
Abstract—
Using a molecular dynamics method, the state of the dimeric thymidine phosphorylase molecule from Escherichia coli in a complex with noncompetitive enzyme inhibitor 3'-azidothymidine and phosphate ion was studied on a trajectory of 50 ns. Previously obtained atomic coordinates of a complex of thymidine phosphorylase with azidothymidine and sulfate at a resolution of 1.52 Å were used as a starting model. It was demonstrated that both subunits of a dimeric enzyme molecule function asynchronously in a given time interval; moreover, each subunit maintains an open conformation. It was found that the nature of ligand at the nucleoside center affects the binding strength of phosphate in the phosphate center. In a complex with an inhibitor, both ligands over the entire time interval remain bound to the enzyme, while the release of phosphate from the active center is observed when simulating the behavior of thymidine phosphorylase in the presence of phosphate and thymidine substrate. The stabilizing effect of azidothymidine on phosphate binding is consistent with the behavior of azidothymidine as a noncompetitive inhibitor of thymidine phosphorylase.
Similar content being viewed by others

INTRODUCTION
Thymidine phosphorylase (TP), related to the family of nucleoside phosphorylase II [1], catalyzes a reversible phosphorolysis of purine and pyrimidine nucleosides and plays a central role in their metabolism, participating in reserve pathways of the synthesis of natural nucleosides [2, 3]. An interest in studying the structure and properties of nucleoside phosphorylases is associated with their involvement in metabolism of anticancer and antiviral agents (nucleoside analogs). Among thymidine phosphorylase inhibitors, analogs of natural nucleosides containing substituents in the carbohydrate component occupy a special place; some of them are used directly as anticancer and antiviral agents [4–6]. For example, 3'-azidothymidine (nucleoside analog) containing an azido group in a ribose ring is used as a drug for the treatment of human immunodeficiency syndrome [7].
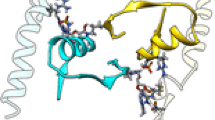
The reaction catalyzed by the enzyme proceeds by nucleophilic attack by phosphate ion located in the phosphate-binding center of the enzyme of the C1 carbon atom of the nucleoside ribose ring located in the nucleoside center. Spatial structures of a number of nucleoside phosphorylases were established by X‑ray diffraction analysis [8–11]. The spatial structure of Escherichia coli thymidine phosphorylase was established for apoenzyme, as well as complexes with substrates and inhibitors [12–14]. Thymidine phosphorylase molecule is a dimer out of identical subunits, each of which has a two-domain structure and consists of small α-helical domain and large α/β-domain (Fig. 1). The active center is located in a cavity between the two domains and includes nucleoside- and phosphate-binding sites. At the same time, the nucleoside site mainly consists of amino acid residues of the small domain, while the phosphate site is located near the C-terminal region of the main β-layer of the α/β-domain and includes amino acid residues of the large domain. The distance between bound substrates (phosphate ion and nucleoside) in the established structures is large enough. The convergence of substrates (required for the reaction) occurs through the movement of domains and is accompanied by the transition of the enzyme from an open conformation to a closed one. The nature of this transition has been insufficiently studied.

Spatial structure of dimeric thymidine phosphorylase molecule. Designations of the elements of secondary structure are given for one of the subunits.
Previously, we studied the kinetic parameters of thymidine phosphorolysis by E. coli thymidine phosphorylase in the presence of 3'-azidothymidine (AZT); it was demonstrated that AZT used as a drug in the treatment of human immunodeficiency syndrome is a noncompetitive enzyme inhibitor [15]. The spatial structure of crystalline complex of thymidine phosphorylase with AZT and sulfate was established at a resolution 1.52 Å [15]. Binding of AZT in the nucleoside center was accompanied by conformational changes, as a result of which the 3'-azido group was immersed in a hydrophobic pocket generated by amino acid residues of both subunit domains. Such type of inhibition was detected for the first time in nucleoside phosphorylases. The position of AZT partially overlapped with the position of the substrate (thymidine), but at the same time, the inhibitor was rotated relative to thymidine by 180° around the axis passing through the 3rd and 6th nitrogen and carbon atoms of the pyrimidine ring. The changed position of the attacked carbon atom of ribose relative to key amino acid residues of the active site for catalysis explains the inhibitory effect of AZT.
In the crystal structure of the complex of thymidine phosphorylase with AZT and sulfate, both subunits are in open conformation; however, open conformation can be caused by the presence of intermolecular contacts in the crystal lattice. The aim of this work was to study changes in the conformation of subunits of the dimeric thymidine phosphorylase molecule in a complex with phosphate and 3'-azidothymidine in the absence of intermolecular contacts. For this, the behavior of TP molecule and ligands bound in a complex was studied using a molecular dynamics method on a time trajectory 50 ns.
RESULTS AND DISCUSSION
When analyzing the spatial structure of the crystal complex of E. coli thymidine phosphorylase with AZT inhibitor and phosphate, it was demonstrated that the complex formation is accompanied by conformational changes and formation of a hydrophobic pocket (in the formation of which the amino acid residues of both domains of the subunits are involved) around the azido group of the inhibitor. At the same time, the open conformation of both subunits is maintained (Fig. 1). However, the open conformation of subunits in a complex TP/AZT/phosphate in the crystal lattice can be stabilized by intermolecular contacts. The analysis of crystal packing of the complex indicates that hydrogen bonds, in which amino acid residues of large domain loops are involved, are formed between the subunits linked by the symmetry operation y + 1/2, x – 1/2, z – 1/4. Several amino acid residues of one of them (mobile loop 367–381) close the cavity of the active site in a closed conformation. Amino acid residues Glu20, Asp65 of small domain and His76, Asn78 of the domain-binding loop are also involved in the formation of intermolecular bonds (symmetry operations –x, –y, –z + 1/2).
The study using a molecular dynamics (MD) method on a time trajectory 50 ns makes it possible to trace how the conformation of subunits of the dimeric enzyme molecule (Fig. 1) changes in a complex with phosphate and AZT in the absence of intermolecular contacts arising in the crystalline complex and to explain the peculiarities of the enzyme inhibition by 3'-azidothymidine.
An MD study of the enzyme dimer demonstrated that RMSD values at the beginning of the experiment tend to increase but starting from the moment of time ~16 ns (Fig. 2), some stabilization of conformational states of the dimer occurs (RMSD values fluctuate in the range ~1.3 Å). By the end of the simulation process, RMSD values begin to decrease (Fig. 2).

Graph of dependence of root-mean-square deviation (RMSD) of Cα-atoms of thymidine phosphorylase dimer on time in a complex with 3'-azidothymidine and phosphate.
Changes in the conformation of subunits in the molecule of TP complex with MD study can be determined by the results of superpositions of coordinates of Сα-atoms of the initial molecule and coordinates at the selected point of the time trajectory of MD 0–50 ns. Superposition of the models of the TF dimer in a complex with AZT and phosphate obtained by the molecular dynamics method on the time trajectory of MD 50 ns with an interval 0.5 ns (100 frames) is demonstrated in Fig. 3. The MD data indicate that in general both the spatial structure of dimer subunits and the positions of ligands (Fig. 3) do not change significantly.

Superposition of thymidine phosphorylase dimer models in a complex with 3'-azidothymidine and phosphate obtained by a molecular dynamics method on MD time trajectory 50 ns with the interval 0.5 ns (100 frames).
The comparison of spatial structures of the subunit A of the TP molecule at different moments of the MD experiment (11 points on the time trajectory with an interval 5 ns) with the structure obtained as a result of an X-ray diffraction experiment (PDB_ID: 4LHM) did not reveal any fundamental differences; RMSD values did not exceed 1.3 Å. It can be assumed that throughout the 50-ns simulation, the dimer subunit (as well as in the crystal structure) maintains an open conformation (Fig. 3). When combining molecules by Сα-atoms of two subunits, RMSD is ~3.0 Å at the initial and end points of the simulation time, which can indicate (taking into account a small RMSD value when comparing A subunits) that there is no synchronization of changes in the conformations of the dimer subunits.
The values of rms fluctuations of Cα-atoms (RMSF) of the dimer subunits (Fig. 4), indicating an instability of the spatial structure of subunits, change within ~2 Å; moreover, the values and nature of fluctuations are different in different dimer subunits. When analyzing the rms fluctuation of Сα-atoms of amino acid residues in the process of MD, it was found that the largest fluctuations are observed in the loops or α-helices and β-ribbons adjacent to them. The loops 66–71, 155–160, and 194–197 (connecting large and small domains) experience significant fluctuations. The loops of the large domain (264–270, 305–334, 365–390, 393–399), amino acid residues of which are involved in intermolecular hydrogen bonds observed in the crystal structure (PDB_ID: 4LHM), also significantly change their position as compared with initial ones, while amino acid residues of these loops change its conformation.

Values of root-mean-square fluctuations of Cα-atoms (RMSF) of thymidine phosphorylase dimer subunits.
A change in the value of distance between amino acid residues located in different domains can be considered as a criterion of displacement of domains relative to each other. These values were tracked on the simulated trajectory characterizing a change in distances between Cα-atoms of amino acid residues Ile173–Ala373, Asp178–Phe210, Leu117–Arg171 and were used to estimate the conformational state of subunits in a complex TP/AZT/phosphate on simulated trajectories. According to data of Pugmire et al. [8], shorter distances between the pairs of amino acid residues Phe210–Asp178 and Ala373–Ile173 (5.14 and 6.64 Å, PDB_ID: 1BRW) confirm the closed conformation; the distances 9.42 and 14.04 Å observed in the crystal structure [15] (PDB_ID: 4LHM) for Phe210–Asp178 and Ala373–Ile173, respectively, confirm the open conformation. The nature of convergence of Leu117 and Arg171residues belonging to different domains of the TP subunit can be traced by a change in the distance Cα_Leu117–Cα_Arg171 (Fig. 5) on the MD trajectory for the structure of TP complex with AZT and the phosphate ion.

Change in distance between Сα-atoms of Leu117 and Arg171 residues during 50-ns simulation for a complex of thymidine phosphorylase with phosphate ion and 3'-azidothymidine.
Judging by the graph (Fig. 5), the distances between Сα-atoms of Leu117, Arg171 residues in the subunits A and B of the TP complex with AZT and the phosphate ion differ significantly at the beginning of the simulation, but the differences decrease over the simulation time ~16–33 ns (the distance value fluctuates mainly within 13–16 Å), and then increase again. Periodic convergences of a pair of Сα-atoms of Leu117 and Arg171 residues to 13 Å (and even to 12 Å in the subunit A at ~15 ns) can be noted in both subunits; the distance between the appropriate atoms observed in the crystal complex of TP with AZT and the phosphate ion (that is, in open conformation) is 14.2 Å. According to the given graph (Fig. 5), no synchronization in changing these distances in enzyme subunits is observed.
The graphs allowing us to estimate the nature of changes in the distances between Сα-atoms of Asp178 (small domain) and Phe210 (large domain) residues are presented in Fig. 6. Periodic convergences of a pair of Сα-atoms in both subunits occur asynchronously, and the processes of domain convergence in the subunits are clearly not consistent, although the nature of periodic convergence of residues becomes almost the same by the end of the process of simulation. The closest distance between Сα-atoms is >7 Å. The distance between homologous residues Phe207 and Asp175 in the crystal structure of pyrimidine nucleoside phosphorylase from the Bacillus stearothermophilus [8] (PDB_ID: 1BRW), where the closed enzyme conformation is observed, is 5.14 Å.

Change in distance between Сα-atoms of Asp178 and Phe210 residues during 50-ns simulation for a complex of thymidine phosphorylase with phosphate ion and 3'-azidothymidine.
Measuring distances between Сα-atoms of Ile173 and Ala373 amino acids located in different domains of the subunit A at different times of simulation with the interval 5 ns (by which it is possible to judge the degree of convergence of domains during the MD experiment) demonstrated that these distances change within 11.0–14.1 Å. These data also confirm that the conformation of the subunit A is open (Table 1).
In the structure of TP/AZT/phosphate complex [15] (PDB_ID: 4LHM), the phosphate ion localized in α/β-domain of the enzyme subunit is directly bound by hydrogen bonds to amino acid residues Ser86, Ser95, Ser113, Thr123, and Lys84, while through water molecules to His85, Lys191, and Asp92. During the MD experiment, the phosphate ion remains in the phosphate binding site, periodically changing its orientation. At the same time, the number of hydrogen bonds changes due to a change in the conformation of some amino acid residues from the coordination sphere of the phosphate ion (Table 2). Sulfate ions at times 0 and 15 ns (time of the closest convergence of domains) and amino acid residues coordinating them are presented in Fig. 7.

Phosphate ion from thymidine phosphorylase subunit A at the moments of time 0 ns (gray color) and 15 ns (black color) surrounded by coordinating amino acid residues.
As demonstrated by Pugmire et al. [13], phosphate ion binding in the active center of pyrimidine nucleoside phosphorylases leads to the formation of a hydrogen bond between His119 and Gly208 residues of the large domain. At the same time, the formation of a hydrogen bond between His119 and Gly208 residues is associated with shifts in the large domain. A trend to periodic convergence of His119 and Gly208 residues to distances corresponding to the hydrogen bond NE2_His119–O_Gly208 is observed on the graph of dependence between the experiment time and NE2_His119–O_Gly208 distance in a complex of TP with AZT and phosphate (Fig. 8) for both subunits throughout the MD experiment. In the subunit A, a change in NE2_His119–O_Gly208 distances occurs from a hydrogen bond length of 2.8 to 5 Å from the beginning of simulation to ~14 ns. In the subunit B, these amino acid residues frequently form hydrogen bonds up to ~25 ns, and then NE2_His119–O_Gly208 distances periodically begin to change significantly, frequently increasing from the value corresponding to the H-bond to 4.5 Å and more (Fig. 8). In the subunit A at 5 and 10 ns (where, according to Ile173–Ala373 and Asp178–Phe210 distances, there is some convergence of domains), the number of hydrogen bonds formed by the phosphate ion decreases (Table 2), while NE2_His119–O_Gly208 distances slightly increase (Table 2). At the next time interval, hydrogen bonds NE2_His119–O_Gly208 are preserved despite the different number of polar contacts formed by the phosphate ion.

Change in distance between O_Gly208 and NE2_His119 atoms during 50-ns simulation for a complex of thymidine phosphorylase with phosphate ion and 3'-azidothymidine.
In a complex with the inhibitor (TP/AZT/phosphate), the hydrogen bond between His119 and Gly208 residues, which confirms the presence of the ion in the phosphate-binding center, is preserved for a longer time over the entire time interval, which distinguishes it from the complex with the substrate [16]. The comparison of simulated trajectories in the complex of TP with an inhibitor and in the complex of TP with a substrate [16] demonstrates that the complex with inhibitor and phosphate is stable over the entire time interval, while the release of phosphate from the binding center was observed in the complex with substrate and phosphate.
The AZT molecule, located in nucleoside-binding site, in the crystal complex TP/AZT/phosphate interacts with amino acid residues from both domains. The nucleoside-binding pocket is limited by amino acid residues of 161–172 (Tyr168 and Arg171) and 179–193 (Ile183, Ser186, and Lys190) helixes that are located on the surface of the α-domain facing the interdomain gap and by residues of 213–231 (Leu220) helix and the β-ribbon 82–86 (His85) of the α/β domain. The N3 atom of the pyrimidine ring of the AZT molecule forms a hydrogen bond with Oγ_Ser186 of the α-domain. O2 and O4 atoms of the pyrimidine ring form hydrogen bonds with guanidine group Arg171 and NZ_Lys190, respectively.
The MD experiment demonstrated that the pyrimidine fragment of the AZT molecule maintains contacts with coordinating amino acid residues throughout the experiment (Table 3). However, the conformation of the AZT molecule during the MD-experiment changes; the orientation of the O5' hydroxyl group relative to the ribose cycle (characterized by C3'-C4'-C5'-O5' torsion angle) remains different during the 50-ns simulation. At 15 ns, some convergence of pairs of residues from different domains (Ile173-Ala373, Asp178-Phe210, Leu117-Arg171) to the distances 11.0, 7.9, and 13.3 Å, respectively, is observed. At the same time, there is a convergence of O_Leu117 and O5'_AZT atoms to 4.9 Å. It should be noted that the side radical of the Leu117 residue from a flexible loop of a large domain of the TP subunit can change its position and orientation during the simulation process, shifting periodically towards the nucleoside, unlike Arg171, which coordinates the pyrimidine fragments of AZT and barely changes its conformation.
When analyzing the spatial structure of the E. coli TP/AZT/phosphate crystal complex [15], it was demonstrated that the complex formation is accompanied by conformational changes and formation of a hydrophobic pocket around the azido group of inhibitor, in the formation of which the amino acid residues of both subunit domains are involved: Leu220 (213–231 helix), Phe210 and Met211 (204–214 loop) of the α/β domain and Val177, Ile183, and Ile187 (179–193 helix of α-domain), Leu117 (1120–122 loop of large domain). Most of these residues are invariant in pyrimidine family II nucleoside phosphorylases. It was demonstrated [15] that the hydrophobic pocket is formed only after AZT binding. At the same time, both subunits are in open conformation.
The azido group of the AZT molecule during the MD experiment occupies a hydrophobic pocket, shifting in its cavity. Hydrophobic residues surrounding this group mostly little change their conformation. The location of the azido group, which is almost maintained during 50-ns simulation, in the hydrophobic pocket (located between large and small domains) can prevent the convergence of reaction centers, which explains the properties of AZT as a noncompetitive inhibitor.
EXPERIMENTAL
The coordinates of spatial structure of E. coli thymidine phosphorylase dimer in a complex with AZT and sulfate solved by a molecular substitution method with a resolution of 1.52 Å (PDB_ID: 4LHM) were a starting model for the TP complex with phosphate and AZT [15]. The sulfate was substituted with phosphate using a Coot program [17]. Molecular modeling was carried out using a GROMACS 2020.2 software complex [18]. The field amber99sb-idln was used as a force field [19]. The modeling area was a rectangular parallelepiped with a size 134.955 × 134.955 × 136 Å. The protein was placed in the cell center, the entire volume of the cell was filled with water; a triatomic model of water (TIP3P) was used. Parametrization of AZT was carried out using an antechamber program [20]. At the first stage, a preliminary energy minimization of the system with AZT and phosphate was performed. After energy minimization, the procedures of system temperature and pressure stabilization were carried out. The temperature value selected was 310 K; pressure value, 1 atm. The following algorithms for maintaining temperature and pressure were used: V-rescale thermostat [21] and Parrinello-Rahman barostat [22], respectively. The systems were modeled on a time interval with a total duration of 50 ns with a step of 2 fs.
The analysis of the TP structure at certain points of the simulated trajectory was carried out using a combination of structural protein models at these points with the dimer structure at the initial point of the simulation process and using a combination of structural protein models in these points with the crystal structure of the protein (PDB_ID: 4LHM). When combining the structures, a PyMol program was used [23].
CONCLUSIONS
In this work, the state of the dimeric thymidine phosphorylase molecule from Escherichia coli in a complex with noncompetitive enzyme inhibitor 3'‑azidothymidine and phosphate ion was studied using a molecular dynamics method on a 50-ns trajectory. Previously obtained atomic coordinates of the thymidine phosphorylase complex with azidothymidine and sulfate with a resolution 1.52 Å was used as a starting model.
The comparison of spatial structures of the enzyme subunit A at different time points of the MD experiment with the structure obtained in the X-ray diffraction experiment revealed no significant differences; therefore, it can be assumed that both dimer subunits maintain an open conformation throughout the 50-ns simulation. The great stability of the TP complex with AZT and phosphate as compared with the TP complex with thymidine and phosphate was demonstrated [16]. The location of the azido group between two domains of the molecules prevents the convergence of domains required for the transition to closed conformation, which explains the noncompetitive nature of inhibition with 3'-azidothymidine.
Data obtained explain some features of the TP function and may be used to develop antitumor and antiviral agents.
REFERENCES
Pugmire, M.J. and Ealick, S.E., Biochem. J., 2002, vol. 361, pp. 1–25. https://doi.org/10.1042/0264-6021:3610001
Friedkin, M. and Roberts, D., J. Biol. Chem., 1954, vol. 207, pp. 245–256.
Leer, J.C., Hammer-Jespersen, K., and Schwartz, M., Eur. J. Biochem., 1977, vol. 75, pp. 217–224. https://doi.org/10.1111/j.1432-1033.1977.tb11520.x
Desgranges, C., Razaka, G., Rabaud, M., Bricaud, H., Balzarini, J., and De Clercq, E., Biochem. Pharmacol., 1983, vol. 32, pp. 3583–3590. https://doi.org/10.1016/0006-2952(83)90307-6
Woodman, P.W., Sarrif, A.M., and Heidelberger, C., Cancer Res., 1980, vol. 40, pp. 507–511.
Schwartz, E.L., Baptiste, N., Megati, S., Wadler, S., and Otter, B.A., Cancer Res., 1995, vol. 55, pp. 3543–3550.
Zhu, L., Yang, F., Chen, L., Meehan, E.J., and Huang, M., J. Struct. Biol., 2008, vol. 162, pp. 40–49.
Pugmire, M.J. and Ealick, S.E., Structure, 1998, vol. 6, pp. 1467–1469. https://doi.org/10.1016/s0969-2126(98)00145-2
Norman, R.A., Barry, S.T., Bate, M.M., Breed, J., Colls, J.G., Ernill, R.J., Luke, R.W.A., Minshull, C.A., McAlister, M.S.B., McCall, E.J., McMiken, H.H.J., Paterson, D.S., Timms, D., Tucker, J.A., and Pauptit, R.A., Structure, 2004, vol. 12, pp. 75–84. https://doi.org/10.1016/j.str.2003.11.018
El Omari, K., Bronckaers, A., Liekens, S., Perez-Perez, M.-J., Balzarini, J., and Stammers, D.K., Biochem. J., 2006, vol. 399, pp. 199–204. https://doi.org/10.1042/BJ20060513
Balaev, V.V., Lashkov, A.A., Gabdulkhakov, A.G., Dontsova, M.V., Seregina, T.A., Mironov, A.S., Betzel, C., and Mikhailov, A.M., Acta Crystallogr. F. Struct. Biol. Commun., 2016, vol. 72, part 3, pp. 224–233. https://doi.org/10.1107/S2053230X1600162X
Walter, M.R., Cook, W.J., Cole, L.B., Short, S.A., Koszalka, G.W., Krenitsky, T.A., and Ealick, S.E., J. Biol. Chem., 1990, vol. 265, pp. 14016–14022. https://doi.org/10.2210/pdb1tpt/pdb
Pugmire, M.J., Cook, W.J., Jasanoff, A., Walter, M.R., and Ealick, S.E., J. Mol. Biol., 1998, vol. 281, pp. 285–299. https://doi.org/10.1006/jmbi.1998.1941
Timofeev, V.I., Abramchik, Yu.A., Fateev, I.V., Zhukhlistova, N.E., Murav’eva, T.I., Kuranova, I.P., and Esipov, R.S., Crystallogr. Rep., 2013, vol. 58, pp. 842–853. https://doi.org/10.1134/S1063774513060230
Timofeev, V., Abramchik, Yu., Zhukhlistova, N., Muravieva, T., Fateev, I., Esipov, R., and Kuranova, I., Acta Crystallogr. D, 2014, vol. 70, pp. 1155–1165. https://doi.org/10.1107/S1399004714001904
Sidorov-Biryukov, D.D., Podshivalov, D.D., Timofeev, V.I., Zhukhlistova, N.E., and Kuranova, I.P., Kristallografiya, 2019, vol. 64, pp. 99–105.
Emsley, P., Lohkamp, B., Scott, W.G., and Cowtan, K., Acta Crystallogr. D, 2010, vol. 66, pp. 486–501. https://doi.org/10.1107/s0907444910007493
Abraham, M.J., Murtola, T., Schulz, R., Pall, S., Smith, J.C., Hess, B., and Lindahl, E., SoftwareX, 2015, vols. 1–2, pp. 19–25. https://doi.org/10.1016/j.softx.2015.06.001
Lindorff-Larsen, K., Piana, S., Palmo, K., Maragakis, P., Klepeis, J.L., Dror, R.O., and Shaw, D.E., Proteins, 2010, vol. 78, pp. 1950–1958. https://doi.org/10.1002/prot.22711
Salomon-Ferrer, R., Case, D.A., and Walker, R.C., WIREs Comput. Mol. Sci., 2013, vol. 3, pp. 198–210. https://doi.org/10.1002/wcms.1121
Berendsen, H.J.C., Postma, J.P.M., van Gunsteren, W.F., Dinola, A., and Haak, J.R., J. Chem. Phys., 1984, vol. 81, pp. 3684–3690.
Parrinello, M. and Rahman, A., J. Chem. Phys., 1982, vol. 76, pp. 2662–2666.
Schrodinger LLC, The PyMOL Molecular GraphicsSystem, version 1.8, 2015. https://github.com/schrodinger/pymol-open-source.
Funding
This work was supported by the Ministry of Science and Higher Education of the Russian Federation within the State Task of the Federal Scientific Research Centre Crystallography and Photonics of the Russian Academy of Sciences.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interests
The authors declare that they have no conflict of interest.
This article does not contain any studies involving human participants or animals performed by any of the authors.
Additional information
Translated by A. Barkhash
Abbreviations: AZT, 3'-azidothymidine; MD, molecular dynamics; TP, thymidine phosphorylase.
Rights and permissions
Open Access. This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Timofeev, V.I., Zhukhlistova, N.E. & Kuranova, I.P. Molecular Dynamics Study of Escherichia coli Thymidine Phosphorylase in a Complex with 3'-Azidothymidine Inhibitor and Phosphate. Russ J Bioorg Chem 47, 1231–1240 (2021). https://doi.org/10.1134/S1068162021060248
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S1068162021060248