
Abstract
Mixed PMoV heteropoly acids with Keggin structures H3+xPMo12–xVxO40 (x = 1–6) and modified sulfide catalysts on their basis were synthesized, their physicochemical characteristics were determined, and their performance in the reactions of hydrodesulfurization (HDS) of dibenzothiophene (DBT) and hydrogenation of naphthalene was evaluated. Modification of the Mo-containing catalysts with vanadium by impregnation with a solution of mixed heteropoly acids was found to enhance their catalytic activity in the studied reactions, as well as to increase the selectivity for the direct hydrodesulfurization route of the DBT HDS reaction.

Similar content being viewed by others

Catalytic hydroprocessing is one of the most important current technologies in petroleum refining industry, which allows removing undesirable, e.g., sulfur-containing compounds from petroleum fractions and obtaining commercial products of better quality [1]. Continuous tightening of environmental requirements for motor fuels requires constant optimization of existing hydrotreatment technologies, which problem is most logically handled by catalyst improvement. There is a need in development of new catalytic systems affording hydrogenates with ultralow sulfur content, even if the quality of the feedstock deteriorates [2].
Heteropoly compounds (HPCs) are a class of inorganic compounds with diversified molecular structures, possessing a broad range of properties, which find application in various fields of science such as chemistry, biology, medicine, and materials science and, in particular, are used in catalysis [3]. Numerous studies have been focused on the redox and acid properties of HPCs, which are directly related to their composition. The most popular are HPCs with Keggin structures which, owing to stability and facile preparation, have been extensively used as active phase precursors for hydrotreating catalysts over several decades [2]. The structural form of α-Keggin anions having the general formula [XM12O40]n–, where X is a heteroatom (most often, P5+, Si4+, or B3+), and M is the addendum atom (most commonly, Mo and W), self-assembles in an acidic aqueous solution and represents the most stable structure of polyoxometalate catalysts [4].
Modern hydrotreating catalysts typically include Co(Ni) and Mo(W) compounds supported on γ-Al2O3 from water-soluble salts such as, e.g., ammonium molybdate and nickel nitrate [5]. It has been repeatedly demonstrated that replacement of conventional precursors by Mo(W)-containing HPCs improves the catalytic performance of hydrotreating catalysts [6–8]. The diversity of HPC structures and compositions enables synthesis of complexes containing both atoms of the main active elements and of the promoter elements by varying their ratios [9]. Another route to increasing the activity of hydrotreating catalysts involves the use of inorganic modifiers [10]. One effective approach to improving the catalytic performance involves the use of Co(Ni)XMoS mixed sulfides, where X is a d metal [11].
The latest published data indicate that modification with small amounts of vanadium increases the activity of hydrotreating catalysts [12–15]. The catalytic activity can be enhanced by using: vanadium as a modifying additive [12], complex metal compositions [13], HPCs as active phase precursors [14], an alternative activation method [15], etc.
Herein, a combination of two methods of improving the performance of the catalysts was implemented via synthesizing mixed HPCs comprising not only the active metal, Mo, but also the modifier metal, V.
The aim of this study was to synthesize sulfide catalysts based on V-containing Keggin HPCs and to examine their physicochemical characteristics and catalytic properties, as well as to compare the efficiencies of the synthesized and conventional catalysts in dibenzothiophene hydrodesulfurization and naphthalene hydrogenation reactions.
EXPERIMENTAL
Complex acids whose anion is constituted by two different acidic oxides (PMoV HPAs) of the composition H3+xPMo12–xVxO40 with Keggin structure (where x = 1, 2, 3, 4, 5, 6) were synthesized according to the procedure described in [16]. In the first stage, V2O5 is dissolved in a cooled H2O2 solution to obtain peroxovanadium compounds, which then spontaneously decompose to form an H6V10O28 solution. The latter is stabilized by addition of H3PO4, yielding an H9PV14O42solution which is added to a boiling H3PO4 + MoO3 aqueous suspension; during its evaporation MoO3 gradually dissolves, forming a PMoV HPA solution. The resulting 0.2 M solution is concentrated by evaporation to give a concentrate which is used for preparation of an impregnating solution.
PMoV catalysts were prepared by single incipient wetness impregnation of the support (γ-Al2O3) with a solution of HPA having the appropriate composition. Reference samples were synthesized on the basis of phosphomolybdic acid and ammonium metavanadate. After impregnation the samples were dried at temperatures of 60 and 80°C for 2 h and at 110°C for 6 h. The choice of the contents of the active metals was dictated by relevant published data and the results of our studies [17, 18]. The metal content in the catalysts synthesized was monitored by X-ray fluorescence (XRF) spectrometry using a Shimadzu EDX800HS analyzer.
The textural characteristics of the prepared catalysts were analyzed by low-temperature nitrogen adsorption–desorption on a Quantachrome Autosorb-1 porosimeter. The specific surface area of the catalysts was determined using the Brunauer–Emmett–Teller (BET) model at a relative partial pressure P/P0 = 0.05–0.3; the total pore volume and the pore diameter distribution were calculated using the Barrett–Joyner–Halenda model.
The structure of the HPAs synthesized was confirmed by X-ray diffraction and Fourier- transform IR spectroscopy. X-ray diffraction patterns were obtained on a Rigaku SmartLab device using CuKα radiation (λ = 1.54 Å, scan speed 2 deg/min) in the 2θ range from 5° to 70°. The IR spectra of the HPA samples were measured in the range of 400–4000 cm–1 on an IRTracer-100 (Shimadzu) instrument with an ATR (attenuated total internal reflection) accessory.
The temperature-programmed reduction analysis of the PMoV samples was performed using a TPDRO 1100 (Thermo Scientific) device under the following conditions: temperature from room temperature to 600°C, heating rate 10°C/min, flow rate of the argon-hydrogen mixture 30 mL/min, hydrogen concentration in the argon-hydrogen mixture 5 vol %.
The catalytic properties of the mixed samples were investigated on a catalytic fixed-bed flow laboratory setup. The reactor was loaded with 0.9 cm3 of the catalyst particles 0.25–0.50 mm in size. The catalysts were subjected to gas-phase sulfidation treatment at 400°C and 1 MPa in an H2S/H2 (10/90 vol %) atmosphere for 2.5 h. A model mixture of DBT (0.86 wt %), naphthalene (3.0 wt %) in toluene with n-hexadecane (1.0 wt %) added as an internal standard served as a feedstock. The catalyst activity was examined under the following conditions: temperature range 320–360°C, hydrogen pressure in the system 3.0 MPa, liquid hourly space velocity 4.5–9 h–1, and volume ratio of H2 to the feedstock 600 nL/L. The hydroconversion process was carried out for at least 8 h after a stable conversion of the reactant was attained.
The composition of the liquid products mixture sampled every hour was determined chromatographically on a Crystal-5000 instrument equipped with a flame ionization detector and a OV-101 nonpolar column (stationary phase dimethylpolysiloxane 30 m × 0.5 mm × 0.5 μm).
The activity of the catalyst in the hydrodesulfurization and hydrogenation reactions was estimated from the apparent reaction rate constant calculated using the pseudo-first-order kinetic model equation:
where F is the reactant (DBT, naphthalene) flow rate, mol/h, W, weight of MoO3, g, and x, reactant conversion, %.
Also, the relative selectivity of the DBT HDS reaction for direct desulfurization and pre-hydrogenation routes was evaluated. The relative selectivity (Sel) was calculated as the ratio of the total concentration of the products obtained by the direct hydrogenation route, i.e., of tetrahydrodibenzothiophene (THDBT), dicyclohexyl (DCH), and cyclohexylbenzene (CHB), to the concentration of biphenyl (BP) produced from the DBT molecule via direct desulfurization route:
where CCHB, CDCH, CTHDBT, and CBP are the respective concentrations of cyclohexylbenzene, dicyclohexyl, tetrahydrodibenzothiophene, and biphenyl.
The apparent activation energy Ea was estimated using the Arrhenius equation, i.e., the experimentally determined temperature dependence of ln k.
RESULTS AND DISCUSSION
Figure 1 shows the X-ray diffraction patterns of selected HPAs synthesized with the composition H3+xPMo12–xVxO40 (x = 1, 2, 3). The most significant X-ray diffraction peaks of the PVMo HPAs are observed in the 2θ ranges of 7°–10°, 18°–23°, and 25°–30°. Upon addition of vanadium the crystallinity and the main diffraction peaks in the 2θ range from 5° to 35° are preserved. According to the literature, the X-ray diffraction patterns of phosphomolybdic acid exhibit well-defined peaks; hence, the Keggin anion structure remains virtually unchanged upon incorporation of one vanadium atom [19].

X-ray diffraction patterns of the synthesized HPAs.
The heteropoly anions formed can be identified by characteristic IR bands in the range of 850–1100 cm–1, which were observed in the spectra of a series of the vanadium-containing HPAs. The absorption bands at 1043–1055 cm–1 are attributable to the vibrations of the P–O bonds, and those at 947–955 cm–1, to M(Mo,V)=O bonds. In the range of 868–889 cm–1 there are also bands associated with the vibrations of the bridging M–O–M bonds. Figure 2 shows that, with an increase in the number of the vanadium atoms in the H3+xPMo12–xVxO40 molecule, each IR band in the spectra of the series of the prepared HPAs was shifted towards lower wavenumbers, in agreement with the published data [20].

IR spectra of the synthesized HPAs: (1–6) H3+xPMo12–xVxO40 (x = 1–6) and (7) H3PMo12O40.
Table 1 presents the composition and the textural characteristics of the synthesized catalysts in the oxide form, and Figs. 3 and 4, the corresponding nitrogen adsorption–desorption isotherms and pore size distributions.

Nitrogen adsorption–desorption curves at 77 K for the support and the catalysts.

Pore size distribution curves for the support and the catalysts.
Impregnation of the support with the HPA solution leads to decreases in the specific surface area (from 309 to 249–277 m2/g) and in the pore volume (from 0.83 to 0.60–0.71 cm3/g), whereas the pattern of the pore size distribution is preserved (Fig. 4). The nitrogen adsorption–desorption curves for the support and the catalyst samples exhibit a type-IV profile characteristic of mesoporous materials (Fig. 3).
The amount of vanadium loaded on the catalyst affects the TPR patterns exhibited by the samples of the catalyst in the oxide form (Fig. 5). The reduction of all the studied samples begins at a temperature of 400–420°C, with the intensity of the TPR peak of the catalysts increasing as PMo12 < PV6Mo6 < PV12 within the temperature range of 490–550°C. Also, an increase in the V loading on the catalyst causes the amount of the hydrogen consumed under the reducing conditions to increase.

Temperature-programmed reduction curves of the samples of the catalysts in the oxide form.

Temperature-programmed reduction curves of the samples of the catalyst in the sulfide form.
Incorporation of V into the catalyst composition significantly affects the TPR patterns (Fig. 6) recorded for the sulfide catalysts. The onset of reduction of the sulfides of all the studied samples is observed at a temperature of 200–220°C. Vanadium loading causes shifting of the TPR peak of the catalysts in the PMo12–V2Mo10–PV6Mo6 series within the temperature range of 320–350°C toward higher values, while the V amount has little effect on the final temperature. The TPR peak area also increases upon vanadium incorporation, suggesting an increase in the number of the metal sulfide-based active sites.
Table 2 presents the range of the conversions of dibenzothiophene and naphthalene under the experimental conditions. For the series of the catalyst samples the DBT conversion at a temperature of 320°C is in the range of 5.6–43.5%, that at 340°C, in the range of 16.4–63.0%, and that at 360°C ranges from 25.4 to 69.6%. The naphthalene conversion at a temperature of 320°C is in the 7.5–54.8% range, that at 340°C, in the 24.7–65.8% range, and that at 360°C ranges from 33.8 to 66.8%. The PV12 reference catalyst is characterized by low conversions of the reactants.
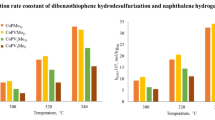
Figures 7 and 8 demonstrate the changes in the catalytic activity exhibited by the samples in dibenzothiophene hydrogenolysis and naphthalene hydrogenation reactions. The rate constants of both reactions tend to increase as the vanadium loading in the precursor increases. For the DBT hydrogenolysis reaction at a temperature of 320°C the reaction rate constant increases in the range of 6.7–17.3, that at 340°C, in the range of 15.0–37.1, and that at 360°C, in the range of 23.2–49.0 mol gMoO3–1 h–1. The rate constant of the reaction of naphthalene hydrogenation at a temperature of 320°C increases in the range of 70.9–127.0, that at 340°C, in the range 126.7–216.0, and that at 360°C, in the range of 173.6– 299.1 mol gMoO3–1 h–1. It should be noted that, with increasing temperature, the reaction rate constants display a more pronounced increase. The reference catalyst PV12 showed low catalytic activity.

Rate constant on the MoO3 weight basis for the DBT HDS reaction at Т = 320, 340, and 360°C.

Rate constant on the MoO3 weight basis for the naphthalene HYD reaction at Т = 320, 340, and 360°C.
Changes in Ea for the Mo-containing catalysts occur in a narrow range (54.7–97.5 kJ/mol for the DBT HDS reaction, and 44.7–77.6 kJ/mol for the naphthalene HYD reaction), as seen from Fig. 9. With an increase in the V loading on the samples the apparent activation energies of the HDS and HYD reactions tend to decrease. It is likely that the active sites on the modified catalyst samples are more energetically favorable to allow the model reactions. The lowest Ea values in the DBT HDS reaction are exhibited by the catalysts with 3 to 5 V atoms, and in the naphthalene HYD reaction, by those with 2 to 4 V atoms. For the V-based catalyst sample (without MoO3) Ea is significantly higher, which may be due to different mechanisms of the reactions considered.

Activation energies of the DBT HDS and the naphthalene HYD reactions in relation to the amount of vanadium loaded on the catalysts.
Incorporation of vanadium into the catalyst composition alters the DBT HDS reaction route selectivity (Table 3). With an increase in the V loading on the catalyst the relative reaction rate along the direct desulfurization route tends to increase; SelHYD/HDS decreases from 2.66 for PMo12 to 1.16 for PV6Mo6 catalyst at 320°C, from 2.35 to 1.31 at 340°C, and from 1.59 to 0.83 at 360°C. The change in the reaction route selectivity is indicative of the alteration of the catalytic properties of the active phase (number and ratio of the active sites for hydrogenation and desulfurization routes) of the catalysts, caused by V incorporation into their composition. Low selectivity of the V-based catalyst compared to the MoV analogs deserves menttioning.
CONCLUSIONS
The present study demonstrated the efficiency of V incorporation into the composition of the molybdenum catalyst from the synthesized HPC. The resultant catalysts exhibit enhanced hydrogenation and hydrodesulfurization activities as compared to the initial molybdenum-based catalyst, as well as lower selectivities in the hydrogenation and desulfurization reactions.
In general, this line of research holds promise for studying modified catalytic systems. The results from these studies may be useful in development of new highly active catalysts to be applied in existing technologies for hydrotreatment of petroleum fractions.
REFERENCES
de León, J.N.D., Kumar, C.R., Antúnez-García, J., and Fuentes-Moyado, S., Catalysts, 2019, vol. 9, no. 1, pp. 1–26. https://doi.org/10.3390/catal9010087
Weng, X., Cao, L., Zhang, G., Chen, F., Zhao, L., Zhang, Y., Gao, J., and Xu, C., Ind. Eng. Chem. Res., 2020, vol. 59, no. 49, pp. 21261–21274. https://doi.org/10.1021/acs.iecr.0c04049
Misono, M., Stud. Surf. Sci. Catal., 2013, vol. 176, pp. 97–155. https://doi.org/10.1016/B978-0-444-53833-8.00004-1
Pope, M.T., in Comprehensive Coordination Chemistry II: Transition Metal Groups 3–6, Wedd, A.G., Ed., New York: Elsevier, 2004, pp. 644–650.
North, J., Poole, O., Alotaibi, A., Bayahia, H., Kozhevnikova, E.F., Alsalme, A., Siddiqui, M.R.H., and Kozhevnikov, I.V., Appl. Catal., A: General, 2015, vol. 508, pp. 16–24. https://doi.org/10.1016/j.apcata.2015.10.001
Tanimu, A. and Alhooshani, K., Energy Fuels, 2019, vol. 33, no. 4, pp. 2810–2838. https://doi.org/10.1021/acs.energyfuels.9b00354
Griboval, A., Blanchard, P., Payen, E., Fournier, M., and Dubois, J.L., Catal. Today, 1998, vol. 45, nos. 1–4, pp. 277–283. https://doi.org/10.1016/S0920-5861(98)00230-2
Lizama, L. and Klimova, T., Appl. Catal., B: Environ., 2008, vol. 82, nos. 3–4, pp. 139–150. https://doi.org/10.1016/j.apcatb.2008.01.018
Okuhara, T., Mizuno, N., and Misono, M., Adv. Catal., 1996, vol. 41, pp. 113–252. https://doi.org/10.1016/S0360-0564(08)60041-3
Berhault, G., in New Materials for Catalytic Applications, Parvulescu, V.I. and Kemnitz, E., Eds., New York: Elsevier, 2016, vol. 10, pp. 313–360. https://doi.org/10.1016/B978-0-444-63587-7.00010-X
Afanasiev, P. and Bezverkhyy, I., Appl. Catal., A: General, 2007, vol. 322, pp. 129–141. https://doi.org/10.1016/j.apcata.2007.01.015
Soogund, D., Lecour, P., Daudin, A., Guichard, B., Legens, C., Lamonier, C., and Payen, E., Appl. Catal., B: Environ., 2010, vol. 98, nos. 1–2, pp. 39–48. https://doi.org/10.1016/j.apcatb.2010.04.024
Betancourt, P., Marrero, S., and Pinto-Castilla, S., Fuel Process. Technol., 2013, vol. 114, pp. 21–25. https://doi.org/10.1016/j.fuproc.2013.03.013
Betancourt, P., Rives, A., Scott, C.E., and Hubaut, R., Catal. Today, 2000, vol. 57, pp. 201–207.
Janssens, J.P., van Langeveld, A.D., and Moulijn, J.A., Appl. Catal., A: General, 1999, vol. 179, nos. 1–2, pp. 229–239. https://doi.org/10.1016/S0926-860X(98)00319-6
Odyakov, V.F., Zhizhina, E.G., Rodikova, Y.A., and Gogin, L.L., Eur. J. Inorg. Chem., 2015, vol. 2015, no. 22, pp. 3618–3631. https://doi.org/10.1002/ejic.201500359
Sun, M., Adjaye, J., and Nelson, A.E., Appl. Catal., A: General, 2004, vol. 263, no. 2, pp. 131–143. https://doi.org/10.1016/j.apcata.2003.12.011
Solmanov, P.S., Maksimov, N.M., Tomina, N.N., Eremina, Y.V., Timoshkina, V.V., Pimerzin, A.A., and Verevkin, S.P., Russ. J. Appl. Chem., 2018, vol. 91, no. 8, pp. 1363–1369. https://doi.org/10.1134/S096554411902018X
Barats-Damatov, D., Shimon, L.J.W., Feldman, Y., Bendikov, T., and Neumann, R., Inorg. Chem., 2015, vol. 54, no. 2, pp. 628–634. https://doi.org/10.1021/ic502541b
Park, D.R., Kim, H., Jung, J.C., Lee, S.H., and Song, I.K., Catal. Commun., 2008, vol. 9, no. 2, pp. 293–298. https://doi.org/10.1016/j.catcom.2007.06.025
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The authors declare no conflict of interest requiring disclosure in this article.
Rights and permissions
Open Access. This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Timoshkina, V.V., Yudintsev, S.V., Frenkel’, E.D. et al. V-Containing Heteropoly Acids with Keggin Structure as Precursors of Sulfide Catalysts: Regularities of the Convertion of Dibenzothiophene and Naphthalene on Nonpromoted Mo–V Catalysts. Pet. Chem. 62, 779–787 (2022). https://doi.org/10.1134/S0965544122050085
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S0965544122050085