
Abstract
The reaction of the tridentate ligand 4-(2,6-bis(5-tert-butyl-1-(2,6-dichlorophenyl)-1H-pyrazol-3-yl)pyridin-4-yl)benzonitrile (L) with iron(II) salt gave the complex [Fe(L)2](BF4)2, which was isolated in a pure state and characterized by elemental analysis, NMR spectroscopy, and X-ray diffraction as two crystal polymorphs differing in the nature of the solvent molecule in the crystal (solvatomorphs I and II). According to the results of X-ray diffraction study (CCDC nos. 2104367 (I), 2104368 (II)), the iron(II) ion in these compounds occurs in different spin states and does not undergo a temperature-induced spin transition, which was previously observed for this complex in solution. The details of supramolecular organization of two solvatomorphs that prevent this transition were studied using the Hirshfeld surface analysis.
Similar content being viewed by others
INTRODUCTION
Development of functional materials the properties of which can be controlled at the molecular level is an important step towards the design of nano-sized devices [1]. One type of such materials is formed by transition metal complexes that can switch their spin state under the action of external stimuli [2] such as a change in the temperature and pressure, application of magnetic or electric field, or even the presence of various analytes [3]. This ability [4] is most often encountered for iron(II) complexes with (pseudo)octahedral environment of nitrogen-containing ligands [5, 6], which undergo transition between the low-spin (LS) diamagnetic and high-spin (HS) paramagnetic states. The accompanying changes in the magnetic and other properties [7, 8] allow the use of these compounds as switchable components of various devices and materials such as optical displays [9], memory devices [10], molecular sensors [11–14], thermometers [15, 16], and contrast agents [17] for medical diagnosis.
Although the spin transition phenomenon has been known for long [2], the rational design of the above-listed materials is markedly complicated by the crystal packing effect [18, 19], which is not always predictable and can either induce sharp spin transition with a hysteresis in the crystalline sample [20, 21] or, conversely, preclude this transition [22, 23]. One class of compounds for which the change in the spin state in solution [24] and in the solid state [25] has been most studied are iron(II) complexes with bis(pyrazol-1-yl)pyridines [26, 27]. The extensive studies of these complexes resulted in a number of recommendations on the chemical modification of ligands in order to control the spin transition parameters [24]; however, no such recommendations are available for isomeric bis(pyrazol-3-yl)pyridines [28, 29]. The key distinction between the two types of ligands is the presence of NH groups that form hydrogen bonds with counter-ions or solvent molecules in solution in the latter case [30, 31]. However, until recently [32], any substituent in position 1 of bis(pyrazol-3-yl)pyridine resulted in the formation of iron(II) complexes stabilized only in the HS state [28]. The combination of these two factors previously precluded the control over the spin state of the metal ion in these complexes by molecular design approaches [28].
Recently, we synthesized the first iron(II) complexes with N,N'-disubstituted bis(pyrazol-3-yl)pyridines that underwent a temperature-induced spin transition, which was attained by selecting the size of ortho-substituents in the N-phenyl groups of ligands of this type [32]. In addition, introduction of p-cyanophenyl substituent into position 4 of the pyridine ring of one ligand (Scheme 1) [33] shifted the transition temperature to room temperature region for the first time, which is necessary for practical applications [34].
In this study, we synthesized the complex [Fe(L)2](BF4) containing electron-donating tert-butyl groups in position 5 of the pyrazol-3-yl ring of N,N'-disubstituted ligand L (Scheme 1), which for this reason undergoes a temperature-induced spin transition in solution around 210 K [33]. The complex exists as two crystal polymorphs (I, II) differing in both the nature of the solvent present in the crystal (i.e., they are solvatomorphs) and the spin state of the iron(II) ion, as was ascertained by X-ray diffraction analysis.

Scheme 1 .
EXPERIMENTAL
All operations related to the synthesis of ligand L and complex [Fe(L)2](BF4)2 were carried out in air using commercially available organic solvents, which were distilled in an argon atmosphere. Iron(II) chloride and NaBF4 (Sigma-Aldrich) were used as received. 2,6-Dichlorophenylhydrazine was synthesized from commercially available 2,6-dichloroaniline using the standard protocol for diazotization followed by SnCl2 reduction [32]. Ligand L and complex I were prepared by the previously described procedure [33]. Elemental analysis for carbon, nitrogen, and hydrogen was carried out using a CarloErba model 1106 microanalyzer. 1H NMR spectra were recorded at room temperature on a Varian Inova 400 spectrometer. The chemical shifts are given in ppm and referred to the residual solvent signal.
Synthesis of diethyl 4-(4-cyanophenyl)pyridine-2,6-dicarboxylate. Acetic acid (2.86 mL, 50 mmol) and pyrrolidine (1.64 mL, 20 mmol) were added at room temperature (25°C) to a solution of 4-cyanobenzaldehyde (6.55 g, 50 mmol) and ethyl pyruvate (11.6 mL, 150 mmol) in acetonitrile (50 mL). At this temperature, the mixture was stirred for 30 h, and then NH4OAc (11.6 g, 150 mmol) and acetic acid (2.86 mL, 50 mmol) were added. The resulting mixture was stirred at the same temperature for 24 h. Then the mixture was poured into a saturated aqueous solution of NaHCO3 (5.0 mL) and extracted with ethyl acetate. The organic layers were combined, dried over Na2-SO4, filtered, concentrated, and purified by column flash chromatography (elution with hexane : ethyl acetate (5 : 1)). The yield was 4.2 g (26%).
1H NMR (CDCl3; 400 MHz; δ, ppm): 1.48 (t, 3JHH = 7.2 Hz, 6H, CH3), 4.53 (q, 3JHH = 7.2 Hz, 4H, CH2), 7.87–7.83 (m, 4H, 2-PhCN + 3-PhCN), 8.49 (s., 2H, 3-Py).
Synthesis of 4-(2,6-bis(5-tert-butyl-1-(2,6-dichlorophenyl)-1H-pyrazol-3-yl)pyridin-4-yl)benzonitrile (L). Pinacolone (0.962 mL, 7.71 mmol) was added to a solution containing potassium tert-butoxide (1.04 g, 9.24 mmol) and diethyl 4-(4-cyanophenyl)pyridine-2,6-dicarboxylate (1 g, 3.08 mmol) in dry THF (100 mL). The mixture was stirred at room temperature for 3 h. The product was dispersed in water (30 mL), and the resulting solution was treated with 1 M hydrochloric acid until it became acidic (pH 5). The resulting precipitate was collected on a filter, washed with water, and dried in a high vacuum. The product was used without further purification. A mixture of 4-(2,6-bis(4,4-dimethyl-3-oxopentanoyl)pyridin-4-yl)benzonitrile (0.863 g, 1.995 mmol) and 2,6-dichlorophenylhydrazine (0.742 g, 4.19 mmol) was dissolved in acetic acid (20 mL). This gave an orange suspension, which was then heated for 8 h at 70°C until a light yellow precipitate formed. The precipitate was collected on a filter, washed with acetic acid, a small amount of DMF, and water, and dried in vacuo. The product was used without further purification. The yield was 1.15 g (81%).
1H NMR (DMSO-d6; 400 MHz; δ, ppm): 1.23 (s, 18H, t-Bu), 7.14 (s, 2H, Pz–CH), 7.64 (t, 3JH,H = 8.5 Hz, 2H, 4-Ph), 7.74 (d, 3JH,H = 8.5 Hz, 4H, 4-Ph), 7.90 (d, 3JH,H = 8.0 Hz, 2H, 2-PhCN), 8.04 (d, 2H, 2‑PhCN), 8.07 (s, 2H, 3-Py).
For C38H32N6Cl4 | |||
Anal. calcd., % | C, 63.88 | H, 4.51 | N, 11.76 |
Found, % | C, 63.99 | H, 4.67 | N, 11.93 |
Synthesis of [Fe(L)2](BF4)2. Ligand L (0.112 g, 0.157 mmol) was suspended in methanol (15 mL) in a 50-mL flask. A solution of anhydrous FeCl2 (0.0099 g, 0.0785 mmol) in methanol (5 mL) was added dropwise to the suspension, and the resulting mixture was refluxed for 1 h. Solid NaBF4 (0.0172 g, 0.157 mmol) was added to the hot solution, and the mixture was stirred for 15 min and cooled down to room temperature. The unreacted ligand was filtered off, and the methanol solution was concentrated. The solid residue was dried in vacuo. The yield was 120 mg (92%).
1H NMR (CD3CN; 600 MHz; δ, ppm): 1.45 (br.s., 36H, t-Bu), 9.95 (br.s., 8H, 3-Ph), 11.12 (br.s., 4H, 2‑PhCN/3-PhCN), 12.91 (br.s., 4H, 2-PhCN/3-PhCN), 19.17 (br.s., 4H, 4-Ph), 51.07 (br.s., 4H, Pz–CH), 66.61 (br.s., 4H, m-Py-H).
For C76H64B2N12F8Cl8Fe | |||
Anal. calcd., % | C, 55.04 | H, 3.89 | N, 10.13 |
Found, % | C, 55.16 | H, 3.96 | N, 9.91 |
X-ray diffraction analysis of solvatomorphs I and II, obtained by slow evaporation in air of a methanol solution of [Fe(L)2](BF4)2 [33] and by gas diffusion of diethyl ether into an acetonitrile solution of the complex, respectively, was carried out on a Bruker APEX2 DUO CCD diffractometer (MoKα radiation, graphite monochromator, ω-scan mode). The structures were solved using the ShelXT program [35] and refined by the full-matrix least-squares method using the Olex2 program [36] in the anisotropic approximation on \(F_{{hkl}}^{2}\). The hydrogen atom positions were calculated geometrically and refined in the isotopic approximation by the riding model. The disordered solvent (water) molecules in solvatomorph I were described as a diffuse contribution to the total scattering using the Solvent Mask option of the Olex2 program [36]. Selected crystallographic data and refinement parameters are summarized in Table 1.
Atom coordinates and complete crystallographic data for solvatomorphs I and II are deposited with the Cambridge Crystallographic Data Centre (CCDC nos. 2104367, 2104368; http://www.ccdc.cam. ac.uk/).
RESULTS AND DISCUSSION
The complex [Fe(L)2](BF4)2 was prepared in a nearly quantitative yield by the reaction of anhydrous iron(II) chloride with N,N'-disubstituted bis(pyrazol-3-yl)pyridine (L) in methanol followed by replacement of the counter-ion by tetrafluoroborate anion, which provided higher solubility of the target product (Scheme 1). The precursor of ligand L was synthesized from diethyl 4-(4-cyanophenyl)pyridine-2,6-dicarboxylate by the procedure that we proposed previously [33], which included the Claisen condensation of this starting compound and pinacolone induced by potassium tert-butoxide in THF and the condensation of the resulting diketone and 2,6-dichlorophenylhydrazine followed by one-step cyclization in acetic acid (Scheme 2).

Scheme 2 .
The complex [Fe(L)2](BF4)2 was isolated in a pure state and characterized by elemental analysis and NMR spectroscopy, which previously showed [33] the presence of a temperature-induced spin transition in a DMF or acetonitile solution for this compound, with the temperature of this transition being approximately 210 K, according to the Evans method traditionally used for this purpose [28, 37].
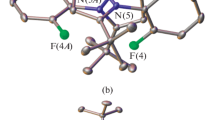
However, attempts to obtain this product as single crystals suitable for X-ray diffraction gave two types of crystals upon slow evaporation of a methanol solution of [Fe(L)2](BF4)2 in air [33] (dark red prisms) and upon diffusion of diethyl ether into acetonitrile solution of the complex (orange needles). The subsequent X-ray diffraction study showed that the crystals were two solvatomorphs I and II, differing in the nature of the solvent present in the crystal (water and acetonitrile), while their color indicated different spin states of iron(II) [2]. Indeed, according to X-ray diffraction data obtained at 120 K (Fig. 1), the complex [Fe(L)2](BF4)2 in solvatomorphs I and II exists in LS and HS states, respectively, as unambiguously follows from analysis of geometric parameters (Table 2). In solvatomorph II, the bond lengths between the iron(II) ion and nitrogen atoms of two N,N'-disubstituited ligands L are in the range typical of HS iron(II) complexes with nitrogen-containing heterocycles (2.0–2.2 Å), while these bonds in solvatomorph I [33] are barely longer than 2 Å, which attests to the LS state [2].

General view of the [Fe(L)2]2+ cation in solvatomorphs (a) I and (b) II with atoms being represented as thermal vibration ellipsoids (p = 20%). The hydrogen atoms and the minor components of disordered moieties are omitted for clarity. The numbering is given only for iron(II) ions and nitrogen atoms coordinated to them.
A similar conclusion can be drawn from comparison of the N(Py)FeN(Py) angle with the angle between the root-mean-square planes of the two ligands L in [Fe(L)2](BF4)2. In the case of an ideal octahedron characteristic of N(6) coordination environment of LS iron(II), these angles are 90° and 180°. The corresponding values were 89.65(11)° and 179.1(4)° for solvatomorph I (89.93(10)° and 179.6(4)° for the second symmetrically independent [Fe(L)2]2+ cation) and 87.36(4)° and 177.21(17)° for solvatomorph II. The distortion of the coordination polyhedron towards a trigonal prism observed in the latter compound is typical of HS iron(II) complexes [2].
This distortion is conveniently quantitatively described using so-called “shape measures” [38]. The smaller the shape measure corresponding to the chosen ideal polyhedron (e.g., octahedron), the more accurate the description of the coordination polyhedron by this ideal polyhedron. The octahedral shape measure estimated from X-ray diffraction data for I and II using the Shape 2.1 program [38] (Table 2) makes it possible to describe the iron(II) coordination polyhedron in both solvatomorphs as a distorted octahedron. However, this distortion, as expected, is more pronounced for the HS complex [Fe(L)2](BF4)2 in solvatomorph II, while the slight deviation of the octahedral shape measure from zero observed for the LS complex in I is caused by rigidity of the tridentate bis(pyrazol-3-yl)pyridine ligands [39].
All of the above-listed characteristics provide the conclusion about different spin states of [Fe(L)2](BF4)2 in the two solvatomorphs, which do not change with temperature. This is indicated, for example, by the retention of the characteristic color of the crystals (dark red and orange [2]) at room temperature. Unfortunately, poor quality of single crystals of solvatomorph I, which could pass to the HS state on heating, precluded obtaining X-ray data for this compound at this temperature.
The absence of temperature-induced spin transition in [Fe(L)2](BF4)2, which does undergo this transition in solution [33], is a consequence of the above-noted crystal packing effect [18, 19], which leads to stabilization of LS and HS states in solvatomorphs I and II, respectively. The key role is evidently played by the nature of the solvent molecules in the crystal. In solvatomorph I, which has two [Fe(L)2]2+ cations in the independent part of the unit cell, this solvent is presumably water, which gets into the crystals during crystallization in air and which had to be described as a diffuse contribution to the total X-ray scattering by the crystal due to pronounced disorder. Conversely, solvatomorph II contains one acetonitrile molecule per symmetrically independent formula unit [Fe(L)2](BF4)2; this gives rise to an absolutely different crystal environment.
A characteristic feature of the supramolecular organization of metal complexes, which has a crucial effect on the spin transition parameters in their crystalline samples [40], is so-called “terpyridine embrace,”) which appears due to stacking interactions between the pyridine or analogous heterocyclic moieties of the ligands [41, 42]. However, these associates are missing in both solvatomorphs I and II, which is consistent with the absence of temperature-induced spin transition. Instead, their crystal packing can be represented as infinite helices of [Fe(L)2]2+ cations connected to one another only by weak intermolecular contacts along the crystallographic a axis in solvatomorph I and b axis in solvatomorph II (Fig. 2). Between them, there are tetrafluoroborate anions and solvent molecules (Fig. 2), which are either water, which stabilizes the LS state of iron(II) in bis(pyrazol-3-yl)pyridine complexes [31], or acetonitrile.

Fragments of the crystal packing of [Fe(L)2](BF4)2 in solvatomorphs (a) I and (b) II, illustrating the formation of infinite helices of [Fe(L)2]2+ cations.
The observed differences in the crystal environments of the [Fe(L)2]2+ cation in the two solvatomorphs can be visualized using the Hirshfeld surfaces [43, 44], which split the crystal into molecular domains with the predominant contribution of certain molecules or ions to the electron density and 2D fingerprint plots of these surfaces [45], reflecting the frequency of implementation of various types of intermolecular interactions as the distance from the point on the Hirshfeld surface to the nearest atom inside (di) or outside (de) this surface. Considering the sums of the van der Waals radii of the pairs of atoms (dnorm), these distances encode the strength of interactions on the Hirshfeld surface with colors: blue regions correspond to interatomic distances shorter than the sum of the van der Waals radii, while red regions, conversely, reflect distances longer than this sum.
On the Hirshfeld surfaces of the [Fe(L)2]2+ cation in solvatomorphs I and II (Figs. 3, 4), the bright red regions are observed where the cation is in contact with the tetrafluoroborate anions, thus forming C–H…F contacts; the partial contributions of these contacts to the Hirshfeld surfaces are 18.2 and 14.0% in I and II, respectively (Table 3). They are reflected by regions with moderate concentrations of di and de points at the edges of 2D fingerprint plots (Figs. 3, 4), while the most populated regions in these plots correspond to the N…H and H…H contacts with partial contributions of 10.5 and 49.7% (11.0 and 47.4% for the second symmetrically independent cation) in I and 12.8 and 47.2%, respectively, in II. The edges of 2D fingerprint plots also accommodate regions of Cl…H contacts, which make approximately equal contributions to the Hirshfeld surfaces in I and II (~9.5%), and C…H contacts, for which the corresponding values are 7.6% (9.2% for the second symmetrically independent cation) and 12.3%. Despite the presence of many aromatic groups, including p-cyanophenyl group in pyridine position 4, in N,N’-disubstituted ligand L, they do not participate in the stacking interactions in either of solvatomorphs. The key difference between the contributions of intermolecular interactions (Table 3) concerns the C–H…F contacts with tetrafluoroborate anions, which differ by 4.1% in the two solvatomorphs, and C…H contacts (including those with the acetonitrile molecules in I), for which the difference is, on average, 3.9%.

(a, c) Hirshfeld surfaces for two symmetrically independent [Fe(L)2]2+ cations in solvatomorph I and (b, d) 2D fingerprint plots of these surfaces created by the Crystal Explorer program [46]. Here and below, the intermolecular contacts are characterized by interatomic distances shorter than, equal to, or longer than the sum of the van der Waals radii and are shown on the Hirshfeld surfaces by red, white, or blue regions, respectively. The green and blue regions in 2D fingerprint plots correspond to higher and lower concentrations of points corresponding to pairs of distances (di, de).

(a) Hirshfeld surface for the [Fe(L)2]2+ cation in solvatomorph II and (b) 2D plot for this surface created by the Crystal Explorer program [46].
Thus, the iron(II) complex [Fe(L)2](BF4)2 with N,N'-substituted 2,6-bis(pyrazol-3-yl)pyridine, described in our previous study [33], which undergoes a temperature-induced spin transition in various solvents, exists in different spin states in the two crystal polymorphs (solvatomorphs) that we found. This is unambiguously indicated by low-temperature X-ray diffraction data for these complexes, first of all, by Fe–N bond lengths and the shape of the coordination polyhedron typical of LS or HS state of the iron(II) ion in the (pseudo)octahedral environment of tridentate heterocyclic ligands [2].
With the spin transition being present in solutions of [Fe(L)2](BF4)2 [33], the absence of this spin transition in the solvatomorphs is obviously related to the effects of crystal packing, including the nature of solvate molecule, which is acetonitrile in the HS solvatomorph II and water in LS analogue I. It is well known that water is able to stabilize the LS state of iron(II) complexes with bis(pyrazol-3-yl)pyridines [31]. According to analysis of Hirshfeld surfaces and their 2D fingerprint plots, the key difference between the contributions of various types of intermolecular interactions in the two solvatomorphs is related to the C–H…F contacts with the tetrafluoroborate anions and C…H contacts, including those with acetonitrile molecules in the crystals of solvatomorph I. These contacts are apparently responsible for stabilization of the [Fe(L)2](BF4)2 complex in different spin states (LS and HS), thus preventing the temperature-induced spin transition in crystalline samples.
REFERENCES
Song, J.-H., Min, S.-H., Kim, S.-G., et al., Int. J. Prec. Eng. Manufactur. Green Tech., 2021, vol. 9, p. 323.
Halcrow, M.A., Spin-Crossover Materials: Properties and Applications, Chichester: Wiley, 2013.
Neville, S.M., Halder, G.J., Chapman, K.W., et al., J. Am. Chem. Soc., 2009, vol. 131, no. 34, p. 12106.
Yamasaki, M. and Ishida, T., J. Mat. Chem., 2015, vol. 3, no. 30, p. 7784.
Guionneau, P., Le Gac, F., Kaiba, A., et al., Chem. Commun., 2007, p. 3723.
Guionneau, P., Costa, J.S., and Létard, J.F., Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2004, vol. 60, no. 11, p. 587.
Molnar, G., Rat, S., Salmon, L., et al., Adv. Mater., 2017, vol. 30, no. 5, p. 1703862.
Senthil Kumar, K. and Ruben, M., Coord. Chem. Rev., 2017, vol. 346, p. 176.
Kahn, O., Kröber, J., and Jay, C., Adv. Mater., 1992, vol. 4, no. 11, p. 718.
Villalva, J., Develioglu, A., Montenegro-Pohlhammer, N., et al., Nat. Commun., 2021, vol. 12, no. 1, p. 1578.
Linares, J., Codjovi, E., and Garcia, Y., Sensors, 2012, vol. 12, no. 4, p. 4479.
Thorarinsdottir, A.E., Gaudette, A.I., and Harris, T.D., Chem. Sci., 2017, vol. 8, no. 3, p. 2448.
Gaudette, A.I., Thorarinsdottir, A.E., and Harris, T.D., Chem. Commun., 2017, vol. 53, no. 96, p. 12962.
Wang, J., Gondrand, C., Touti, F., and Hasserodt, J., Dalton Trans., 2015, vol. 44, no. 35, p. 15391.
Thorarinsdottir, A.E., Gaudette, A.I., and Harris, T.D., Chem. Sci., 2017, vol. 8, no. 3, p. 2448.
Jeon, I.-R., Park, J.G., Haney, C.R., and Harris, T.D., Chem. Sci., 2014, vol. 5, no. 6, p. 2461.
Tsukiashi, A., Min, K.S., Kitayama, H., et al., Sci. Rep., 2018, vol. 8, no. 1, p. 14911.
Elhaïk, J., Kilner, C.A., and Halcrow, M.A., CrystEngComm, 2005, vol. 7, no. 23, p. 151.
Kershaw Cook, L.J., Kulmaczewski, R., Cespedes, O., and Halcrow, M.A., Chem.-Eur. J., 2015, vol. 22, no. 5, p. 1789.
Brooker, S., Chem. Soc. Rev., 2015, vol. 44, no. 10, p. 2880.
Halcrow, M.A., Chem. Lett., 2014, vol. 43, no. 8, p. 1178.
Nikovskiy, I.A., Polezhaev, A.V., Novikov, V.V., et al., Crystals, 2021, vol. 11, no. 8, p. 922.
Melnikova, E.K., Aleshin, D.Y., Nikovskiy, I.A., et al., Crystals, 2020, vol. 10, no. 9, p. 793.
Kershaw Cook, L.J., Kulmaczewski, R., Mohammed, R., et al., Angew. Chem., Int. Ed. Engl., 2016, vol. 55, no. 13, p. 4327.
Halcrow, M.A., Capel Berdiell, I., Pask, C.M., and Kulmaczewski, R., Inorg. Chem., 2019, vol. 58, no. 15, p. 9811.
Halcrow, M.A., Coord. Chem. Rev., 2005, vol. 249, no. 25, p. 2880.
Halcrow, M.A., Coord. Chem. Rev., 2009, vol. 253, no. 21, p. 2493.
Halcrow, M.A., Crystals, 2016, vol. 6, no. 5, p. 58.
Kershaw Cook, L.J., Mohammed, R., Sherborne, G., et al., Coord. Chem. Rev., 2015, vol. 289, p. 2.
Barrett, S.A. and Halcrow, M.A., RSC Adv., 2014, vol. 4, no. 22, p. 11240.
Barrett, S.A., Kilner, C.A., and Halcrow, M.A., Dalton Trans., 2011, vol. 40, no. 45, p. 12021.
Nikovskiy, I., Polezhaev, A.V., Novikov, V.V., et al., Chem.-Eur. J., 2020, vol. 26, p. 5629.
Aleshin, D.Y., Nikovskiy, I., Novikov, V.V., et al., ACS Omega, 2021, vol. 6, no. 48, p. 33111.
Khusniyarov, M.M., Chem.-Eur. J., 2016, vol. 22, no. 43, p. 15178.
Sheldrick, G.M., Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, vol. 64, p. 112.
Dolomanov, O.V., Bourhis, L.J., Gildea, R.J., et al., J. Appl. Crystallogr., 2009, vol. 42, p. 339.
Evans, D.F., J. Chem. Soc., 1959, p. 2003.
Alvarez, S., Chem. Rev., 2015, vol. 115, p. 13447.
Craig, G.A., Costa, J.S., Roubeau, O., et al., Chem.-Eur. J., 2012, vol. 18, no. 37, p. 11703.
Pritchard, R., Kilner, C.A., and Halcrow, M.A., Chem. Commun., 2007, p. 577.
Scudder, M.L., Goodwin, H.A., and Dance, I.G., New J. Chem., 1999, vol. 23, no. 7, p. 695.
McMurtrie, J. and Dance, I., CrystEngComm, 2010, vol. 12, no. 10, p. 2700.
McKinnon, J.J., Mitchell, A.S., and Spackman, M.A., Chem. Eur. J., 1998, vol. 4, no. 11, p. 2136.
Spackman, M.A. and Jayatilaka, D., CrystEngComm, 2009, vol. 11, no. 1, p. 19.
Spackman, M.A. and McKinnon, J.J., CrystEngComm, 2002, vol. 4, no. 66, p. 378.
Wolff, S.K., Grimwood, D.J., McKinnon, J.J., et al., Crystal Explorer. Version 3.1, Perth (Australia): Univ. of Westaern Australia, 2012.
ACKNOWLEDGMENTS
X-ray diffraction studies were carried out using the research equipment of the Center for Investigation of Molecular Structure of the Nesmeyanov Institute of Organoelement Compounds, Russian Academy of Sciences, under the support from the Ministry of Science and Higher Education of the Russian Federation.
Funding
This study was supported by the Russian Science Foundation (project no. 17-13-01456).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The authors declare that they have no conflicts of interest.
Additional information
The issue is dedicated to the 70th birthday of Academician V.I. Ovcharenko
Translated by Z. Svitanko
Rights and permissions
Open Access. This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mel’nikova, E.K., Nikovskii, I.A., Polezhaev, A.V. et al. Solvatomorphs of Iron(II) Complex with N,N'-Disubstituted 2,6-Bis(pyrazol-3-yl)pyridine with a Temperature-Induced Spin Transition in Solution. Russ J Coord Chem 48, 493–503 (2022). https://doi.org/10.1134/S1070328422080048
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S1070328422080048