 3a, but the use of the micropipette presents some advantages. First, the use of the micropipette in place of the glass coverslip as linking surface allows minimizing possible unspecific interactions between the coverslips and the studied molecule or the optically trapped particle, since the micropipette can be easily located at the centre of the main fluidic channel, far from the chamber walls. Moreover, torsional stress can be applied to the molecule through a controlled-rotation of the micropipette [65]. Finally, in the micropipette configuration, the two beads can be held quite close together without both of them jump into the same optical trap, and this allows investigating also short DNA/RNA molecules.
The replacement of the coverslip with the micropipette was pioneered by the group led by C. Bustamante, who also introduced major improvements to the basic setup by using two counter-propagating beams to create a single optical trap [64]. In Fig. 4, a simplified scheme of the instrument is reported, which presents symmetric focusing and recollecting paths for the two beams. Each of the two near-infrared diode lasers is connected to an optical fibre, whose end is supported by a wiggler system. The wiggler is a mechanical device that exploits piezoelectric actuators to gently move the optical fibre tip and direct the light beam at will. Then, a pellicle beam-splitter (BS) is used to split the beam into two parts and a small amount of light (\(\sim \) 4%) is sent to a Position Sensitive Detector (PSD), which allows monitoring the movements of the optical trap in the plane perpendicular to the beam propagation direction. The remaining light is collimated by a lens and directed to a Polarizing Beam-Splitter (PBS) and a quarter-wave plate; then the circular-polarized beam is focused by a high numerical aperture objective, thus creating the optical trap inside the microfluidic chamber used for the experiments. The light exiting the trap is collected by a second identical objective, that redirects the light through two polarizing beam splitters to a photodiode, equipped with a bulleyes filter, and a PSD: these detectors are used to monitor the forces applied to the trapped bead in the z and x-y directions, respectively. Moreover, the micro-fluidic chamber is mounted on a three-axis motorised stage equipped with piezoelectric actuators, which allow translating the whole experimental area with respect to the two fixed objectives (see Fig. 4). Finally, a CCD camera is usually employed, but only to display the experimental area during the experiment. The two beams follow an identical optical path and the two objective lens are exploited to both focus one beam and to collect the light coming from the other beam behind the optical trap. The main advantage of such complex setup relies in the reduced axial scattering force that is achieved, so that a wider force range (up to 200 pN) and a more stable optical trap can be obtained.
4 Single-molecule experiments with Optical Tweezers
In this section, some single-molecule experiments performed with the Optical Tweezers geometries discussed above will be presented. As it is known, the existing literature on this topic is enormously wide and we have not the ambition to provide a complete and exhaustive discussion on all the possible biological systems which have been investigated through single-molecule optical tweezers methods. Our intent is rather that of presenting a few experimental results to highlight the potentialities offered by this technique in the field of manipulations at the single-molecule level and, at the same time, intrigue and give inspiration for new researches.
4.1 DNA pulling experiments
Since the creation of the first optical tweezers instrument at the end of the 1980s, DNA molecules have been extensively studied with the aim of determining their elastic properties and better understanding their biological functions. As well known since the extraordinary discover of DNA structure [66,67,68], this molecule consists of two polynucleotide chains that coil around each other forming a double helix (double-stranded DNA). The subsequent nucleotides of one chain are joined together by covalent bounds, whereas they form hydrogen bonds with the nucleotides lying in the opposite chain, creating the so-called base-pairs. DNA is a key player in molecular biology. Inside cells it constantly undergoes bending, twisting and stretching in several processes, like transcription performed by molecular motors or DNA-packaging inside the cell nucleus. It is therefore easy to understand why so many efforts have been devoted to understand how DNA complies the mentioned mechanical stress and in which way the presence of an external force could affect its elastic parameters.
In this scenario, pioneering works with optical tweezers were performed by Smith et al. [69] and Wang et al. [70] at the end of ’90s, by exploiting the single-beam geometries discussed in Sect. 3.2 and depicted in Figs. 3a and 4, respectively. By varying the distance between the bead in the optical trap and that on the micropipette tip (or the coverglass), it was possible to apply a mechanical stimulus to the DNA molecule under investigation and to record the corresponding force to extension (FE) curve. This kind of measurements are also known as pulling experiments and in Fig. 5 an experimental FE curve obtained for a 9000 base-pair(bp) long dsDNA is reported. Pulling experiments show that DNA in its B-form (i.e. DNA in its typical right-handed double helix conformation) has an elastic behaviour dominated by entropic effects at forces below 5 pN. At these low forces, DNA can be treated as a semi-flexible rod of length \(L_0\) (also called contour length), that bends smoothly due to thermal fluctuations. In particular, the response of DNA molecules to an external mechanical stress is well described by the Worm-Like-Chain(WLC) model [69, 71], where the correlation between the rod local direction is considered to decrease exponentially with a decay length P, defined as the persistence length of the chain. The stiffer the chain, the longer the persistence length. However, above 10 pN, enthalpic contributions cannot longer be neglected and the experimental data can be satisfactorily fitted only if a further elastic parameter is taken into account, that is the stretch modulus S. In this case, the molecule is considered as a stretchable rod, whose contour length \(L_0\) increases linearly with the applied force. Usually, up to 50 pN the FE curves are well fitted by the modified Marko-Siggia model, also referred as extensible Worm-Like-Chain model [70, 72, 73], which includes both entropic and enthalpic contributions to the DNA elasticity. In this model the relation between the force F applied to the molecule and its extension x is given by
where \(k_\mathrm{B}\) and T are the Boltzmann constant and the absolute temperature, respectively. Wang and co-workers fitted their experimental data with Eq. 3, deriving a stretch modulus of S \(\sim \) 1100 pN and a persistence length of P \(\sim \) 48 nm for a lambda-phage molecule stretched in low-sodium buffer (10mM \(Na^+\)); these values perfectly agreed with those reported in Smith et al. [69].

The overstretching (OS) transition of DNA under tension. (left) Experimental force-extension (FE) curve (black line) of a 9kbp-long DNA molecule, where it is possible to observe the OS transition occurring at about 65 pN. The measurement was performed at the Physics and Astronomy dept. of the University of Padua with the setup described in Fig. 4; the molecules were prepared by prof. F. Ritort’s group (Small Biosystems Lab, University of Barcelona, Dept. of Condensed Matter Physics). The FE curves predicted by the inexensible (blu line) and the extensible (red line) Worm-Like-Chain (WLC) models are reported as comparison. The values of persistence lengths used for the simulation are those reported in [69] and [70]; the contour length \(L_0\) was calculated as \(L_0\,=\,n_\mathrm{b}\,\cdot \,d_{nn}\), where \(n_\mathrm{b}\) is the number of base pairs of the poliptide chain and \(d_{nn}\) is the rise per base-pair, assumed equal to 0.34nm [74]; for the stretch modulus S the value of 1100 pN [52] was used. (right) Schematic representation of the three models used to describe the DNA overstretching. In its B-form a DNA molecule has a base-pair (bp) rise of 0.34nm and a number of base-pairs per turn (helical pith) equal to 10.5. When the DNA is overstretched three different scenarios can occur: (1) the transformation of B-DNA into S-DNA, which has a bp-rise of 0.58nm and an helical pitch of 35 [74]; (2) the formation of melting bubbles (i.e. underwound melted segments where the base pairing is broken); (3) the peeling off of one DNA strand
A striking feature of the force-extension curve of a DNA molecule is the overstretching (OS) transition, that represents a highly cooperative conformational change where DNA dramatically increases its extension with apparently no force resistance. This regime is identified in the FE curve by a plateau at about 65 pN, where the DNA molecule elongates of \(\sim \) 70% beyond its contour length, under an increase in the pulling force of only 2 pN. The presence of the overstretching regime can be observed for both torsionally unconstrained (relaxed) molecules and torsionally constrained ones, even if it occurs at different force values. In torsionally relaxed DNA, the molecule is free to swivel about its anchored ends so that no torsional constraints can be applied to it; this was also the case of the dsDNA molecules investigated in Smith’s experiments [69] discussed before. For such molecules the overstretching occurs at 65pN, as shown in Fig. 5(left). Usually, these molecules have both strands of one end attached to one bead, whereas on the opposite end one of the two strands (3’ or 5’) is left free and the other is immobilized to the second bead (or surface); alternatively, only one of the strands on either side of the DNA is linked to the beads. On the contrary, in torsionally constrained DNA both strands are typically attached at either end of the molecule and no nicks are present along the peptidic chain; in this case the molecule is prevented from releasing energy by unwinding during the pulling experiment and its overstretching force increases from 65 pN to about 110 pN. The structural changes occurring at the overstretching transition has been debated for several years and the large variety of experiments performed to investigate this phenomenon had sometimes caused controversy [75]. For both torsionally unconstrained and constrained DNA molecules, experimental data revealed the presence of single-stranded DNA (ssDNA) in the OS transition, confirming that DNA gradually melts at those forces [76]. However, some other evidences were put against this interpretation, as the observation that the two DNA strands do not immediately separate when pulled beyond the OS transition [69] or the demonstration that at this regime the DNA base pairing can be preserved with an helicity of about 33 base pairs per turn [65]. A detailed description of all these phenomena have been achieved only in recent times and nowadays three mechanisms are considered to be involved in the DNA over-stretching process: the strand peeling, the localized breaking of molecule base pairs and the formation of the S-form DNA, as depicted in Fig. 5(right). In the peeling process, one strand of the dsDNA breaks the base-pair bounds with its complementary strand and retracts; in this way the molecule progressively converts into two single-stranded DNAs. In the second model, localized sections of the dsDNA molecule denatures (melts) by breaking the base pairing and these finite ssDNA domains are referred as “melting bubbles”. Finally, the third model considers the transition of DNA from its B-form to a new structure, where the base-pairing is preserved and a cooperative strand unwinding occurs: this conformation is called S-form DNA.
Recently, a clear answer to the long-running debate over the structure of overstretched DNA was provided by King [77] and van Mameren [78], whose results were later supported by molecular dynamics simulations [79] and exploited for studying stretching investigations also on RNA and hybrid RNA/DNA systems [80]. In both works, the authors combined a dual-optical tweezers setup, as that shown in Fig. 3b, with fluorescence microscopy, in order to directly visualize the structural changes of a single DNA molecule undergoing the overstretching transition. In particular, they used in the buffer an intercalating dsDNA-specific dye and ssDNA-binding proteins as fluorescent markers, then recorded fluorescence images during pulling measurements with optical tweezers. In this way, they were able to distinguish in real-time between portions of the DNA molecule that presented a single-stranded or a double-stranded conformation, as well as discriminate between DNA in the B- or S-form. They investigated torsionally constrained molecules and unconstrained ones, confirming that in both cases a force-induced melting of the dsDNA into ssDNA unambiguously occurs in the overstretching transition. Moreover, they clearly demonstrated the presence of residual dsDNA segments in the molecule even after the whole OS process has been completed, thus explaining how DNA can transiently remain intact and be pulled also at force above 65 pN. They also noticed that the presence of free ends or nicks in the polynucleotide chain act as nucleation points for DNA melting, which starts at those positions. These results demonstrated that the molecule topology (i.e. the presence of free ends or single-stranded nicks in the inalteraded molecule) strongly affects the mechanisms through which dsDNA regions melt into ssDNA, preventing or promoting a peeling transition with respect to melting bubble formation. Nevertheless, the topology is not the only parameter to take into account, and also salt concentration in the buffer (often referred as ionic strength), as well as nucleotides composition of the chains play a key role in determining the balance between the three mechanisms involved in the OS transition. These dependences were carefully investigated in [77], where Kings and co-workers investigated torsionally unconstrained molecules presenting all the four strand ends linked to the two optically trapped beads (see Fig. 6a) or one free end (Fig 6b). As expected, for both attachment geometries an overstretching transition at 65 pN was observed, but at low ionic strength (50 mM NaCl) the topologically open molecules exhibit a wide force hysteresis during DNA retraction of the end-to-end distance. This phenomenon confirms the presence of peeling mechanisms with stick-slip dynamics during the stretching of topologically open molecules, whereas in topologically closed systems a localized breaking of base pairs occurs (formation of melting bubbles), which gradually recovers when the molecules are relaxed. It was also demonstrated that base-pair breaking mechanisms (i.e. the formation of melting bubble or the peeling) preferentially take place in the less stable regions of the polynucleotide chain, that is in correspondence of AT-base pairs. As a matter of fact, AT-base pairs have a lower binding energy with respect to CG-ones, so the former bonds can be more easily broken during the overstretching. Besides topology and sequence, overstretching is also very sensitive to ionic strength. As a matter of fact, at high salt concentrations (above 250 mM NaCl) both peeling and melting bubbles are progressively less favoured, till being completely prevented in 1M NaCl buffer solution. This is due to the fact that dissolved ions shield the negative charges of DNA, thus an higher salt concentration decreases the electrostatic repulsion between the two DNA strands and makes the double helix more stable. At high ionic strengths the conversion of canonical B-DNA into the extended S-DNA conformation is dominant both in open and closed topology, so the overstretched molecule exhibits base-pairing interactions arranged in an unwound double-helix structure and stick-slip features are not observed in pulling investigation. It is worth noting that the same results have been confirmed almost at the same time by Zhang et al. [81], by means of pulling experiments performed on single molecules with a calorimetric magnetic tweezers setup.

a–d Stretching experiments performed at low ionic strength (50 mM NaCl) on torsionally unconstrained DNA molecules by combining dual-trap OT geometry and fluorescence microscopy. a Schematic representation of a topologically closed molecule tethered between two optically trapped beads. The pulling and relaxing force-extension curves do not present relevant hysteresis, so that peeling effects are disregard. b A topologically open molecule presenting some nicks is pulled and relaxed and the corresponding force-extension curves exhibit a pronounced hysteresis, demonstrating the presence of stick-slip dynamics typical of peeling mechanisms. c, d Corresponding fluorescence images of the ssDNA-binding protein RPA and the dsDNA binding agent Sytox. In c the coexistence of double-stranded and single-stranded areas are highlighted by the uniform fluorescence of both RPA and Sytox, which is consistent with the formation of melting bubbles along the DNA chain; on the contrary in d the two fluorescence markers are localized in separated segments and the peeled domains are highlighted by orange arrows. e Pictorial representation (adapted from [82]) of the different mechanisms involved in the stretching process of torsionally constrained DNA molecules, where the base-pairing and its tilt are not represented. At low forces, the DNA is in its B-form, whereas at 50–100 pN the mechanical force applied to the molecule induces the appearance of unwounded segments, which could correspond to S-DNA regions or melting bubbles, at higher or lower salt concentration in the buffer, respectively. Starting from about 110 pN, underwound (S-DNA/melted-bubbles) and overwound domains coexist: the overwound segments correspond to the P-DNA, which presents a bp rise of 0.54nm and an helical pitch of 2.6 [74].The images are taken from Graeme A. King et al. PNAS 2013;110:10:3859-3864 [77]
Although torsionally relaxed molecules have allowed obtaining valuable insights into the DNA elastic response, also the study of torsionally constrained systems is fundamental to understand the role played by DNA elasticity in cellular processes. As a matter of fact, often in genomic processes (like transcription, translation and compaction) DNA is subjected to torsional stress, so also the investigation of torsionally constrained molecules is relevant and could help to better understand the mechanics of DNA-protein binding. In [82] the authors exploit once more a standard dual-trap optical tweezers setup combined with fluorescence spectroscopy to quantify and explain the influence of torsional constraints on the structural stability of DNA under applied tension. Their results show that torsionally constrained molecules present a heterogeneous plasticity as the pulling force is increased, as depicted in Fig. 6b. In particular, in the range \(\sim \) 50–110 pN the molecules exhibit locally underwound domains, whose structure strongly depends on buffer ionic strength: the base-pairing is disrupted at low salt concentration leading to the formation of underwound bubble-melted, whereas at high salt contents these underwound regions exist as S-DNA. However, it was also demonstrated that at these forces the linking number of the molecules, that is this the sum of the twist number (number of turns of the double helix) and the writhe one (number of times the double helix crosses over on itself), is maintained constant. This implies that such local unwinding in the double-helix must be accompanied by small overwounded segments which are still in the B-form. Finally, at forces above 110 pN, they provided evidence of the co-existence of underwound bubble-melted/S-form DNA domains with overwound sections in a \(\sim \) 20:80 ratio. In particular, these overwound DNA regions present a conformation different from the typical B-form, which is called P-DNA and consists in backbones so much tightly wrapped around one another to cause the base-pairs to flip out (for more details about P-DNA structure, the linking number and the supercoiling DNA the reader is referred to [83] and [84]). The observation of P-structure confirms previous theoretical works predicting both its presence in the overstretching regime and the coexistence of unwinding and overwinding mechanisms at high forces. Moreover, this work clearly highlights as, despite its good agreement with experimental data, the extensible WLC model overlooks some important aspects of the DNA elasticity, as the enthalpic twist rigidity and the twist-stretch coupling. To take into account these contributions, a twistable worm-like-chain model has been proposed by Gross [85] and co-workers to fit experimental data, showing as the introduction of the force-dependent twist-stretch coupling correctly quantifies how DNA complies to external applied forces. In particular, the authors demonstrated as the DNA overwinds under tension at forces below 30 pN, leading to a negative twist-stretch coupling coefficient, which become positive at about 35 pN, when the DNA has been proved to start to unwind. These results confirm those obtained by Gore in [52], but they also put in evidence the need for more theoretical and experimental efforts to provide an exhaustive explanation to the non-trivial behaviour of DNA force-dependent elasticity.
4.2 Protein folding
Together with nucleic acids, proteins are fundamental life’s building blocks, whose functions are involved in a great variety of biological processes. Proteins are linear polymers made of monomer units called amino acids, which arrange into two-dimensional (\(\beta \)-sheets and \(\alpha \)-helices) and three-dimensional (protein folding) structures, to perform a designed function. The protein structure can be thought like a puzzle, whose unique solution is represented by the well-defined 3D structure to which the polypeptide chain spontaneously folds. What is surprising is the time required by the unfolded polypeptide chain to achieve this final stable state (known as native state), which is usually on the order of seconds, milliseconds or even faster. As a matter of fact, if we consider the unfolded aminoacid chain, it has an enormous number of possible conformational configurations and a random search of the native state among all them is unfeasible in the biological timescales mentioned before (Levinthal’s paradox). This observation introduces the so-called protein folding problem, which has represented a grand challenge for scientists since the last century: it aims to understand which mechanisms guide proteins organization into their 3D structure helping them to avoid misfolded states and possible aggregation, leading to pathologies as in the case of Alzheimer’s and Parkinson’s diseases [86]. For a long time the folding and unfolding mechanisms of protein have been investigated by means of conventional bulk methods, like differential scanning microscopy or spectroscopic techniques, which greatly contributed to our understanding on these complex phenomena. However, these ensemble approaches provide information averaged over a large number of molecules, so they are often inadequate to characterize heterogeneous process as that represented by protein folding. Indeed, although proteins may present average features, it is nowadays clear that any single protein can take a different path towards the native state, along which intermediate configurations are often formed. These intermediates can be on-pathway, that is productive to protein folding, or off-pathway, which means that they do not lead directly to the native state, but their usual short lifetimes (down to ms or even less) make them hardly detectable with standard bulk techniques (as an example see Fig. 7).
Almost 20 years ago, single molecule experiments together with computational techniques started to revolutionise the research field related to protein folding, by offering the possibility to observe rare events and identify sub-populations within protein ensembles. In particular, single-molecule force spectroscopy offers a unique tool to investigate the available folding and unfolding pathways of a protein and to determine its free energy landscape. This latter represents a complete picture of the free energy and kinetic rates of each possible conformational state between the native and denatured (unfolded) state of the polypeptide chain, so it is extremely useful to study proteins structure and stability. In force spectroscopy measurements on single proteins, the molecule is attached between two surfaces, whose relative distance is varied, leading to the application of a force on the polypeptide chain. By increasing the tensile force, the unfolding of the protein is promoted, whereas the force release allows the protein to refold. The determination of the energy landscape under mechanical stress is undoubtedly biologically relevant, considering that also for protein mechanical forces are strictly involved while performing their molecular functions. At the beginning, the major role was played by Atomic Force Spectroscopy, which is nowadays widely used to study polyprotein complexes and phenomena related to protein aggregation. However, as explained in Sect. 2, this technique is characterised by high stiffness values, so it is mainly limited to investigate unfolding/refolding behaviours at high forces; for the same reason, often with this method it is difficult to observe folding events at low forces or to monitor the equilibrium between the folded and unfolded states. On the contrary, Optical Tweezers allow overcoming these limits, thanks to their lower stiffness and loading rates, which make them more sensitive to folding events and intermediate states.

Single-molecule unfolding experiment performed on 169-aminoacids apo-Flavodoxin protein. The protein molecules have been prepared in collaboration with the groups led by prof F. Ritort (University of Barcelona), prof. G. Zanotti (University of Padua) and prof. J. Sancho (University of Zaragoza) [87]. By moving the bead in the optical trap with respect to the fixed micropipette it was possible to stretch the protein (at 100 nm/s) and record the corresponding force values as a function of the trap position(main graph). By subtracting the bead contribution, the force-molecular extension curve is derived (inset below), where the force jump corresponds to the complete unfolding of the protein and consequently to the release of all the aminoacids. At the standard sample rate (1kHz) of the instrument (main graph), no intermediate states are observed during unfolding measurements; on the contrary, by implementing a data acquisition board with high temporal resolution (up to 100 kHz) intermediate conformations with ms-lifetimes (or even shorter) are easily detectable (upper inset)

Misfolding pathways observed in neuronal calcium sensor-1 (NCS-1). a Structure of NCS-1, with the N-domain (gray) and C-domain (blue). b Schematic representation of the OT setup employed for the measurements. The protein is tethered between the two polystyrene beads via DNA handles; one bead is kept fixed by air suction at the tip of the micropipette, the other is optically trapped. c Force-extension curves relative to the stretch (red) and release (black) of NCS-1. The dashed grey lines are fit of the experimental data with the Worm-Like-Chain model. Besides the native (N) and unfolded (U) states, also the two intermediates I1 and I2 are present. d Force-release curves that highlight the presence of the misfolded states M1 (blue) and M2 (green) during protein folding process. e Example of a rescue transition: once the protein has folded into a misfolded state (black line), it may happen (1%) that during the subsequent stretching event the nonnative contacts of the misfolded configuration are broken and the molecule first snaps to the I2 intermediate state and then goes to the native one before completely unfolds. f Fraction of the observed misfolded proteins as a function of both the speed used during the force release and the calcium concentration in the buffer. The images are taken from Pétur O. Heidarsson et al., PNAS, 111, (2014) 13069-74 [88]
In protein folding investigations with OT, the biological system is typically tethered between the two surfaces employing two long dsDNA handles [89], which are attached to the protein via cysteine residues. These long handles (usually 500 base-pairs or more) allow also the manipulation of small proteins, avoiding non specific interactions between them and the linking surfaces. This protocol was implemented for the first time by Cecconi et al. [90] and it allowed extending the use of OT also to proteins with nanometric diameter. Indeed, till then OT were mainly used to stretch micrometric-long proteins, as the first works on the muscle protein titin [91, 92] at the end of ’90s. Even if the use of such long handles is quite expensive and limited to forces below the DNA overstretching regime [89], it is also true that in this force range it is possible to observe the unfolding/folding events of a large number of proteins, above all if low loading rates are used during pulling experiments. As a way of example, in Fig. 7 the unfolding force to distance curve of an apo-Flavodoxin protein is reported. The individual molecules were tethered between the two beads by means of 500bp-long DNA handles and they were manipulated by using the OT setup available at the Physics and Astronomy dept. of the University of Padua, which is the same as that described in Fig. 4. The force jump relative to the unfolding of the 169 aminoacids is evident, and at the standard sample rate of 1kHz the presence of intermediates states are not observable; on the contrary, by using a higher temporal sampling (up to 100kHz) it was possible to highlight the occurrence of an intermediate state in the unfolding pathway of the protein. Very recently new biological methods have been presented [93] to create DNA-protein bounds without the need for cystein residues in the polypeptide chain, thus broadening the range of proteins which can be investigated by OT.
From force spectroscopy measurements on a single protein, it is possible to derive valuable information on both the kinetics and the thermodynamics of the aminoacids chain, which are used to reconstruct the main features of its free energy landscape (FEL). In their pioneering work at the beginning of the new century, Cecconi et al. [89] exploited the optical tweezers geometry depicted in Fig. 3c to study the protein RNase H (E. coli protein ribonuclease H Q4C/V155C), which has a single-domain structure. By moving the micropipette with respect to the optical trap it was possible to stretch and release the protein several times and derive the force-extension curves, where the force jumps correspond to the protein transitions from the folded to the unfolded state or vice-versa (as the case of Fig. 8). The elastic rigidity of the polypeptide chain is well described by the Worm-Like-Chain model discussed in the previous section, which allows calculating the variation in contour length relative to each conformational transition and the corresponding number of aminoacids which are released (or refolded). Their results indicated the presence of an intermediate state along the folding pathway, which involves the refolding of only a part of the peptides sequence. Moreover, by keeping the protein at constant force (force-clamp experiments), it was possible to observe hopping phenomena between the intermediate and denatured (or native) states, depending on the value of the applied force. From the observed lifetimes, the kinetic rates of each state were derived, which helped to define the FEL of the protein. The authors clearly identified that the observed intermediate state was an on-path conformation, an obligatory step in the trajectory of the molecule from its denatured state to the native one. This work definitely paved the way for the use of OT in studying protein folding, but it also showed the extraordinary relevance of mechanical statistics in interpreting the results achieved by single-molecule experiments. A similar approach was used also to study the folding and unfolding mechanisms of the protein Barnase [94]. In this work, the authors used the Bell-Evans and Dudko-Hummer-Szabo models to analyse the force-induced protein unfolding, assuming the existence of a single energetic barrier between the native and the denatured states, that the protein has to overcome during its conformational transitions. The distributions of unfolded forces allowed deriving the force-dependent kinetic rates of the protein and, consequently, the height of the energetic barrier relative to the transitional state. An advantage of force spectroscopy in studying the unfolding/folding of single molecules with respect to other bulk methods relies on the fact that it provides more intuitive reaction coordinates to describe the free energy landscape, as for instance the number of released aminoacids along the polypeptide chain. This reaction coordinate was also used in [94] to study the movement of the energetic barrier between the native and the unfolded states: it was demonstrated that the barrier approaches the native state by increasing the applied forces, satisfying the Leffler-Hammond postulate. The high sensitivity of the instrument also permitted to investigate the folding events, occurring at low forces (< 4 pN). The pulling experiments relative to both unfolding and folding processes were used to calculate the corresponding irreversible work distributions, which in turns allowed deriving the protein free energy at zero force by exploiting the Jarzynski equality (more details on nonequilibrium thermodynamics and fluctuation theorems applied to nucleic acids and protein systems can be found in [46, 95]).
Optical Tweezers can also be useful to investigate the folding cooperativity in proteins with multiple domains. This feature is crucial in molecular biology, since a lower cooperative communication between the different domains of a protein could increase the population of partially folded proteins and consequently promote misfolding diseases. In [90], the authors studied the cooperative behaviour between the two domains of the protein T4 lysozyme, which are defined as C-domain and N-domain. First, the protein was stretched from the ends of the whole polypeptide chain and the pulling curves clearly showed a complete unfolding of the protein. As second step, the attachment points of the DNA handles were changed and located in correspondence of the ends of the only N-domain. Also in this case the unfolding of the entire polypeptides sequence was observed, although the mechanical force was applied only to the N-domain. This is a clear indication of the existence of a high degree of coupling between the two parts of the protein. In order to identify the key element which acts as a bridge between the two domains and promotes their cooperation, the authors used a circulant permutant T4 lysozyme, where the 12-residues helix initially located at the N-domain was attached to the C-one. A lack of cooperativity was observed in this second protein architecture, thus confirming the crucial role played by the protein topology (i.e. the order of structural elements along the sequence) in preserving cooperativity among different domains.
Recently, the interest in protein misfolding processes at the single molecule level has significantly increased. The renewed attention to this biological topic has been fuelled also by the technological developments achieved in the last years, which allowed single-molecule techniques to achieve the spatial and temporal resolution necessary to investigate misfolded states that can nucleate aggregation. Heidarsson et al. [88] have recently investigated the neuronal calcium sensor-1 (NCS-1), a protein that works as calcium sensors and whose biological functions are associated with cognitive processes, such as learning and memory, and also linked to severe neuro-degenerative disorders. By means of pulling and releasing experiments, the authors were able to identify the presence of two misfolded states besides the two intermediate configurations which were already known, as shown in Fig. 8c, d. While the protein unfolding occurs in a three-state mechanism, during the folding process two possible misfolding pathways can originate from one of the two on-pathway intermediate states. The kinetics of the various transitions as well as the free energy landscape of the molecule were determined by using a Hidden Markov Model algorithm [96] and the Bell model. However, one of the most worthwhile result of this work has been the discovery that the probability to populate such misfolded states depends on the calcium concentration in the buffer. In fact, already at calcium contents compatible with physiological conditions (hundreds \(\mu \)M), a relevant percentage of misfolded proteins was revealed (see Fig. 8f) and their amount was observed to increase for higher \(\hbox {Ca}^{+}\) concentrations. Considering that increased calcium concentrations as well as the presence of misfolded NCS proteins have been independently related to major neuronal disorders, this study could represent the first demonstration at the single-molecule level of an existing link between these two effects.
Besides the remarkable results presented above, optical tweezers are also employed in many other single-molecule investigations that concern proteins, recently performed both at the outer membrane and inside living cells [97], as for instance the ligand-receptor binding [98]. Especially interesting is also the case of the protein-chaperone interactions during the protein arrangement into its three-dimensional structure [86, 99]. During the folding mechanism, proteins are often at risk of forming unwanted reactions, which cause an improper folding or the formation of aggregates. In this scenario, a key role is played by the molecular chaperones, that are specialized proteins that assist other client proteins during the folding process or assembly mechanisms, reducing the probability that unappropriated interactions with other molecules could occur in the crowded environment of the cell. The variety of molecular chaperones is wide and they perform a multitude of tasks with both nascent and fully synthesized proteins, often collaborating and acting on the same protein at different stages of its folding process. For instance, some chaperones prevent the client protein from forming aggregates when it is unfolded due to stress conditions, whereas other ones directly influence the conformational search of the protein for its native state. Thanks to their ability to directly follow the stepwise process of protein folding, optical tweezers and other single-molecule techniques have recently contributed to shed light on the mechanisms of chaperon activities, giving access to information that would be hardly obtained with traditional ensemble methods, due to the heterogeneous and transient nature of the chaperone-protein interactions. Among the others, the Heat Shock Proteins (Hsp) received special attention. These chaperones assist the client protein during the refolding process and ensemble techniques have already established their crucial role in protecting the protein against misfolding events and aggregations. However, a deeper understanding of the mechanisms underlying the functions of heat shock proteins was achieved only recently, thanks to single-molecule experiments. Several excellent works have been carried out on this topic by using optical tweezers (as further reading we suggest the review by Choudhary et al. [100] and the references therein reported). As a way of example, we report on the work by Mashaghi et al. [101], who employed OT to investigate the bacterial Hsp70 homologue (DnaK) from Escherichia coli and its role in guiding the folding/unfolding cycles of maltose proteins (MBP). The authors used the experimental configuration illustrated in Fig. 3 to pull complexes made by four maltose proteins linked head-to-tail, thus mimicking a molecular crowded situation, where maltose proteins are known to be prone to aggregate. During the refolding process, in absence of chaperones the single units of tetramer interacts with each other forming aggregates, a situation which is clearly inhibited if the experiments are carried out in presence of the DnaK system (DnaK, DnaJ, GrpE chaperones) and ATP. These results confirm the well-known chaperon ability to bind to unfolded segments of the peptide chain preventing undesired contacts and aggregation between them, which was first demonstrated by ensemble techniques. However, in this work the authors were able to gain new insights on the Hsp70 chaperone activity, that bulk experiments could not observe. By performing pulling protocols on single maltose proteins in buffers containing different amounts of chaperones and ATP, they clearly demonstrated that DnaK can also bound to client proteins that are partially folded or in a near-native state, stabilizing them against subsequent force unfolding or aggregation. The stabilization mechanisms of the DnaK was observed to be modulated by the ATP hydrolysis cycle and the experiments confirmed that both the lid and the groove of the chaperone contribute in conveying stability. More interestingly, the results suggest that the activity of DnaK is more varied than expected. In fact, the chaperones may be also able to discriminate between partially folded (native) and misfolded structures of the client protein, by accompanying the first in the late folding stages till a near-native configuration has been achieved and, in contrast, by selectively destabilizing the latter.
Another class of outstanding experiments with OT on proteins regards the protein-based molecular motors, which will be briefly presented in the next section.
4.3 Molecular motors: the RNA polymerase case
One of the major achievements of Optical Tweezers is the possibility to monitor the movements and conformational changes of one single molecule over biologically relevant forces, distances and times. This has allowed following individual molecular motors in real time while they perform their specific function, contributing to advance our knowledge in many biological processes. Molecular motors are highly efficient enzymes which usually convert chemical energy, coming for instance from ATP, into mechanical work; these machines are responsible for the majority forms of movement in cells. The most famous molecular motors studied with optical tweezers are kinesin and myosin [46, 102]. Kinesin is a processive motor that moves along microtubules up to several microns, while transporting vescicular cargos. Block et al. have deeply characterized this tiny machine [103, 104] and most of the experiments were carried out with the single-trap geometry: the kinesin was attached to the bead in the optical trap and the force acting on the system was monitored during the kinesin walk on the microtubule. The measurements proved that kinesin moves by 8-nm steps in a hand-over-hand fashion, exhibiting also backward motions and unidirectional rotation during the walk [59, 105]. In a similar manner, myosins are proteins responsible for muscular cell contraction that walk along actin filaments, showing stepping movements. This molecular motor has often been studied by employing a dual-trap configuration, where the actin filament is tethered between the optically trapped beads while the myosin is attached to a third bead fixed to the coverglass [4, 102, 106,107,108].
Another class of molecular motors is represented by the RNA-Polymerase (RNApol). RNA polymerases are enzymes fundamental in the transcription of DNA, which represents the first step in the expression of genetic information. There are different types of RNA polymerases, depending on the host organism and the specific RNA synthesis that they carry out; however, their functions can be described in terms of common general stages. During transcription, the enzyme moves stepwise along the dsDNA template and creates a complementary ssRNA chain, converting chemical energy into mechanical work. Specific transcription factors act together with RNA polymerase enzyme to regulate the three main steps of the transcription mechanism, which are: the initiation, where the enzyme binds to the DNA template to start the RNA synthesis; the elongation, during which the enzyme moves along the DNA chain and produces the complementary RNA molecule; and the termination, which represents the dissociation of the RNA polymerase from the DNA template and the final release of the transcribed RNA molecule. Single-molecule techniques have significantly contributed to our actual understanding of transcription mechanisms by RNA polymerase. Techniques as single-molecules fluorescence, atomic-force microscopy, magnetic and optical tweezers have been mainly employed to this aim, complementing results previously obtained with bulk assays and contributing to unravel the complete picture of the various phenomena involved in this process [109]. In particular, the right combination of temporal, spatial and force resolutions characterizing optical tweezers makes them an ideal tool to investigate this highly complex molecular motor [110]. The two most studied RNA polymerase are the bacterial Escherichia coli (E. coli) RNA polymerase and the more complex eukaryotic RNA polymerase II (Pol II).
The first experiments with OT on RNA polymerase molecules were carried out at the end of ’90s by the groups of Block and Gelles [60, 111], who used a single-beam geometry (see Fig. 3a) to study the influence of an applied force on the transcription mechanism. In those pioneering investigations, a single molecule of E. coli RNA polymerase was attached to a glass coverslip, while the DNA template was tethered between it and the bead in the optical trap. During the transcription phenomenon, the DNA was pulled by the fixed polymerase and a force was applied to the template, whose intensity was derived from the bead displacement with respect to the centre of the focused laser beam. The authors also applied calibrated forces in the opposite direction to the translocation one, finding that for sufficiently high forces (15-25 pN) the polymerase could be stalled, in many cases reversibly. Moreover, they observed a complex and heterogeneous polymerase dynamics, with the presence of pauses during the translocation rate.
In the last 20 years, the technological developments have led to more sensitive optical tweezers instruments, which allowed shedding light on many more details of the whole DNA transcription process. Recently, Fazal et al. [62] focused on the initiation stage of DNA transcription by RNA Polymerase II. In the initiation stage, the polymerase molecule binds to a specific promoter sequence of the DNA template and unwinds part of the DNA chain (forming a bubble) with the help of one or more transcription factors; after forming this new complex (called open promoter complex, OPC) the polymerase starts the synthesis of a short part of the RNA sequence; this initial elongation complex is quite unstable and often leads to abortive mechanisms, where the polymerase spontaneously releases the short RNA chain and restart the synthesis. Once a stable elongation complex has been created and the transcription factors have been released, the subsequent elongation stage takes place. In [62], the authors used a dual-trap configuration, where the complex made by the polymerase and the transcription factors where attached to one bead, whereas the DNA template was bound to the second bead. Once the initiation stage started, the authors monitored in real time the distance between the two optically trapped bead, following with sub-nanometric resolution any displacement due to polymerase action. They found that, contrary to previous measurements, the unwound DNA bubble is quite long, in average 85 base-pair(bp), and that an RNA sequence of the same length is transcribed before starting with the elongation stage.
Many more investigations have been carried out to characterise the high complexity of the elongation stage, which consists of a period where nucleotides are progressively added to the RNA chain. In each nucleotide addition cycle, the polymerase has to bind the correct nucleotide to the new RNA sequence and in doing this it moves not only forward but also backward, or it reversibly enters paused states [112]. In 2005, Abbondanzieri et al. [61] employed a dual-beam optical tweezers setup to detect movements of E. coli RNA polymerases at the angström scale: they clearly observed stepwise advancement of the molecule with a step length of 3.4 Å, which is consistent with crystallographic spacing between subsequent base pairs in B-DNA. Moreover, in this work the authors provided evidences that the heterogeneous movement of polymerase (made by pauses and backtracking) can be described by a Brownian ratchet model, where translocation and nucleotide binding can occur in either order. An unfortunate drawback of these experiments is that a great amount of data have to be discarded due to instrumentation noise, which also limits the duration of the molecular trajectory recording. Recently, a significant improvement has been achieved by Righini et al. [113] by using a “time-shared” dual-optical trap, where the two traps are formed by a single laser beam whose direction is switched at high rates by an acousto-optic deflector. The employed experimental configuration is depicted in Fig. 9a: the polymerase is attached to one bead by means of a long DNA handle, while the DNA template is linked to the second bead. This new approach, combined with a Hidden Markov Model algorithm, allows reducing the noise typically present in dual-beam optical tweezers and to fully uncover the molecular trajectory of the single polymerase molecule over long distances and times (see Fig. 9b).
Key features of the elongation stage are the pausing events, whose prominent mechanism is represented by backtracking, which is essential for proofreading and overcoming nucleosome obstacles ([113,114,115,116] and references therein). Pauses play a crucial role in regulating the whole gene expression, but a complete understanding of such phenomena requires instruments with high spatio-temporal resolution, so its characterization has represented a challenge in single-molecule experiments for a long time. New insights have been recently achieved by Gabizon et al. [116], who investigated the dynamics of pausing down to the \(\sim \) 100 ms time scale. They employed the same time-shared dual-optical trap instrument and experimental configuration used in [113], combining the remarkable performances offered by this instrument with improved data analysis methods. In this way, they were able to accurately determine in real time the position of the E. coli polymerase on the DNA template, to monitor sequence-dependent pausing events, as well as the time spent at each pause site. Besides the evidence that pause sequences promote the polymerase entrance into long-lived pausing states by reducing its forward transcription rate, the authors also observed novel details in the backtracking dynamics. In particular they fully characterised how far the molecule backtracks (backtrack depth), the time-lapse between the starting of the pause event and that of the backtracking and the overall backtrack duration. Backtrack depths down to only 2 bp , i.e. \(\sim \) 7 Å, were identified. Moreover, it was demonstrated that the polymerase enzyme does not backtracks immediately after entering into a paused state, but it usually waits a period ranging from 1s to 10s before moving in backward direction. Thus the backtracking phenomenon occurs in two steps: a first stabilization period during which the enzyme stops or backtracks at most by 1bp and a second stage where it enters in a longer paused state.

Characterization of full molecular trajectories of E. coli RNA polymerase during the DNA transcription process. a Schematic representation of the used experimental configuration: the DNA template is attached to the lower bead, whereas the single polymerase molecule is bound to the upper bead via 1.5kbp-long DNA handle; the red line represents the nascent transcribed RNA sequence b Distance versus time curves recorded during the trascription process. Experimental data (blue line) have been fitted (orange line) by exploiting a Large State Space Hidden Markov Model algorithm. The two insets shows an enlargement of the regions marked by black rectangles: the grey vertical lines indicate the time limits of the steps, whereas the grey horizontal lines indicate the spatial position of the fitted steps, which are separated by \(\sim \) 0.33 nm, i.e. almost one base-pair. The images are taken from M. Righini et al., PNAS 115, (2017) 1286-1291 [113]
Concerning the termination stage, this has been less investigated and the available results mainly refer to the simpler prokaryotic RNA polymerases (such as the E. coli RNA polymerase). In this last case, the termination process usually occurs in two possible ways: the Rho-independent process where a terminator sequence in the template induces the formation of a stem-loop structure in the nascent RNA chain, which in turns causes the disruption of the DNA-RNA-polymerase complex; the Rho-dependent process, where the protein Rho factor binds to the new RNA sequence, translocates along the polynucleotide chain towards the polymerase and finally stimulates the dissociation of the transcriptional complex. Both cases have been investigated by Block’s group [117, 118] for E.coli RNA polymerase. They used a smart experimental configuration based on a dual-trap geometry: the template-polymerase complex is linked to one optically trapped bead, where the already transcribed RNA chain is attached at one end to the polymerase and at the other end to the second bead. Once a Rho protein present in the buffer binds to the RNA chain, it starts to translocate towards the bead labelled with the polymerase, while a force is applied to the system by varying the relative beads position. The rips observed in the force-extension curves represent the Rho detachment from the RNA chain: from the number of released nucleotides it was possible to demonstrate that the termination factor Rho binds to the native RNA sequence via two possible states (configurations), involving 57 or 85 nucleotides, respectively.
5 Concluding remarks
In this review, we have presented the main features and some potentialities offered by optical tweezers in the field of single-molecule manipulation. After a brief comparison between optical tweezers and other force spectroscopy techniques widely employed in this scientific field, i.e. atomic force spectroscopy and magnetic tweezers, we have focused on the main physical mechanisms involved in optical manipulation and the experimental configurations most commonly used to investigate individual molecules. Finally, a selection of experiments performed by optical tweezers on different bio-polymers has been discussed, including the elastic properties of double-stranded DNA molecules, the folding and unfolding mechanisms of proteins and the investigations of the molecular motor RNA polymerase. It goes without saying that the highlighted topics do not cover all the relevant single-molecule applications made possible by OT, but only a tiny part of the ever growing number of research lines which are nowadays active in this field.
In our view, there are three main factors that will confirm the continuous spread of optical tweezers as the major tool in single-molecule experiments: its inter-disciplinary nature, the never stopping hardware development and the advances in the methods of data analysis. Indeed, OT applied to single-molecule manipulation represent a bridge between chemistry, biology and physics. This obviously concerns not only the procedures used to prepare the biological assays, but also the topics that OT allow investigating, which often lie in between these major scientific areas. The cooperation between people with different backgrounds and expertises has always represented an added value, thus their future common efforts will certainly lead to a progressive enlargement of the OT in single-molecule investigations, also to fields which are unthinkable now. Moreover, the works published in the last years have definitively established the importance of data analysis tools based on statistical mechanics in optimizing the results that can be derived from optical tweezing measurements. If on one side the enhanced performance of OT instruments permits to investigate more complex systems and observe details with unprecedented resolution, on the other this implies new challenges in data analysis. Examples of such efforts include the algorithms based on the Hidden Markov Model, which have been successfully implemented to analyze single molecule force spectroscopy trajectories [113, 119], or the Jarzynski equality and Crook fluctuation theorems applied to non-equilibrium small systems [46, 120]. These results suggest that statistical methods will be a support and a stimulus of future improvements for single-molecule investigations.
Finally, concerning the technological breakthroughs, not only they have allowed to further enhance the spatial and temporal resolution of OT experiments and widen the temperature range at which investigations can be carried out [113, 121], but also they have contributed to open new exciting research areas, such as the manipulation of single molecules within their native living cells [97, 122]. Recently, the scientific community has spent great efforts in researching innovative and efficient methods able to overcome the limitations of conventional OT, recognizing also the potentialities offered by the combination of optical trapping with other technologies and experimental methods [55]. A prime example is represented by the union of optical manipulation with fluorescence-based methods, which expands the capability of single-molecule experiments. In fact, the one-dimensional nature of the experimental data obtained with OT is compensated by the use of fluorescence microscopy, which adds valuable visualization capabilities to optical trapping, as already introduced in Sect. 4.1 by discussing the work by King et al. [77]. The strength of this marriage is undoubtedly represented by the variety of possible experimental configurations that are offered both by fluorescence microscopy (epi-illumination, total internal reflection fluorescence, confocal microscopy) and OT setups, whose combination results in a very powerful single-molecule technique for both in vitro and in vivo experiments [123]. The possibility to visualize binding events in DNA, molecular machines and conformational changes of proteins [55, 123] are only a part of the experiments made possible by the mentioned hybrid systems, whose great potentialities have fuelled the development of new sophisticated instruments which recently are emerging also on the market. Another progress in the field of optical trapping has been achieved by replacing the continuous-wave lasers with ultrashort femtosecond lasers. These light sources have been successfully exploited to directly manipulate cells, such as red blood and prokaryotic cells [124], without damaging the biological sample, thanks to the short duration of the light pulses which limits the working energy to few nanojoules. Also optical fibres have been used to replace standard cw lasers in optical manipulation experiments. Strong intensity gradients have been obtained at the tips of optical fibres by micro-machining them with techniques typical of material science, such as chemical etching, photo-polymerization and polishing [125], which permit to realize increasingly complex configurations of fibre-based trapping. Anyway, the most attractive property of this approach is its capability of performing optical manipulations without the need for external bulk optical elements, which represents a first step towards the integration of optical tweezers with Lab-on-a-chip (LOC) functionalities. As a matter of fact, the possibility to combine the tools typical of OT with the potentialities offered by LOC systems would significantly expand the horizons of optical manipulation, contributing to realize compact and user-friendly devices for single-target analysis in biological and biomedical applications. A remarkable progress in this direction is represent by nano-optical tweezers (as further reading we suggest the review by Maragò et al. [25] and the references therein reported), which on one side permit to overcome one of the major limitation of conventional OT, that is the manipulation of nanometer-sized objects, and, on the other side, are extremely promising for realizing optical manipulation stages to be implemented in LOC platforms. Among the others, two approaches are emerging within the field of nanophotonic trapping to realize fixed-position traps and force spectroscopy measurements for single-molecule studies [55, 126]: Photonic Crystal Optical Tweezers and the Plasmonic Tweezers. Contrary to microscope-based optical traps, where the objects are trapped at the beam focus, in photonic crystals and plasmonic optical tweezers evanescent near-fields located at the surface of micro- and nano-structures are used to trap objects below the diffraction limit of conventional OT. Photonic crystals are periodic dielectric structures that allow or forbid the propagation of light of certain frequency ranges and they can be properly engineered to realize waveguides and resonators, which exhibit strong gradients of the optical field at the boundary surface, resulting in optical forces able to move and trap nanoscopic objects. With this technique it has been possible to directly trap proteins [127] as well as living cells [128] without damaging them, demonstrating the feasibility of using photonic crystals OT to manipulate biological material in vivo. In plasmonic optical tweezers, the laser illumination of nanoscale metallic structures is used to excite surface plasmons, whose resulting electric fields decay quickly with the distance from the conductor surface. The field gradients can be further enhanced by using properly designed nanostructures, which give rise to strong optical gradient forces able to trap nanometric particles and biomolecules [129]. Recent works have demonstrated that plasmonic optical tweezers are efficient also in characterizing the properties of the trapped object, rather than just trap it. Especially, it is worth mentioning the work presented in [130], where authors used double-nanoholes optical tweezers to trap individual 10bp DNA-hairpins and investigate their interaction with the tumour suppressor p53 protein, paving the way for the use of this technique also in the field of biosensors. Moreover, the majority of the works discussed in [55, 126, 129, 130] have been realized by using plasmonic or photonic crystals OT already implemented in microfluidic platforms, a fact that confirms the high compatibility of nano-tweezers with LOC devices and demonstrate the considerable advances made in this direction. Of course, several barriers have still to be overcome to enhance the performance of nanotweezers in biophysics, as the control of photothermal effects and the accuracy in the control of individual molecules, but the progress made so far in this research area undoubtedly give them a prominent place in the scenario of the next generation optical manipulation techniques for single-molecule analysis.
References
A. Ashkin, Phys. Rev. Lett. 24, 156 (1970)
A. Ashkin, Phys. Rev. Lett. 25, 1321 (1970)
A. Ashkin, J.M. Dziedzic, J.E. Bjorkholm, S. Chu, Opt. Lett. 11, 288 (1986)
P.H. Jones, O.M. Maragò, G. Volpe, Optical Tweezers: Principle and Applications (Cambridge University Press, Cambridge, 2015)
M.J. Padgett, J.E. Molloy, D. McGloin, Optical Tweezers: Methods and Applications (CRC Press, Boca Raton, 2010)
M. Ribezzi-Crivellari, F. Ritort, Nat. Phys. 15, 660 (2019)
G. Pesce, A.C. De Luca, G. Rusciano, P.A. Netti, S. Fusco, A. Sasso, J. Opt. A: Pure Appl. Opt. 11, 034016 (2009)
P.Y. Chiou, A.T. Ohta, M.C. Wu, Nature 436, 370 (2005)
L. Lucchetti, K. Kushnir, A. Zaltron, F. Simoni, J. Eur. Opt. Soc. 11, 16007–1 (2016)
J. Villarroel, H. Burgos, Á. García-Cabañes, M. Carrascosa, A. Blázquez-Castro, F. Agulló-López, Opt. Express 24, 24320 (2017)
M. Esseling, A. Zaltron, W. Horn, C. Denz, Laser Photon. Rev. 9, 98 (2015)
D.G. Grier, Nature 424, 810 (2003)
M. Padgett, R. Di Leonardo, Lab Chip 11, 1196 (2011)
J. Curtis, B.A. Koss, D.G. Grier, Opt. Commun. 207, 169 (2002)
J.C. Crocker et al., Phys. Rev. Lett. 73, 352–5 (1994)
A. Curran, M.P. Lee, M.J. Padgett, J.M. Cooper, R. Di Leonardo, Phys. Rev. Lett. 108, 240601 (2012)
L. Damet, G.M. Cicuta, J. Kotar, M. Cosentino Lagomarsino, P. Cicuta Soft Matter 8, 8672 (2012)
G. Volpe, G. Volpe, S. Gigan, Sci. Rep 4, 3936 (2014)
Y. Roichman, D.G. Grier, Opt. Express 13, 5434 (2005)
S. Bianchi, G. Vizsnyiczai, S. Ferretti, C. Maggi, R. Di Leonardo, Nat. Commun. 9, 4476 (2018)
M.P. MacDonald, Science 296, 1101 (2002)
G. Volpe, L. Kurz, A. Callegari, G. Volpe, S. Gigan, Opt. Express 22, 18159 (2014)
A. Atajanov, A. Zhbanov, S. Yang, Micro Nano Syst. Lett. 6, 2 (2018)
A. Barroso, S. Landwerth, M. Woerdemann, C. Alpmann, T. Buscher, M. Becker, A. Studer, C. Denz, Biomed. Microdevices 17, 26 (2015)
O.M. Maragò, P.H. Jones, P.G. Gucciardi, G. Volpe, A.C. Ferrari, Nat. Nanotech. 8, 807 (2013)
M. Ploschner, T. Cizmar, M. Mazilu, A. Di Falco, K. Dholakia, Nano Lett. 12, 1923 (2012)
M. Dienerowitz, M. Mazilu, P.J. Reece, T.F. Krauss, K. Dholakia, Opt. Express 16, 4991 (2008)
F. Wang, W.J. Toe, W.M. Lee, D. McGloin, Q. Gao, H.H. Tan, C. Jagadish, P.J. Reece, Nano Lett. 13, 1185 (2013)
R. Agarwal, K. Ladavac, Y. Roichman, G. Yu, C. M. L., D.G. Grier, Opt. Express 13, 8906 (2005)
A. Irrera, P. Artoni, R. Sajia, P.G. Gucciardi, M.A. Iatì, F. Borghese, Nano Lett. 11, 4879 (2011)
P.J. Reece, W.J. Toe, F. Wang, Nano Lett. 11, 2375 (2011)
L. Jauffred, A.C. Richardson, L.B. Oddershede, Nano Lett. 8, 3376 (2008)
O.M. Maragò, ACS Nano 4, 7515–23 (2010)
C.W. Twombly, J.S. Evans, I.I. Smalyukh, Opt. Express 21, 1324 (2013)
L. Tong, K. Ramser, M. Käll, Optical Tweezers for Raman Spectroscopy (Springer, Berlin, 2012)
E. Messina, E. Cavallaro, A. Cacciola, R. Saija, F. Borghese, P. Denti, B. Fazio, C. D’Andrea, P.G. Gucciardi, M.A. Iatì, M. Meneghetti, G. Compagnini, V. Amendola, O.M. Maragò, J. Phys. Chem. C 115, 5115 (2011)
P.J. Reece, S. Paiman, O. Abdul-Nabi, Appl. Phys. Lett. 95, 1109 (2009)
F. Svedberg, Z. Li, H. Xu, M. Käll, Nano Lett. 6, 2639 (2006)
D.T. Crouse, J. Chem. Educ. 84, 803–9 (2007)
B.D. Hoffman, C. Grashoff, M.A. Schwartz, Nature 475, 316–23 (2011)
T.T. Perkins, Laser Photon. Rev. 3, 203–20 (2009)
Y. Shitamichi, M. Ichikawa, Y. Kimura, Chem. Phys. Lett. 479, 174 (2009)
A. Ebbesen, H. Bruus, J. Neurosci. Methods 209, 168 (2012)
D.J. Stevenson, F. Gunn-Moore, K. Dholakia, J. Biomed. Opt. 15, 041503 (2010)
P. Hinterdorfer, A.M. van Oijen, Handbook of Single Molecule Biophysics (Springer, New York, 2009)
F. Ritort, J. Phys.: Condens. Matter 18, 531–83 (2006)
K.C. Neumann, A. Nagy, Nat. Methods 5, 491–505 (2008)
X. Zhang, L. Ma, Y. Zhang, Yale J. Biol Med. 86, 367–83 (2013)
Y.F. Dufrêne, T. Ando, R. Garcia, D. Alsteens, D. Martinez-Martin, A. Engel, C. Gerber, D.J. Müller, Nanotech. 12, 295 (2017)
M. Li, D. Dang, L. Liu, N. Xi, Y. Wang, Sensors 17, 200 (2017)
T. Lionett, S. Joubaud, R. Lavery, D. Bensimon, V. Croquette, Phys. Rev. Lett. 96, 178102 (2006)
J. Gore, Z. Bryant, M. Nöllmann, M.U. Le, N.R. Cozzarelli, C. Bustamante, Nature 442, 836 (2006)
M. Capitanio, F.S. Pavone, Biophys. J. 105, 1293–303 (2013)
M.G. Donato, S. Vasi, R. Sayed, P.H. Jones, F. Bonaccorso, A.C. Ferrari, P.G. Gucciardi, O.M. Maragò, Opt. Lett. 37, 3381 (2012)
D. Choudhary, A. Mossa, M. Jadhav, C. Cecconi, Biomolecules 9, 1 (2019)
A. Ashkin, Biophys. J. 61, 569 (1992)
P. Polimeno, A. Magazzu, M.A. Iati, F. Patti, R. Saija, C.D. Boschi, M.G. Donato, P.G. Gucciardi, P.H. Jones, G. Volpe, O.M. Marago, J. Quant. Spectrosc. Radiat. Trans. 218, 131 (2018)
K.C. Neumann, S.M. Block, Rev. Sci. Instrum. 75, 2787–809 (2004)
A. Ramaiya, B. Roy, M. Bugiel, E. Schäffer, PNAS 114, 10894 (2017)
M.D. Wang, M.J. Schnitzer, H. Yin, R. Landick, J. Gelles, S.M. Block, Science 282, 902 (1998)
E.A. Abbondanzieri, W.J. Greenleaf, J.W. Shaevitz, R. Landick, S.M. Block, Nature 438, 460 (2005)
F.M. Fazal, C.A. Meng, K. Murakami, R.D. Kornberg, S.M. Block, Nature 525, 274 (2015)
J. Jiao, A.A. Rebane, L. Ma, Y. Zhang, Methods Mol. Biol. 1486, 357 (2017)
S.B. Smith, Y. Cui, C. Bustamante, Methods Enzymol. 361, 134 (2003)
Z. Bryant, M.D. Stone, J. Gore, S.B. Smith, N.R. Cozzarelli, C. Bustamante, Nature 424, 338 (2003)
J.D. Watson, F.H.C. Crick, Nature 171, 737 (1953)
M.H. Wilkins, A.R. Stokes, H.R. Wilson, Nature 171, 738 (1953)
R.E. Franklin, R.G. Gosling, Nature 171, 740 (1953)
S.B. Smith, Y. Cui, C. Bustamante, Science 271, 795 (1996)
M.D. Wang, H. Yin, R. Landick, J. Gelles, S.M. Block, Biophys. J. 72, 1335 (1997)
F.J. Marko, E.D. siggia, Macromolecules 28, 8759 (1995)
C. Bustamante, S.B. Smith, J. Liphardt, D. Smith, Curr. Opin. Struct. Biol. 10, 279 (2000)
Y.M. Tseytlin, Advanced Mechanical Models of DNA Elasticity (Academic Press Press, London, 2016)
F. Kriegel, N. Ermann, J. Lipfert, J. Struct. Biol. 197, 26 (2017)
D. Marenduzzo, E. Orlandini, F. Seno, A. Trovato, Phys. Rev. E 81, 051926 (2010)
M.C. Williams, J.R. Wenner, I. Rouzina, B.A. Bloomfield, Biophys. J. 80, 874 (2001)
G.A. King, P. Gross, U. Bockelmann, M. Modesti, G.J.L. Wuite, E.J.G. Peterman, PNAS 110, 3859 (2013)
J. van Mameren, P. Gross, G. Farge, P. Hooijman, M. Modesti, M. Falkenberg, G.J.L. Wuite, E.J.G. Peterman, PNAS 43, 18231 (2009)
L. Bongini, V. Lombardi, P. Bianco, J.R. Soc, Interface 11, 20140399 (2014)
L. Melkonyan, M. Bercy, T. Bizebard, U. Bockelmann, Biophys. J. 117, 509 (2019)
X. Zhang, H. Chen, S. Le, I. Rouzina, P.S. Doyle, J. Yan, PNAS 110, 3865 (2013)
G.A. King, E.J.G. Peterman, G.J.L. Wuite, Nat. Commun. 7, 11810 (2016)
Y. Seol, K.C. Neumann, Biophys Rev. 8, 101 (2016)
G.A. King, F. Burla, E.J.G. Peterman, G.J.L. Wuite, PNAS 116, 26534 (2019)
P. Gross, N. Laurens, L.B. Oddershede, U. Bockelmann, E.J.G. Peterman, G.J.L. Wuite, Nat. Phys. 7, 731 (2011)
C.M. Dobson, Nature 426, 884–90 (2003)
J. Fernandez-Recio, C.G. Genzor, J. Sancho, Biochemistry 40, 15234 (2001)
P.O. Heidarsson, N.M. Naqvi, M.R. Otazo, A. Mossa, B.B. Kragelund, PNAS 111, 13069 (2014)
C. Cecconi, E.A. Shank, S. Marqusee, C. Bustamante, in DNA Nanotechnology, ed by Zuccheri G., Samorì B. (Humana Press, 2011)
C. Cecconi, E.A. Shank, C. Bustamante, S. Marqusee, Science 309, 2057 (2005)
M.S.Z. Kellermeyer, S.B. Smith, H.L. Granzier, C. Bustamante, Science 276, 1112 (1997)
M.S.Z. Kellermeyer, S.B. Smith, C. Bustamante, H.L. Granzier, J. Struct. Biol. 122, 197 (1998)
M. Synakewicz, D. Bauer, M. Rief, L.S. Itzhaki, Sci. Rep. 9, 13820 (2019)
A. Alemany, B. Rey-Serra, S. Frutos, C. Cecconi, F. Ritort, Biophys. J. 110, 63 (2016)
J. Comunas-Soler, A. alemany, F. Ritort, Science 355, 412 (2017)
I. Sgouralis, S. Pressé, Biophys. J. 112, 2021 (2017)
C. Arbore, L. perego, M. sergides, M. Capitanio, Biophys. Rev 11, 765 (2019)
T. Surren, D. Rutz, P. Mößmer, U. Merkel, J. Buchner, M. Rief, PNAS 115, 11688 (2018)
P. Bechtluft, R.G.H. van Leeuwen, M. Tyreman, D. Tomkiewicz, N. Nouwen, H.L. Tepper, A.J.M. Driessen, S.J. Tans, Science 318, 1458 (2007)
D. Choudhary, L. Mediani, S. Carra, C. Cecconi, Cell Stress and Chaperones 25, 615 (2020)
A. Mashaghi, S. Bezrukavnikov, D.P. Minde, A.S. Wentink, R. Kityk, B. Zachmann-Brand, M.P. Mayer, G. Kramer, B. Bukauand, S.J. Tans, Nature 539, 448 (2016)
J.A. Spudich, S.E. Rice, R.S. Rock, T.J. Purcell, H.M. Warrick, Cold Spring Harb Protoc. 11, 1305 (2011)
K. Svoboda, C.F. Schmidt, B.J. Schnapp, S.M. Block, Nature 365, 721 (1993)
S.M. Block, L.S. Goldstein, B.J. Schnapp, Nature 348, 348 (1990)
C.L. Asbury, A.N. Fehr, S.M. Block, Science 302, 2130 (2003)
M. Capitanio, M. Canepari, P. Cacciafesta, V. Lombardi, R. Cicchi, M. Maffei, F.S. Pavone, R. Bottinelli, PNAS 103, 87 (2006)
E.J. Molloy, J.E. Burns, J. Kendrick-Jones, R.T. Tregear, D. C. S. White, Nature
J.T. Finer, R.M. Simmons, J.A. Spudich, Nature 368, 113 (1994) 378, 209 (1995)
K.M. Herbert, W.J. Greenleaf, S.M. Block, Annu. Rev. Biochem. 77, 149 (2008)
A. Lisica, S.W. Grill, BioMol. Concepts 8, 11 (2017)
H. Yin, M.D. Wang, K. Svoboda, R. Landick, S.M. Block, J. Gelles, Science 270, 1653 (1995)
M.H. Larson, R. Landick, S.M. Block, Mol. Cell 4, 249–62 (2011)
M. Righini, A. Lee, C. Cañari-Chumpitaz, T. Lionberger, R. Gabizon, Y. Coello, I. Tinoco Jr., C. Bustamante, PNAS 115, 1286 (2017)
J.W. Shaevitz, E.A. Abbondanzieri, R. Landick, S.M. Block, Nature 426, 684 (2003)
C. Hodges, L. Bintu, L. Lubkowska, M. Kashlev, C. Bustamante, Science 325, 626 (2009)
R. Gabizon, A. Lee, H. Vahedian-Movahed, R.H. Ebright, C.J. Bustamante, Nature Comm. 9, 2930 (2018)
M.H. Larson, W.J. Greenleaf, R. Landick, S.M. Block, Cell 132, 971 (2008)
D.J. Koslover, F.M. Fazal, R.A. Mooney, R. Landick, S.M. Block, J. Mol. Biol. 423, 664 (2012)
J. Stigler, M. Rief, Chem. Phys. Chem. 13, 1079–86 (2012)
M. Ribezzi-Crivellari, F. Ritort, PNAS 111, 3386 (2014)
S. de Lorenzo, M. Ribezzi-Crivellari, J.R. Arias-Gonzalez, S.B. Smith, F. Ritort, Biophys. J. 108, 2854 (2015)
K. Norregaard, L. Jauffred, K. Berg-Sørensenb, L.B. Oddershede, Phys. Chem. Chem. Phys. 16, 12614 (2014)
M.H. Shabestari, A.E.C. Meijering, W.H. Roos, G.J.L. Wuite,E.J.G. Peterman, Single-molecule enzymology: nanomechanical manipulation and hybrid methods; ed by M. Spies and Y.R. Chemla (Elsevier Academic Press Inc: San Diego, 2017)
M. Zhou, H.F. Yang, J. Di, E.L. Zhao, Chin. Opt. Lett. 6, 919 (2008)
R.S. Rodrigues Ribeiro, O. Soppera, O. A. G, A. Guerreiro, P.A.S. Jorge, J. Light. Technol 33, 3394 (2015)
J.E. Baker, R.P. Badman, M.D. Wang, WIREs Nanomed. Nanobiotechnol. 10, 1477 (2018)
Y.F. Chen, X. Serey, R. Sarkar, P. Chen, D. Erickson, Nano Lett. 12, 1633 (2012)
P. Jing, J. Wu, G.W. Liu, E.G. Keeler, S.H. Pun, L.Y. Lin, Sci. Rep. 6, 19924 (2016)
K.B. Crocier, Light Sci. Appl 8, 35 (2019)
A. Kotnala, R. Gordon, Biomed. Opt. Express 5, 1886 (2014)
F. Wang, P.J. Reece, S. Paiman, Q. Gao, H.H. Tan, C. Jagadish, Nano Lett. 11, 4149 (2011)
Acknowledgements
We acknowledge funding from Padua University Supply Award 2015 No. 6710927028 and Fondazione Cariparo Visiting Programme 2018 (project Time-resolved Force Spectroscopy of single DNA molecules). A. Z. thanks prof. Felix Ritort for his kind hospitality in these last years at his lab (Small Biosystems Lab, University of Barcelona, Department of Condensed Matter Physics) and for the continuous support and willingness to help her.
Funding
Open access funding provided by Universitá degli Studi di Padova within the CRUI-CARE Agreement.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zaltron, A., Merano, M., Mistura, G. et al. Optical tweezers in single-molecule experiments. Eur. Phys. J. Plus 135, 896 (2020). https://doi.org/10.1140/epjp/s13360-020-00907-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1140/epjp/s13360-020-00907-6