
Abstract—
We have developed an alternative method for the synthesis of an analog of natural retinal, which contains the p-fluorophenyl fragment instead of the trimethylcyclohexene ring. The proposed scheme for the synthesis of the target all-E-isomer of the target retinoid consists of using C5-phosphonate that contains the terminal nitrile group under Horner–Emmons reaction conditions. It has been shown that this scheme is more efficient and provides a higher total yield of the target product than the previously described variant. The procedure has been developed for the preparation of an analog of microbial proteorhodopsin ESRh from Exiguobacterium sibiricum, which contains a modified chromophore. It has been found that, as in the case of bacterioopsin from Halobacterium salinarum, the replacement of the trimethylcyclohexene ring in the natural chromophore by the p-fluorophenyl fragment does not prevent the formation of the artificial pigment F-Phe-ESRh from proteorhodopsin ESRh, which preserves the cycle of photochemical reactions. Certain differences have been found between the properties of native recombinant ESRh and its analog F-Phe-ESRh including a shift in the absorption maximum to the short-wavelength region, the formation of M intermediate at lower pH values, the presence of “long-lived M,” and a general slowdown in the photocycle. The reduced stability of the resulting proteorhodopsin analog F-Phe-ESRh to prolonged exposure to visible light has been also demonstrated.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
Retinal-containing proteins play a key role in many important biological processes, i.e., vision, light-dependent transport of protons and other ions (chlorine, sodium), and phototaxis. Retinal-containing proteins are divided into several families, the main of which are visual pigments of rods and cones of higher animals and microbial rhodopsins. More than 1000 representatives of these chromoproteins have been found in all biological kingdoms from higher animals to archaea, fungi, algae, eubacteria, and viruses [1–3].
Retinal-containing proteins have the following common features: (1) they are membrane proteins that consist of seven α-helical transmembrane strands (7TM) connected by loops; (2) as a chromophore group, they contain a certain isomer of vitamin A aldehyde, retinal (all-E-isomer in microbial pigments and 11Z-isomer in visual pigments), which is connected to the protein through the protonated aldimine bond with the ε-amino group of the Lys residue; (3) their common function is associated with the conversion of light quantum energy into various chemical or physiological responses [1–6].
In 2022, 17 years have passed since the beginning of the “era of optogenetics” (neurobiophotonics), the purpose of which is to use genetically encoded photosensitive proteins to regulate the activity of human and animal cells. To develop this promising area of research, it is necessary to create technologies for obtaining photoswitchable components for optogenetics. These new compounds with specified and/or predicted spectral-kinetic parameters can be based on modified microbial rhodopsins and other photosensitive proteins [5, 6].
Directed changes in the photochemical properties of retinal-containing proteins can be achieved by using the following approaches: (1) substitution of one or more amino acid residues in certain positions of the protein molecule by site-specific mutagenesis methods; (2) substitution of a natural retinal fragment with its various analogs; (3) a combination of the above methods.
One of the possible ways to study the structure-function relationship in retinal-containing proteins is the replacement of the natural chromophore with its analogs and a comprehensive investigation of the properties of new hybrid products [7–14]. Analogs of the native chromophore have already provided valuable structural, spectroscopic, and functional information about the structure of the ground state of the chromophore in microbial rhodopsins before high-resolution 3D structures of these molecules became available. These analogs continue to be widely used to determine the structure of intermediates of their photocycles. The directed modification of the chromophore makes it possible to vary widely the position of the absorption maximum of the ground state of the pigment and other important photochemical parameters.
Thus, the replacement of natural retinal with its analogs is one of the most promising approaches to studying the features of the chromophore-binding site in microbial rhodopsins of various families. The successful application of this method has been demonstrated for a well-studied light-dependent proton pump of bacteriorhodopsin (BRh) from the extremely halophilic microorganism Halobacterium salinarum. Using this object, a wide arsenal of modern research methods has been tested over the past 40 years to determine the structure-function relationship. Modifications of the BRh chromophore molecule made it possible to obtain its artificial analogs [7, 8].
The object of this study, proteorhodopsin (ESRh), is a new representative of retinal-containing proteins from the psychrotrophic microorganism Exiguobacterium sibiricum, whose gene was isolated from permafrost soil samples aged three million years [15]. Proteorhodopsin (ESRh) is the membrane protein that functions as a light-dependent proton pump. The spatial 3D structure of ESRh with a resolution of 2.3 Å (PDB:4HYJ) was first studied by the V. Gordeliy group (Institute of Structural Biology, Grenoble, MIPT). It was found that ESRh has a structure typical of the retinal-containing protein family including the presence of seven α-helical segments and a retinal molecule in an all-E configuration covalently bound to the K225 residue by a protonated aldimine bond [16].
The structural features of proteorhodopsin ESRh include (1) the presence of a nonstandard proton donor for the Schiff base, the K96 residue, and its location in the hydrophobic cavity closer to the protein surface; (2) violation of the α-helical structure in the middle part of the F spiral; (3) the presence of the hydrogen bond between the H57 and D85 residues; (4) the presence of several specific features of the ESRh photocycle including proton release in its late stages, accelerated formation of an M-like intermediate, and its pK shift to the alkaline pH region; (5) the absence of spectral differences in ESRh forms adapted to light and dark.
A large number of works have been devoted to studying how the ESRh and its mutant variants function [15–22]. However, the replacement of the chromophore group has not been studied before, and this work is pioneering.
The existence of some differences in the mechanisms of the photocycle and proton transport for ESRh from other retinal-containing proteins makes it interesting and relevant to compare the effect of the type of modification of their chromophore groups on the spectral parameters of these proteins and their functioning [8, 22].
To study the influence of the nature of the ESRh chromophore group on its functioning and structure, we present (1) an alternative method for the synthesis of the fluorophenyl analog of retinal (II); (2) the results of studying the interaction of the retinal (II) analog with the ESR-opsin apoprotein from E. sibiricum; (3) photochemical characteristics of the synthesized artificial pigment.
RESULT AND DISCUSSION
Synthesis of the fluorophenyl analog of retinal (II). We chose the fluorophenyl analog of retinal (II) for modification of ESRh because this compound was previously successfully used to obtain the fluorophenyl analog of BRh from H. salinarum [23]. In addition, the presence of the fluorine atom in the p-fluorophenyl fragment opens up the possibility of using 19F-NMR spectroscopy to study the structure of this protein. The study of the photoreaction cycle of the fluorophenyl analog of BRh [24] showed the delay in the reverse reaction of a light–dark adaptation by several orders of magnitude, although this artificial pigment retained the efficiency of the photocycle at the level of ~50% of that of the natural BRh cycle.
In works [22, 23], all-E- and 13Z-isomers of the fluorophenyl analog of retinal (II) were synthesized by the four-stage procedure for elongation of the retinoid polyene chain. As a key stage, the authors used the Wittig olefination reaction of carbonyl C10 precursor (IV) with ilide, which was generated from 4-fluorobenzyltriphenylphosphonium bromide (III), with a total yield of 41% (Scheme 1).

Scheme 1 . Synthesis of target retinoid (II) using Wittig olefination reaction of carbonyl compounds.
In this paper, we proposed and studied an alternative classical variant of the retinoid polyene chain extension by olefination of the initial aldehyde (XIV) using C5-phosphonate anion (XIIIa, b) under the conditions of the Horner–Emmons reaction (Schemes 2 and 3) [7, 25–29].

Scheme 2 . Synthesis of С5-phosphonate (XIIIa, b) with the terminal nitrile group.
Comparison of two synthetic approaches to obtaining the target retinoid (II). Earlier [22, 23] and in this study, two variants of olefination of carbonyl compounds were used as key reactions to create a system of conjugated double bonds in the polyene chain of the retinoid (II) molecule, i.e., Wittig olefination reaction (Scheme 1) and Horner–Emmons olefination reaction (Scheme 3).

Scheme 3 . Synthesis of target retinoid (II) by the Horner–Emmons olefination of carbonyl compounds.
The following is a critical assessment of the advantages and disadvantages of these two synthetic approaches to obtaining target retinoid (II).
Scheme 1 (the key stage is the Wittig olefination reaction):
(1) A high yield.
(2) A low stereoselectivity of the newly formed double bond; the formation of a mixture of E- and Z-isomers (1 : 1).
(3) The formation of the difficult-to-separate by-product, triphenylphosphine oxide.
(4) The need for the use of preparative HPLC.
(5) The need for additional research because of the loss of regio- and stereoselectivity during the procedure of semireduction of the triple bond.
(6) The need for an additional stage, i.e., isomerization of the 13Z-isomer under the action of iodine.
Scheme 3 (the key stage is the Horner–Emmons olefination reaction):
(1) A high yield.
(2) A high stereoselectivity of the newly formed double bond (98% of the E-isomer):
(3) The need for the use of preparative HPLC.
(4) The need to develop and search for an optimal procedure for the synthesis of C5-phosphonate (XIIIa, b).
Thus, synthesis Schemes 1 and 3 are characterized by the presence of a high yield of the desired product and the need for preparative HPLC to obtain individual compounds. The advantages of Scheme 3 are the high stereoselectivity of the newly formed double bond (98%) and the development and optimization of only one synthetic procedure. The significant disadvantages in Scheme 1 are the low stereoselectivity of the newly formed double bond (E-/Z- = 1 : 1), the presence of a difficult-to-separate byproduct (triphenylphosphine oxide), and the development of two synthetic procedures with subsequent optimization.
Synthesis of C5-phosphonate (XIIIa, b) that contains the terminal nitrile group. The synthesis of the initial C2-phosphonate (IX) was carried out by the Arbuzov reaction of commercially available chloroacetonitrile with triethylphosphite (XII). The yield of compound (IX) after distillation was 87%.
For the synthesis of C5-phosphonate (XIIIa, b), two pathways were studied (Scheme 2). As haloketone, we used commercially available chloroacetone (VII) or bromoacetone (VIII); the latter was synthesized in one stage by bromination of acetone in a yield of 50%.
Condensation of ketone (VII) with C2 phosphonate (IX) under Horner–Emmons reaction conditions yields an isomeric mixture of halonitrile (Xa, b) with a total yield of 52%. Condensation of ketone (VIII) with C2-phosphonate (IX) under the same conditions leads to the formation of an isomeric mixture of the halogen derivative (XIa, b) with a total yield of 30%. The low yield of the reaction in the second variant, via bromoketone (VIII), is explained by its low stability. Therefore, it is more profitable to use chloroacetone (VII). Moreover, bromine-containing nitrile (XIa, b) has less thermal stability than the corresponding chloro-substituted nitrile (Xa, b). The resulting halonitriles (Xa, b) and (XIa, b) were used in further transformations without additional purification. The Arbuzov condensation of compound (Xa, b) with triethylphosphite (XII) gives, after distillation, an isomeric mixture of C5-phosphonate (XIIIa, b) with a yield of 81%. The ratio of isomers (XIIIa, b) (E-/Z- = 60 : 40) was evaluated by the 1H-NMR data by comparing the integral intensity of signals of the methyl groups at 1.94 and 2.04 ppm for Z- (XIIIb) and E- (XIIIa) isomers, respectively. The homogeneity of the isomeric mixture of (XIIIa, b) was evaluated by the 31P-NMR spectroscopy. The 31P-NMR spectrum contains two signals related to pentacoordinated phosphorus at 23.8 and 23.0 ppm in a weak field (relative to the external standard, 85% H3PO4) for E- (XIIIa) and Z-(XIIIb) isomers, respectively.
Unfortunately, C5-synthon (XIIIa, b) could not be obtained based on bromonitrile (XIa, b) because of the tendency of the latter to polymerization under conditions of the Arbuzov reaction.
Thus, we have shown that the most efficient option for the synthesis of C5-phosphonate (XIIIa, b) is the two-stage pathway from commercially available chloroacetone (III). The parameters of the 1H- and 13C‑NMR spectra for the E- and Z-isomers of phosphonate (XIIIa, b) are given in the Experimental section.
The key stages of the synthesis of the fluorophenyl analog of retinal (II) are shown in Scheme 3. In the first stage, we carried out the Horner–Emmons olefination of the initial 4-fluorobenzaldehyde (XIV) with the anion of C5-phosphonate synthon (XIIIa, b) that contained the terminal polar nitrile group. NaH in THF was used as the base for generating the C5-phosphonate anion. It was shown that, as a result of the Horner–Emmons reaction, the newly formed C=C bond in product (XV) had an E configuration, which was confirmed by the values of the spin-spin interaction constants (16.2 Hz). This was followed by a stage of reduction of the nitrile function with DIBAH at a temperature from –70 to –80°С.
Repetition of the specified sequence of operations, i.e., olefination of aldehyde (XVI) by Horner–Emmons and subsequent reduction of the nitrile function of compound (XVII), led to the synthesis of target retinoid (II) with a total yield of 47% relative to the initial aldehyde (XIV). The individual all-E-isomer of retinoid (II) was isolated with a 98–99% purity using preparative HPLC. The structure of the all-E-isomer of compound (II) was confirmed by the physicochemical methods (UV, 1H-NMR spectroscopy, and mass spectrometry). The presence of a complete set of doublets of the polyene chain protons (6.05–7.15 ppm), a signal of the proton of the aldehyde group (doublet, 10.1 ppm; J, 8.2 Hz), and two proton signals of two methyl groups (2.10 and 2.35 ppm) in the 1H-NMR spectrum along with the proton signals corresponding to the p-substituted aromatic fragment (7.03 and 7.42 ppm) strictly confirmed the structure of the target aldehyde (II).
The mass spectrum contained a molecular ion ([M+] 256.1) corresponding to the calculated value.
Thus, it was shown that the scheme of the synthesis of the all-E-isomer of retinoid (II) (Scheme 3) using C5-phosphonate (XIIIa, b) that contained the terminal nitrile group is more effective than the synthesis variant we used earlier [22, 23] (Scheme 1).
Proteorhodopsin (ESRh) and its analog F-Phe-ESRh. To obtain recombinant ESRh, we used the previously constructed expression system in Escherichia coli cells, which ensures the incorporation of the protein into the inner membrane of bacteria [15]. The isolation of the target ESRh protein or its derivative F-Phe-ESRh was carried out after the addition of natural chromophore all-E-retinal (I) or its fluorophenyl analog (II) to the membrane fraction of bacterial cells in the micelles of the nonionic detergent n-dodecylmaltoside (DDM), which ensured the preservation of the native conformation and functional properties of the samples. Purification of the protein that contained the C-terminal hexahistidine sequence was performed by metal-affinity chromatography on the nickel-containing resin (Ni-Sepharose FastFlow).
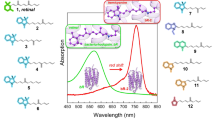
Electronic absorption spectra of ESRh and its analog F-Phe-ESRh. The samples of recombinant ESRh and its analog F-Phe-ESRh were synthesized in two alternative ways. The first method included the addition of the 1.5 molar excess of all-E-retinal (I) or its analog (II) in the ethanol solution (no more than 0.01% by volume to minimize side processes, i.e., denaturation and aggregation) to apoprotein ESRh solubilized in buffer A (0.2% DDM, 50 mM NaH2PO4, and 200 mM NaCl, pH 8.0) at 20°C. The second method included the addition of the 1.5 molar excess of all-E-retinal (I) or its analog (II) in the ethanol solution to the culture medium of E. coli BL21(DE3)pLysS, followed by the isolation of ESRh and its analog using solubilization in buffer A (pH 8.0) and metal-chelate chromatography. For the spectral studies, we changed the ratio of the protein and retinoid derivative to 1 : 0.8 when synthesizing the samples of ESRh and its analog (Fig. 1a, curves 3 and 4).

(a) Electron absorption spectra of fluorophenyl analog of retinal (II) in ethanol (1); recombinant ESRh in DDM micelles (buffer А, рН 8.0) (2); F-Phe-ESRh sample, which was prepared under conditions of 0.8 eq. of fluorophenyl analog of retinal (II) in DDM micelles (buffer А, рН 8.0) (3); F-Phe-ESRh sample, which was prepared under conditions of 1.5 eq. of fluorophenyl analog of retinal (II) in DDM micelles (buffer А, рН 8.0) (4). (b) Electron absorption spectra of F-Phe-ESRh sample, which was prepared under conditions of 0.8 eq. of fluorophenyl analog of retinal (II) in DDM micelles (buffer А) at different pH values (рН 5.0–9.0).
The absorption spectra of the resulting preparations of ESRh and its analog F-Phe-ESRh were recorded using stationary and pulsed spectroscopy in the range of 250–700 nm. The analysis of the formation of artificial pigments based on ESRh and F-Phe-ESRh has shown that there are almost no differences in the kinetics of embedding the all-E isomer of retinal and its fluorophenyl analog in ESRh-opsin solubilized in DDM micelles (τ ≤ 2 min). At the same time, when the all-E-isomer of retinal or its fluorophenyl analog is added to the preparation of apomembranes that contain bacterioopsin from H. salinarum, the kinetics of these processes differ significantly (τ ~30 min and ~6 h, respectively) [8, 22].
These differences can probably be explained by structural differences in the immediate environment of the ESRh and BRh chromophore binding sites [3]. The H57 residue, which is located at a short distance from the Schiff base in the ESRh molecule and is absent in the BRh molecule, determines the properties of the primary acceptor [3, 17]. In addition, the side chain of the R82 residue in ESRh is oriented oppositely compared to that in BRh and does not significantly affect the properties of the protein. To determine the exact reasons for the observed effects, further studies are needed including those with mutant forms of ESRh.
The absorption spectrum of resulting F-Phe-ESRh at pH 7.0 contains the maximum at 496 nm, which is 36 nm shorter than that for recombinant ESRh (532 nm) [15–20]. When changing pH to 9.0, this maximum shifts to the short-wavelength region to 490 nm (Fig. 1b). Comparison of the absorption spectra of F-Phe-ESRh (λmax = 496 nm) with the spectra of F-Phe-BRh (\(\lambda _{{\max }}^{{{\text{DA}}}}\) = 524 nm and \(\lambda _{{\max }}^{{{\text{LA}}}}\) = 510 nm) showed that the maximum in the absorption spectrum of the ESRh protein template is shifted to the short-wavelength region by 28 nm [8, 24]. Similar shifts of the absorption maximum to the short-wavelength region were found for aromatic derivatives of most BRhs and several BRhs with the modified trimethylcyclohexene chromophore ring [8, 30].
The opsin shift was calculated by the formula:
The opsin shifts for the F-Phe-ESRh and F-Phe-BRh samples were 1963 and 3040 cm–1, respectively [8].
Model aldimine chlorohydtrate of retinal analog (II) with n-butylamine (SBH+) has λmax = 452 nm in methanol.
The F-Phe-ESRh and ESRh samples adapted to light and dark (the phenomenon of light–dark adaptation) also show no differences in the spectra, which is characteristic of natural BRh from H. salinarum and most of its analogs [8, 30].
The shelf life of the F-Phe-ESRh preparations at 4°C was at least six months without a noticeable change in their characteristics.
Features of the photocycle of the proteorhodopsin analog, F-Phe-ESRh. The photocycle of F-Phe-ESRh was studied by pulsed laser spectroscopy (flash photolysis). The changes in kinetic curves of the F-Phe-ESRh absorption were revealed at four characteristic wavelengths in a suspension of DDM micelles at pH values of 7.0 and 9.0 (Figs. 2a, 2b).

Features of the photocycle of recombinant F-Phe-ESRh. The kinetics of light-induced changes in absorption of the protein in DDM micelles at pH 7.0 (a) and 9.0 (b) at characteristic wavelengths. The inset in (a) shows the changes in absorption at 390 nm because of the formation of M-intermediate.
A flash of light at pH 7.0 leads to the rapid (τ = 50–70 μs) formation of a short-lived M-intermediate, which corresponds to the deprotonated Schiff base with an absorption maximum at 390 nm (Fig. 2a). This fact distinguishes the F-Phe-ESRh photocycle from the wild-type ESRh that contains all-E-retinal, in which the formation of the M-intermediate is barely detected in DDM micelles at pH 7.0 [15–21]. Thus, it can be argued that the pKa value of the M-intermediate formation in the case of the F-Phe-ESRh derivative is shifted to the acidic pH region compared to that of wild-type ESRh. In addition, the formation of a more long-lived M-form (τ = 34 ± 7 ms) in the second fraction of the pigment (~40%) is observed.
As a result of the decay of the short-lived M-intermediate (τ = 7.7 ms), the Schiff base is reprotonated, and the part of the pigment is transformed to a state similar to the N/O intermediate of the BRh photocycle with an absorption maximum at 550 nm. The other part, presumably, does not generate a long-wave form but is involved in the reversible reprotonation of the Schiff base by the D85 proton acceptor. As a consequence, the presence of a significant proportion of the long-lived form of the M-intermediate is observed in F-Phe-ESRh up to the end of the photocycle, the total duration of which is significantly slowed down to ~27 s. An even more significant deceleration of the photocycle is observed at pH 9.0 (up to ~100 s) without noticeable accumulation of the N/O intermediate (Fig. 2b).
Stability and accessibility of the aldimine bond in F‑Phe-ESRh under the action of various conditions and reagents. In a classic work, Shkrob et al. [31] developed a whole set of chemical tests in combination with spectroscopic methods to determine the structure of the interaction products of a series of arylpolyene aldehydes with apomembranes that contained bacterioopsin from H. salinarum.
To assess the availability of the protonated aldimine bond to the action of various stimuli and reagents, four series of experiments were performed: (1) illumination of F-Phe-ESRh samples with visible light of ThorLabs OSL1-EC halogen lamp (ZhS-12 light filter, λ ≥ 400 nm) for 2–10 min; (2) exposure of the F-Phe-ESRh sample to a solution of 50-µM NaBH4, pH 9.0, in the dark and in the light; (3) the substitution of the F-Phe-ESRh sample with the all-E-retinal solution in ethanol (5 mM) in the dark and in the light; (4) exposure of the F-Phe-ESRh sample to NH2OH solution (50 mM) in the dark and in the light.
The results have shown that prolonged illumination of the F-Phe-ESRh sample by the visible light of a halogen lamp leads to partial hydrolysis of its protonated aldimine bond with the appearance of a decomposition product, which is spectrally identical to fluorophenylretinal (II) in aqueous buffer A. When the illuminated F-Phe-ESRh sample was kept in the dark for more than 2–3 h, a partial reconstruction of the pigment was revealed by an increase in the optical absorption of the F-Phe-ESRh at 496 nm. We observed a fine structure of the absorption spectrum of the decomposition product, i.e., the main band at 374 nm and two shoulders at 355 nm and 398 nm (Fig. 3a, curves 2, 3). Additional proof of the structure of the decomposition product was its transformation into the corresponding alcohol by the action of a 50-µM NaBH4 solution, pH 9.0, in the dark. The reduced decomposition product was obtained by the treatment of fluorophenylretinal (II) with NaBH4 in aqueous ethanol. The absorption spectrum of this product in buffer A was very close to the spectrum of the reference sample of fluorophenylretinol (main peak at 343 nm and two shoulders at 328 and 361 nm). A control reduction of the F-Phe-ESRh sample with NaBH4 in the dark showed that the protonated aldimine bond in the F‑Phe-ESRh sample is sufficiently stable to the action of NaBH4 in the dark (τ1/2 ≈ 10 min). After illumination of the “sample after NaBH4 treatment” for 2 min, the pigment band almost completely disappeared.

Electron absorption spectra of F-Phe-ESRh. (a) Initial sample, which was prepared under conditions of 1.5 eq. of fluorophenyl analog of retinal (II) (1); the same sample after 2-min illumination with visible light at >450 nm (ZhS-12 filter) (2); the same sample after 12-min illumination with visible light at >450 nm (ZhS-12 filter) (3); the sample treated with 50-µM NaBH4 (рН 9.0) after 15-min illumination (4). (b) Initial sample, which was prepared under conditions of 1.5 eq. of fluorophenyl analog of retinal (II) (1); “dark” initial sample 15 min after the addition of 50-µM NaBH4 (рН 9.0) (2); the sample treated with 50‑µM NaBH4 (рН 9.0) after 2-min illumination with visible light at >450 nm (3).
At the same time, the recombinant ESRh sample in DDM micelles (buffer A, pH 8.0) proved to be resistant to the effects of visible light illumination [15–21].
The substitution reaction of the chromophore analog in F-Phe-ESRh with an all-E-retinal solution in the dark proceeded very slowly (τ1/2 > 18 h). This fact suggests that there are no free sites of the chromophore binding to the ε-amino group of the K225 lysine residue or other possible lysine residues capable of forming the protonated aldimine bond in the protein.
The F-Phe-ESRh sample was also sufficiently stable in the dark in the substitution reaction with hydroxylamine (τ1/2 > 3 h).
EXPERIMENTAL
We used chloroacetonitrile and triethylphosphite (Fluka, Switzerland), chloroacetone and 4-fluorobenzaldehyde (Merck, Germany), and domestic reagents and solvents of the “chemically pure” and “pure for analysis” grades.
Thin-layer chromatography was carried out on Kieselgel 60 F254 plates (Merck, Germany) in a solvent system A (hexane/ether, 1 : 1). The compounds on the plates were detected by iodine vapor. Preparative flash chromatography was performed on Kieselgel 60 silica gel (Merck, Germany). Diethyl ether and tetrahydrofuran were purified by distillation over lithium alumohydride; methanol was absolutized using magnesium shavings.
Preparative HPLC was performed on a SmartLine 1000 HPLC chromatograph (Knauer, Germany) in isocratic mode (Knauer Eurosphere 100-10 Si column, 20 × 250 mm; eluent, hexane/diethyl ether, 7 : 1 v/v; flow rate, 5 mL/min). Optical absorption was detected on a K-2500 UV detector at 370 nm.
All operations with reagents sensitive to moisture and oxygen were carried out in thoroughly dried equipment in an atmosphere of dry argon. Evaporation of solutions was carried out on a rotary evaporator at a temperature no higher than 35°С and a pressure of 12 mm Hg.
All spectral studies were carried out at 20°. The 1H‑NMR (500 MHz), 13C-NMR (126 MHz), and 31P-NMR (203 MHz) spectra of solutions in deuterochloroform were recorded on an Avance III-500 spectrometer (Bruker, Germany). Chemical shifts are given in ppm relative to the internal standards for 1H‑NMR (tetramethylsilane, 0.00 ppm or deuterochloroform, 7.25 ppm) and 13C-NMR (deuterochloroform, 77.2 ppm) and the external standard for 31P-NMR (85% orthophosphoric acid in D2O). The values of the spin-spin interaction constants are given in Hz.
The numbering of atoms in polyene aldehyde (II) is the same as that in the polyene chain of natural retinal (I). When describing the NMR spectra, the following abbreviations are used: s, singlet; d, doublet; t, triplet; q, quartet; m, multiple.
Mass spectra were recorded on a Finnigan 4021 spectrometer (United States) with a direct sample insertion and electron impact ionization (EI 70 eV).
Spectral and photochemical characteristics of solutions of compounds and pigments were measured in quartz cuvettes with a thickness of 10 mm on a UV-2140PC spectrophotometer (Shimadzu, Japan) and a special stand created on the basis of a set of fiber-optic spectrophotometric equipment (Ocean Optics, United States). The samples were irradiated with visible light (λ ≥ 400 nm) using an OSL1-EC halogen lamp (ThorLabs, United States, 25 W) in combination with an ZhS-12 light filter.
Photoreactions of the ESRh and F-Phe-ESRh samples were studied by flash photolysis on a pulsed single-beam differential spectrophotometer with double monochromatization for measuring light [17, 19, 20]. The Nd:YAG laser LS 2131M (LOTIS TII, Belarus, 532 nm, 8 ns, 5 mJ) was used as a light excitation source. To improve the signal-to-noise ratio, 100 single signals were accumulated and averaged using an Octopus CS 8327 analog-to-digital converter (GaGe Applied Technologies, United States).
To obtain a complete kinetic picture of the F-Phe-ESRh photocycle, the measurements were carried out at four wavelengths, which are characteristic of transformations of various intermediates, i.e., 390, 480, 520, and 550 nm. A set of kinetic curves in a logarithmic time scale was analyzed using the Mathematica program (Wolfram Research, United States) by the global fitting method with the selection of 4–5 characteristic exponential components.
Mixture of E- and Z-isomers of 3-methyl-4-chloro-2-butenonitrile (Xa, b). An eighty percent suspension of NaH (3.8 g, 0.13 mol) in mineral oil was placed into a four-neck reactor (250 mL) with a dropping funnel and washed with absolute hexane (2 × 5 mL), followed by the addition of freshly distilled THF (30 mL) under stirring in the argon flow. The reaction mixture was cooled to 0°С, followed by the dropwise addition of C2-phosphonate (IX) (15 mL, 9.9 mmol) under intense stirring. The reaction mixture was stirred for 30 min at the same temperature up to complete dissolution of NaH, followed by the dropwise addition of chloroacetone (VII) (8.6 mL, 0.11 mol) under stirring. The reaction mixture was kept at 20°С for 1.5 h, followed by the addition of H2O (30 mL), diethyl ether (20 mL), and 0.1 N HCl to pH 6.0. After extraction with ether (3 × 100 mL), the combined ether fractions were washed with water to pH 7.0 and dried over Na2SO4. The desiccant was filtered out, the solvent was removed, and the remaining reaction mixture was distilled in a vacuum (0.1 mmHg). The resulting product, (Xа, b): yellow-brown oily liquid (5.94 g, 52%); the isomer mixture E-/Z-, 75 : 25; boiling point, 45–60°С (0.1 mmHg.). The 1Н-NМR spectrum (δ, ppm): E‑isomer (Xa): 2.12 (3Н, d, J 0.5, 3-СН3), 4.05 (2Н, s, –СН2Cl), 5.49 (1Н, dd, J 3.0/1.5, 2-СН); Z-isomer (Xb): 2.03 (3Н, d, J 1.5, 3-СН3), 4.24 (2Н, s, ‒СН2Cl), 5.27 (1Н, dd, J 3.0/1.5, 2-СН).
Mixture of E- and Z-isomers of diethyl(2-methyl-3-cyano-2-propenyl)phosphonate (XIIIa, b). The isomer mixture of chloronitrile (Xa, b) (11.55 g, 0.1 mol) and freshly distilled (EtO)3P (XII) (16.84 g, 0.1 mol) were placed into a three-neck reactor (250 mL) equipped with a highly efficient reverse condenser and a distillation nozzle with a thermometer and a Liebig condenser, followed by the gradual heating of the reaction mixture to 150°С under intense stirring making sure that there was no rapid foaming. The completion of the reaction was controlled by the end of the release of ethyl chloride. The residue was distilled in a vacuum (0.1 mm Hg). Resulting С5-phosphonate (XIIIa, b): 17.58 g, 81%; the isomer mixture E-/Z-, 60 : 40; boiling point, 78–98°С (0.1 mm Hg). The 1Н-NMR spectrum (δ, ppm): E-isomer (XIIIa): 1.14 (6Н, t, J 7.0, (‒ОСН2СН3)2), 2.04 (3Н, dd, J 3.4/1.3, –СН3), 2.55 (2Н, d, J 23.5, –СН2), 3.95 (4Н, q, J 7.0, (‒ОСН2СН3)2), 5.12 (1Н, m, =СН); Z-isomer (XIIIb): 1.15 (6Н, t, J 7.0, (–ОСН2СН3)2), 1.94 (3Н, dd, J 3.8/1.7, –СН3), 2.81 (2Н, d, J 24.0, –СН2), 3.96 (4Н, q, J 7.0, (–ОСН2СН3)2), 5.12 (1Н, m, =СН).The 13С-NMR spectrum (δ, ppm): E-isomer (XIIIa): 16.2 (s, (–ОСН2СН3)2), 22.0 (s, –ССН3), 36.8 (d, J 81.4, –СН2), 62.3 (s, (–ОСН2СН3)2), 116.1 (s, –ССН3), 155.6 (d, J 11.1, –CN); Z-isomer (XIIIb): 16.1 (s, (–ОСН2СН3)2), 23.9 (s, –ССН3), 34.1 (d, J 81.4, –СН2), 62.2 (s, (–ОСН2СН3)2), 115.9 (s, ‒ССН3), 155.2 (d, J 11.1, –CN).The 31Р-NMR spectrum (δ, ppm): E-isomer: 23.84; Z-isomer: 23.01.
Standard procedures of Horner–Emmons olefination of carbonyl precursors (XIV) and (XVI) by C5-phosphonate (XIIIa, b) and subsequent DIBAH reduction of the nitrile group in intermediate nitriles (XV) and (XVII). An eighty percent suspension of NaH (0.6 g) in mineral oil was placed into a three-neck reactor (100 mL) in an argon atmosphere and washed with absolute hexane (3 × 3 mL). С5-Phosphonate (XIIIa, b) (0.3 mL, 1.56 mmol) in abs THF (10 mL) was added to sodium hydride under intense stirring. The reaction mixture was stirred for 1 h to the complete dissolution of NaH, followed by the gradual syringe addition of aldehyde (XIV) or (XVI) (1.3 mol) in absolute THF (10 mL). At the end of the reaction, H2O (5 mL) and Et2O (10 mL) were added drop by drop, and the reaction mixture was neutralized with 0.1 N HCl to pH 6.0. The organic layer was removed, and the residual was extracted with Et2O (3 × 50 mL). The ether extracts were combined with an organic layer, washed with water to pH 7.0, and dried over anhydrous Na2SO4. The solvent was removed, and the residue was chromatographed on a column with 10 g of silica gel using a gradient of Et2O in hexane (0→15%) for elution.
Nitrile-containing fractions (XV) or (XVII) were combined, the solvent was removed; the residue was dried in a vacuum for 1 h at 0.1 mmHg, dissolved in absolute toluene (10 mL), and placed in a three-neck reactor (100 mL) in an argon atmosphere. The reaction mixture was cooled to a temperature in the range from –70 to –80°С, followed by the gradual syringe addition of 20% DIBAH solution (1.5 eq.) in toluene. The mixture was kept until the reaction temperature reached 20°С, followed by stirring with wet silica gel for 30 min. The reaction mass was filtered through a celite layer (1 cm), and the sorbent was washed with Et2O (50 mL). The filtrate was evaporated to dryness, the residue was chromatographed on a silica gel column (10 g) using a gradient of Et2O in hexane (0→10%) for elution. Fractions that contained a mixture of intermediate or target aldehyde isomers ((XVI) or (II)) were combined, the solvent was removed, and the residue was dried in a vacuum for 1 h at 0.1 mm Hg.
all-E-Isomer of target aldehyde (II) was isolated by preparative HPLC on a SmartLine 1000 chromatograph (Knauer, Germany) on a Knauer Eurospher 100-10 Si column (20 × 250 mm) using a hexane/diethyl ether (7 : 1, v/v) mixture as an isocratic eluent at a flow rate of 5 mL/min. The compounds were detected using a UV K-2500 detector at 370 nm. The yield of all-E-isomer of target aldehyde (II) was 47% relative to initial aldehyde (XIV). Rf 0.44 (A). The 1Н-NMR spectrum (CDCl3, δ, ppm): 2.10 (3H, s, 9-СН3), 2.35 (3H, d, J 1.5, 13-СН3), 6.05 (1H, d, J 8.2, 14-Н), 6.36 (1H, d, J 11.5, 10-Н), 6.43 (1H, d, J 15.5, 12-Н), 6.68 (1H, d, J 16.2, 8-Н), 6.82 (1H, d, J 16.2, 7-Н), 7.03 (2H, dd, J2(4)H,F 8.5, J 8.5, 2,4-Н), 7.15 (1H, dd, J 15.5, J 11.5, 11-Н), 7.42 (2H, J1(5)H,F 5.5, J 8.5, 1,5-Н), 10.12 (1H, d, J 8.2, 15-Н). The UV spectrum (methanol): λmax, nm, [ε, M–1 cm–1]): 387.5 [47 700]. The mass spectrum (m/z) ([М+] 256.1).
Synthesis of aldimine of retinal (II) analog with butylamine. n-Butylamine (1 mL) and molecular sieves 3 Å (10 mg) were added to the solution of aldehyde (II) (3 mg) in dry methanol (0.1 mL), and the reaction mixture was kept for 24 h at 0°С in a dark in the argon atmosphere. The sieves were removed, the solution and the excess of n-butanol were evaporated at 20°С and pressure of 0.1 mmHg, and the residue was dissolved in methanol (0.2 mL) and stored at ‒10°С. The spectral characteristics (methanol, λmax, nm, [ε, M–1 cm–1]) of retinal (II) analog with n-butylamine: 369 [43 500]; aldimine chlorohydrate of retinal (II) analog with n-butylamine: 452 [56 000].
Synthesis of recombinant proteorhodopsin ESRh and its analog F-Phe-ESRh. We used the E. coli BL21(DE3)pLysS strain (Novagen Merck, Germany) transformed by the pET-ESRh plasmid to synthesize a preparative amount of recombinant ESRh that contained a natural or modified chromophore [15]. A night culture of the strain was seeded in the LB medium (200 mL) that contained ampicillin (100 µg/mL) up to OD560 = 0.15. The culture was incubated at 37°C on an Innova shaker (New Brunswick Scientific, United States) at 250 rpm to OD560 = 0.8, followed by the addition of a solution of isopropyl-β-D-1-thiogalactopyranoside up to 0.2 mM and one of the retinal derivatives up to 6–7 µM, and cultivation continued at 30°C for another 24 h. For solubilization, a solution of 10% n-dodecyl-β-D-maltopyranoside (DDM) and a cocktail of a protease inhibitor (Sigma, United States) were added up to 1% and 0.3%, respectively, to the membrane fraction. The suspension was incubated at room temperature on a shaker for 3 h, followed by centrifugation for 15 min at 30 000 g. The isolation of the protein after solubilization was carried out by metal-affinity chromatography on Ni-Sepharose FastFlow (GE Healthcare, United States), as described in [15]. Recombinant ESRh was synthesized with a high degree of purity (at least 90%) and a yield of 10–15 mg/L.
CONCLUSIONS
We have proposed an alternative two-stage variant for the elongation of the polyene chain of the target retinoid, which consists of the Horner–Emmons olefination reaction of the initial 4-fluorobenzaldehyde with the C5-phosphonate anion (the first stage) and the subsequent DIBAH reduction of the nitrile function in the intermediate nitriles to the formyl group at temperatures from –70 to –80°С (the second stage). It has been shown that the scheme of the synthesis of the all-E-isomer of retinoid (II) using C5-phosphonate that contains the terminal nitrile group is more efficient and gives a higher total yield of the target retinoid (II) compared to the previously described variant of the synthesis [22, 23].
It has been found that the replacement of the trimethylcyclohexene ring of the natural chromophore of recombinant proteorhodopsin ESRh by the p-fluorophenyl fragment leads, as in the case of bacterioopsin from H. salinarum, to the formation of the artificial pigment F-Phe-ESRh, which preserves the cycle of photochemical reactions. Certain differences were found in the properties of the native recombinant ESRh and its analog F-Phe-ESRh including a shift of the absorption maximum to the short-wavelength region, the formation of an M-intermediate at lower pH values, the presence of a long-lived M, and a general deceleration of the photocycle. The reduced stability of the synthesized analog F-Phe-ESRh to prolonged exposure to visible light was also demonstrated.
The results confirm the previously described requirements for the structure of the chromophore of microbial rhodopsins including the presence of the terminal formyl group, a certain length of the polyene chain and its configuration, the absence of strict spatial restrictions in the trimethylcyclohexene ring region, and the presence of certain obstacles in the region of the C13=C14 double bond [8–13, 23, 29, 30].
REFERENCES
Kandori, H., Biophys. Rev., 2020, vol. 12, pp. 355–361. https://doi.org/10.1007/s12551-020-00645-0
Ernst, O.P., Lodowski, D.T., Elstner, M., Hegemann, P., Brown, L.S., and Kandori, H., Chem. Rev., 2014, vol. 114, p. 126. https://doi.org/10.1021/cr4003769
Gushchin, I. and Gordeliy, V., Membrane Protein Complexes: Structure and Function, Subcellular Biochemistry, Harris, J.R. and Boekema, E.J, Eds., Singapore: Springer, 2018, vol. 87, ch. 2, pp. 19–56. https://doi.org/10.1007/978-981-10-7757-9_2
Oesterhelt, D. and Stoeckenius, W., Nat. New Biol., 1971, vol. 233, pp. 149–152. https://doi.org/10.1038/newbio233149a0
Deisseroth, K., Nat. Neurosci., 2015, vol. 18, pp. 1213–1225. https://doi.org/10.1038/nn.4091
Govorunova, E.G. and Koppel, L.A., Biochemistry (Moscow), 2016, vol. 81, pp. 928–940. https://doi.org/10.1134/S0006297916090029
Chemistry and Biology of Synthetic Retinoids, Dawson, M.I. and Okamura, W.H., Eds., Boca Raton: CRC Press Inc., 2017. https://doi.org/10.1201/9781351070638
Khodonov, A.A., Belikov, N.E., and Demina, O.V., Properties of Artificial Bacteriorhodopsin Analogs, Version 2, 2020. From 1975 to 2019, Moscow: IBCP/MIPT, Russia. http://biochemphysics.ru/assets/upload/documents/docs/BRDT_v2.pdf
Mitsner, B.I. and Khodonov, A.A., Svetochuvstvitel’nye biologicheskie kompleksy i opticheskaya registratsiya informatsii (Light-Sensitive Biocomplexes and Optical Information Registration), Ivanitskii, G.R., Ed., Pushchino: Akad. Nauk SSSR, 1985, pp. 38–49.
Mitsner, B.I., Khodonov, A.A., Zvonkova, E.N., and Evstigneeva, R.P., Bioorg. Khim., 1986, vol. 12, pp. 5–53.
Mitsner, B.I., Khodonov, A.A., Zvonkova, E.N., and Karnauchova, E.N., in Retinal Proteins, Ovchinnikov, Yu.A., Ed., Utrecht: VNU Press, 1989, pp. 561–569.
Khodonov, A.A., Eremin, S.V., Lokshin, Dzh.L., Shvets, V.I., Demina, O.V., Khitrina, L.V., and Kaulen, A.D., Bioorg. Khim., 1996, vol. 22, pp. 745–776.
Barachevsky, V.A., Khodonov, A.A., Belikov, N.E., Laptev, A.V., Lukin, A.Yu., Demina, O.V., Luyksaar, S.I., and Krayushkin, M.M., Dyes and Pigments, 2012, vol. 92, pp. 831–837. https://doi.org/10.1016/j.dyepig.2011.05.009
Khodonov, A.A., Laptev, A.V., Lukin, A.Yu., Belikov, N.E., Fomin, M.A., Demina, O.V., Skladnev, D.A., Tyurin, S.A., and Shvets, V.I., Vestnik MITKhT, 2011, vol. 6, pp. 15–36.
Petrovskaya, L.E., Lukashev, E.P., Chupin, V.V., Sychev, S.V., Lyukmanova, E.N., Kryukova, E.A., Ziganshin, R.H., Spirina, E.V., Rivkina, E.M., and Khatypov, R.A., FEBS Lett., 2010, vol. 584, pp. 4193–4196. https://doi.org/10.1016/j.febslet.2010.09.005
Gushchin, I., Chervakov, P., Kuzmichev, P., Popov, A.N., Round, E., Borshchevskiy, V., Ishchenko, A., Petrovskaya, L., Chupin, V., Dolgikh, D.A., Arseniev, A.S., Kirpichnikov, M., and Gordeliy, V., Proc. Natl. Acad. Sci. U.S.A., 2013, vol. 110, pp. 12631–12636. https://doi.org/10.1073/pnas.1221629110
Balashov, S.P., Petrovskaya, L.E., Lukashev, E.P., Imasheva, E.S., Dioumaev, A.K., Wang, J.M., Sychev, S.V., Dolgikh, D.A., Rubin, A.B., Kirpichnikov, M.P., and Lanyi, J.K., Biochemistry, 2012, vol. 51, pp. 5748–5762. https://doi.org/10.1021/bi300409m
Dioumaev, A.K., Petrovskaya, L.E., Wang, J.M., Balashov, S.P., Dolgikh, D.A., Kirpichnikov, M.P., and Lanyi, J.K., J. Phys. Chem. B, vol. 117, pp. 7235–7253. https://doi.org/10.1021/jp402430w
Petrovskaya, L.E., Balashov, S.P., Lukashev, E.P., Imasheva, E.S., Gushchin, I., Dioumaev, A.K., Rubin, A.B., Dolgikh, D.A., Gordeliy, V.I., Lanyi, J.K., and Kirpichnikov, M.P., Biochemistry (Moscow), 2015, vol. 80, pp. 688–700. https://doi.org/10.1134/S000629791506005X
Siletsky, S.A., Mamedov, M.D., Lukashev, E.P., Balashov, S.P., Dolgikh, D.A., Rubin, A.B., Kirpichnikov, M.P., and Petrovskaya, L.E., Biochim. Biophys. Acta, 2016, vol. 1857, pp. 1741–1750. https://doi.org/10.1016/j.bbabio.2016.08.004
Smitienko, O.A., Feldman, T.B., Petrovskaya, L.E., Nekrasova, O.V., Yakovleva, M.A., Shelaev, I.V., Gostev, F.E., Cherepanov, D.A., Kolchugina, I.B., Dolgikh, D.A., Nadtochenko, V.A., Kirpichnikov, M.P., and Ostrovsky, M.A., J. Phys. Chem. B, vol. 125, pp. 995–1008. https://doi.org/10.1021/acs.jpcb.0c07763
Belikov, N.E., Melnikova, I.A., Demina, O.V., Petrovskaya, L.E., Kryukova, E.A., Dolgikh, D.A., Kuzmichev, P.K., Chupin, V.V., Lukin, A.Yu., Shumsky, A.N., Chizhov, I., Levin, P.P., Kirpichnikov, M.P., Varfolomeev, S.D., and Khodonov, A.A., Mendeleev Commun., 2018, vol. 28, pp. 406–408. https://doi.org/10.1016/j.mencom.2018.07.022
Mitsner, B.I., Khodonov, A.A., Zvonkova, E.N., and Evstigneeva, R.P., Bioorg. Khim., 1987, vol. 13, pp. 238–251.
Drachev, L.A., Zorina, V.V., Mitsner, B.I., Khitrina, L.V., Khodonov, A.A., and Chekulaeva, L.N., Biokhimiya, 1987, vol. 52, pp. 1559–1569.
Walker, B.J., in Organophosphorus Reagents in Organic Synthesis, Cadogan, J.I.G, Ed., London: Academic Press, 1979, ch. 3., pp. 155–206.
Courtin, J.M.L., Verhagen, L., Biesheuvel, P.L., Lugtenburg, J., van der Bend, R.L., and van Dam, K., Recl. Trav. Chim. Pays-Bas, 1987, vol. 106, pp. 112–119.
Ernst, L., Hopf, H., and Krause, N., Org. Chem., 1987, vol. 52, pp. 398–405.
Groesbeek, M., de Vries, E.F.J., Berden, J.A., and Lugtenburg, J., Recl. Trav. Chim. Pays-Bas, 1993, vol. 112, pp. 303–308.
Mironova, E.V., Lukin, A.Y., Shevaykov, S.V., Alexeeva, S.G., Shvets, V.I., Demina, O.V., Khodonov, A.A., and Khitrina, L.V., Biochemistry (Moscow), 2001, vol. 66, pp. 1323–1333. https://doi.org/10.1023/A:1013147722255
Khodonov, A.A., Belikov, N.E., Lukin, A.Yu., Petrovskaya, L.E., Chupin, V.V., and Demina, O.V., Aktual’nye Voprosy Biologicheskoi Fiziki i Khimii, 2020, vol. 5, pp. 91–100.
Shkrob, A.M., Rodionov, A.V., and Ovchinnikov, Yu.A., Bioorg. Khim., 1981, vol. 7, pp. 1169–1194.
Funding
The work was supported by the Russian Foundation for Basic Research (grant no. 20-03-00139а).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The authors state that there is no conflict of interest.
This article does not contain any studies on the use of animals as objects of research.
Additional information
Translated by A. S. Levina
Abbreviations: BRh, bacteriorhodopsin; ESRh, microbial rhodopsin ESRh from Exiguobacterium sibiricum; F-Phe-ESRh, artificial rhodopsin ESRh, in which the natural retinal is replaced by a fluorophenyl analogue; DIBAH, diisobutylaluminum hydride; DDM, n-dodecyl-β-D-maltopyranoside.
Rights and permissions
Open Access. This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Belikov, N.E., Petrovskaya, L.E., Kryukova, E.A. et al. Interaction of the Fluorophenyl Analog of Retinal with Proteorhodopsin from Exiguobacterium sibiricum. Russ J Bioorg Chem 48, 1190–1201 (2022). https://doi.org/10.1134/S1068162022060073
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S1068162022060073