
Abstract
A study is performed of the possibility of using gas–liquid chromatography (GLC) to determine the composition of solutions of lithium salts in aprotic dipolar solvents and solvate ionic liquids. The objects of study are solutions of lithium perchlorate and lithium trifluoromethanesulfonate in sulfolane and solvate complexes of lithium perchlorate with sulfolane obtained in two ways: direct interaction of the initial components in a given molar ratio and interaction of the components in a common solvent with its subsequent removal via evaporation. It is shown that GLC is a convenient way of determining the content of a solvating solvent in the composition of solutions and solvate ionic liquids. The presence of lithium salt in the analyzed solutions does not affect the period of retention; instead, it raises the degree of asymmetry of the chromatographic peak of the solvent and manifestation of the tailing effect. It is found that the presence of salt in the considered system also does not reduce the accuracy of determining the solvent content. The error in determining the content of solvent in solutions of lithium salts and solvate complexes by GLC is no greater than 1%.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
INTRODUCTION
The popularity of lithium-ion batteries (LIBs), the type with the highest specific energy, continues to grow in many areas of modern technology. The mass use of LIBs requires a considerable increase in their explosion and fire safety. The safety of LIBs is largely determined by the properties of the electrolyte systems that are used. Traditional liquid electrolytes are solutions of lithium salts in aprotic dipolar solvents (ADSes), usually mixtures of organic carbonates with a salt content of ⁓1 mol/L [1, 2]. The main disadvantages of such electrolyte solutions are low electrochemical stability and high danger of fire and explosion [3]. Highly concentrated non-aqueous solutions in which the content of lithium salts is ⁓3 mol/L or more are considered promising alternative liquid electrolytes for lithium–ion and lithium batteries [4–6]. At such salt concentrations, almost all solvent molecules are bound in the solvate shells of the lithium cation, and the fraction of free solvent molecules is so small that the electrolyte system is essentially not a “salt in a solvent,” but a “solvent in a salt” [4]. This new class of electrolytes, which are in the liquid phase at temperatures close to room temperature, is called solvate ionic liquids [1, 4, 5]. Solvate ionic liquids (SILs) are, by their chemical nature, solvate complexes of lithium salts with ADSes. The absence of a free solvent means that the electrochemical and thermal stability of SILs are higher than those of the initial components (solvents and lithium salts) [6–8].
Solvate ionic liquids can be synthesized in several ways: via direct interaction of a lithium salt and a solvent in a given molar ratio; interaction between lithium salts and ADSes in solutions of auxiliary solvents with their subsequent removal by evaporation at atmospheric or reduced pressures. Partial removal of the main electrolyte solvent is possible when obtaining and drying SILs, which can alter their composition. A simple and reliable procedure is therefore needed to establish the actual composition of solvate ionic liquids. Gas–liquid chromatography could be such a quantitative analytical way of determining the content of solvents in SILs.
The aim of this work was to assess the possibility of using gas–liquid chromatography to determine the composition of solvate ionic liquids. The objects of study were a 1 M solution of lithium perchlorate in sulfolane and solvate ionic liquids based on sulfolane with a 1 : 4 molar ratio of salt : solvent. The compositions of sulfolane solutions of lithium trifluoromethanesulfonate were also studied to determine the effect of the nature of the supporting salt anion via gas–liquid chromatography.
The choice of sulfolane as an object of study was due to it being a promising solvent for electrolyte systems intended for use in lithium and lithium-ion batteries based on high-potential cathode materials, since it has high anodic stability [9, 10]. However, the low cathodic stability of sulfolane limits the possibility of its use in electrolyte systems for batteries with lithium metal electrodes [11]. This disadvantage of sulfolane electrolyte systems can be overcome by increasing the concentration of lithium salts. SILs based on solvate complexes of lithium salts with sulfolane are therefore of great interest [12].
EXPERIMENTAL
Tetramethylene sulfone (sulfolane, 99%, Sigma-Aldrich) purified and dried via double vacuum distillation was used in this work. According to results from coulometric titration in Fischer’s reagent medium, the content of water was 40 ± 5 ppm. Ethanol (GOST 18300-87) was dried by double or triple treatment with freshly calcined CuSO4 to a residual water content of 0.1 wt %. Chemically pure (GOST 110097-86) acetonitrile (C2H3N) was purified via distillation and dried by treating it with 4 Å zeolites.
Lithium perchlorate (95%, Acros Organics) was purified via recrystallization from an aqueous solution and dried in vacuum at 100°С using 4 Å zeolites. Lithium trifluoromethanesulfonate (triflate) (99.995%, Sigma-Aldrich) was used without additional purification or drying.
Calibration solutions of sulfolane (SL) in acetonitrile (AN) were prepared according to volume and weight. The content of sulfolane in the calibration solutions was varied from 6.6 to 30.2 vol %. Electrolyte solutions of lithium salts in sulfolane were prepared and stored in a glove box filled with dry air (dew point, −56°С).
Solvate ionic liquids (solvate complexes of lithium perchlorate with sulfolane) of composition LiClO4⋅nSL were obtained in two ways: (1) via direct interaction of the initial components in a salt : solvent molar ratio of 1 : 4; (2) interaction of the initial components in a molar ratio of salt : solvent equal to 1 : 4 in an auxiliary solvent (anhydrous ethanol). After complete dissolution of all components, ethanol was distilled off on an IKA®RV 8 rotary evaporator (Germany) at temperatures of 30–50°C and a vacuum of 10 mm Hg.
According to results from coulometric titration in Fischer’s reagent medium, the content of water in the resulting solvate ionic liquids was 180–200 ppm. The SILs were subjected to additional azeotropic drying by adding dehydrated benzene to the system with subsequent distillation of the azeotropic benzene–water mixture on a rotary evaporator. After additional drying, the content of water in the SILs fell to 80–90 ppm.
Coulometric titration of organic solvents and electrolyte solutions in Fischer’s reagent was performed using a TitroLine®7500 KF trace automatic titrator (SI Analytics).
The thermal stability of sulfolane, lithium salts, and electrolyte solutions was determined via thermogravimetry. Thermogravimetric analysis (TGA) was performed on an upgraded MOM-1000 derivatograph (Hungary) at atmospheric pressure in a closed corundum crucible, by heating the samples at a rate of 5 K/min.
Chromatographic studies (GLC analysis) were performed on a Khromos GH-1000 gas chromatograph (Chromos Engineering, Russia) equipped with a flame ionization detector. The chromatograph was packed by steel column 2 m long with an internal diameter of 3 mm. Inerton AW with grain sizes of 0.125–0.160 mm was used as the solid support, and Carbowax 20M (10% of the support mass) was used as a liquid phase.
The velocity of the carrier gas (helium) was 60.0 ± 0.1 mL/min; that of hydrogen and air, 5.0 ± 0.1 mL/min. A sample of each analyzed sample in amounts of 1.0 ± 0.02 μL was injected into the chromatograph evaporator using a microsyringe (Hamilton, United States). Chromatography was applied to 10% solutions of the objects of study in acetonitrile. Processing results from GLC analysis (determining the area of the chromatographic peak of sulfolane) were made using a chromatograph integrator. Absolute calibration and comparisons to standard samples were used to quantitatively determine the content of sulfolane in electrolyte systems via GLC [13].
The asymmetry of the chromatographic peaks of sulfolane was estimated from the coefficients of asymmetry [14, 15], which were calculated for 10 and 50% of the peak height using the formula
where Kas is the coefficient of asymmetry of the chromatographic peak; μr is the rear half-width of the peak at 10 or 50% of the height; and μf is the half-width of the peak front at 10 or 50% of the height.
The number of theoretical plates (N) was calculated using the formula [13–16]
where tR is the period of sulfolane retention, min; μ0.5 is the sulfolane peak width at half maximum, min.
Five to six chromatograms were obtained for each sample to ensure the required accuracy. The accuracy of determining the content of sulfolane was 0.1–0.8%.
RESULTS AND DISCUSSION
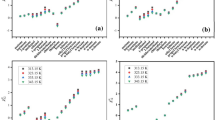
As a working parameter, temperature is of great importance in GLC analysis [15, 16]. On the one hand, the temperature of chromatography must ensure effective separation of the components of a given system. On the other, there should be no thermal decomposition during analysis. To achieve a balance between temperature and period of retention, we must choose the optimum conditions for performing GLC analysis (column oven, evaporator, and detector temperatures). In order to establish the optimum temperature parameters of chromatography, we determined the thermal stability of the initial components (lithium salts and sulfolane) and electrolyte solutions via TGA (Fig. 1).

Curves of (a, b) mass loss and (c, d) thermal effects: (1) LiClO4 (solid), (2) sulfolane, (3) 1 M solution of LiClO4 in sulfolane, (4) solvate complex LiClO4⋅4SL, (5) LiSO3CF3 (solid), and (6) 1 M solution of LiSO3CF3 in sulfolane.
Thermal decomposition of lithium salts (lithium perchlorate and triflate) in the solid state occurs at temperatures of 400–450°C. Sulfolane and solutions of lithium salts in sulfolane are thermally stable up to a temperature of 200°C (Figs. 1a, 1b). The loss of mass of electrolyte solutions and the solvate complex is accompanied by an endoeffect with a maximum temperature of ⁓250°C (Figs. 1c, 1d). This indicates the evaporation of sulfolane in the 150 to 300°C range of temperatures, rather than the thermal decomposition of electrolytes.
Results from thermogravimetric studies showed there was no thermal decomposition of individual compounds or components of electrolyte solutions when heated to 200–250°C. Temperature regimes were therefore set for chromatographic analysis: the temperature of the thermostat of the column was 230°С, the temperature of the evaporator was 250°С, and the temperature of the detector was 240°С.
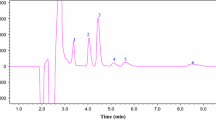
Chromatographic studies showed that regardless of the composition of the studied solutions, the period of sulfolane retention was 8.7 ± 0.1 min (Figs. 2a, 2b). However, the composition of the studied solutions affected the shape of the chromatographic peaks.

Chromatograms of acetonitrile solutions of (a) sulfolane (CSL = 10.3 vol %) and (b) solvate ionic liquid LiClO4⋅4SL (CSL = 15.79 vol %).
The chromatographic peak corresponding to sulfolane had pronounced asymmetry (Fig. 3). The lithium perchlorate in the analyzed solution raised the asymmetry of the sulfolane peaks slightly and changed their shape. Blurring of the lower part of the rear of the peak the so-called tailing of chromatographic peaks appears on the chromatograms of solutions containing an inorganic salt. As the concentration of salt rises, the width of the peak front remains unchanged, while the rear width of the peak grows (Fig. 3).

Normalized peaks of sulfolane in chromatograms of (a) acetonitrile solutions of sulfolane, 1 M solution of LiClO4 in sulfolane, solvate complex LiClO4⋅4SL and (b) sulfolane solutions of LiSO3CF3.
Compared to calibration solutions, changes in the coefficient of asymmetry of the sulfolane peak in solutions containing lithium salt are barely noticeable at the half maximum of the peak (Table 1). The largest increase in the width of the rear of the peak (μr) when a lithium salt was introduced into the analyzed solution appears at 10% of its height, while the width of the frontal part of the peak (μf) remains practically unchanged (Table 1).
Introducing the salts into the studied solutions of sulfolane lowered the number of theoretical plates (Table 1), testifying to deterioration of the chromatographic column’s efficiency of separation. The increase in the degree of asymmetry of the sulfolane peak upon raising the concentration of the electrolyte salt can be explained from the viewpoint of the kinetic theory of chromatography.
Sulfolane molecules in calibration solutions are in a free state. In solutions containing lithium salts, some of the sulfolane molecules are bound in solvate shells of the lithium cation, and some remain free. The proportion of bound sulfolane molecules grows along with the concentration of salt in the solution, and the rate of the bound sulfolane’s evaporation from the stationary phase during chromatographic analysis is slower than that of free sulfolane. The presence of salts in the studied solutions therefore increases the width of the rear area of the chromatographic peak.
The patterns of changes in the width of chromatographic peaks as the concentration of salt grew in the test solution are in good agreement with results from thermogravimetric analysis, since the rates of change in the mass of sulfolane during its evaporation differ (Fig. 1a). Blurring of the sulfolane peak does not affect the accuracy of determining its area or the reproducibility of the chromatography results.
A comparison of the ways of determining the content of sulfolane showed that absolute calibration and comparison to a standard sample gave similar results that were in good agreement with the calculated composition of the considered system (Table 2).
The content of sulfolane in electrolyte solutions, determined via gas–liquid chromatography, coincided with the calculated one within the achieved accuracy of chromatographic analysis (Table 3).
GLC analysis of the resulting solvate complexes showed that part of the main electrolyte solvent (sulfolane) evaporated during synthesis of the solvate complex of lithium perchlorate with sulfolane when using an auxiliary solvent, since the content of sulfolane in the synthesized solvate complex is lower than the calculated value (Table 3).
CONCLUSIONS
Solutions of lithium salts in sulfolane and solvate complexes lithium salt (sulfolane) were used to show that gas–liquid chromatography is a convenient way of determining their composition. The presence of lithium salt in the analyzed solutions did not affect the period of retention; instead, it increased the asymmetry of the solvent’s chromatographic peak and the tailing effect. The presence of salt in the studied system also did not reduce the accuracy of determining the solvent content. The error in determining the solvent content in solutions of lithium salts and solvate complexes by GLC was no greater than 1%.
REFERENCES
T. R. Jow, K. Xu, O. Borodin, et al., Electrolytes for Lithium and Lithium Ion Batteries, Vol. 58 of Modern Aspects of Electrochemistry (Springer Science, New York, 2014). https://doi.org/10.1007/978-1-4939-0302-3
K. Xu, Chem. Rev. 114, 11503 (2014). https://doi.org/10.1021/cr500003w
S. Chen, J. Zheng, L. Yu, et al., Joule 2, 1548 (2018). https://doi.org/10.1016/j.joule.2018.05.002
M. Watanabe, K. Dokko, K. Ueno, et al., Bull. Chem. Soc. Jpn. 91, 1660 (2018).https://doi.org/10.1246/bcsj.20180216
Y. Yamada and A. Yamada, J. Chem. Lett. 46, 1056 (2017). https://doi.org/10.1246/cl.170284
A. Nakanishi, K. Ueno, D. Watanabe, et al., J. Phys. Chem. C 123, 14229 (2019). https://doi.org/10.1021/acs.jpcc.9b02625
K. Yoshida, M. Nakamura, Y. Kazue, et al., J. Am. Chem. Soc. 133, 13121 (2011). https://doi.org/10.1021/ja203983r
Y. Yamada and A. Yamada, J. Electrochem. Soc. 162, A2406 (2015). https://doi.org/10.1149/2.0041514jes
F. Wu, H. Zhou, Y. Bai, et al., ACS Appl. Mater. Interfaces 7, 15098 (2015). https://doi.org/10.1021/acsami.5b04477
Y. Wang, L. Xing, W. Li, et al., J. Phys. Chem. Lett. 4, 3992 (2013). https://doi.org/10.1021/jz401726p
C. Li, P. Wang, S. Li, et al., ACS Appl. Mater. Interfaces 10, 25744 (2018). https://doi.org/10.1021/acsami.8b05125
J. Alvarado, M. A. Schroeder, M. Zhang, et al., J. Mater. Today 21, 341 (2018). https://doi.org/10.1016/j.mattod.2018.02.005
Quantitative Analysis Using Chromatographic Techniques, Ed. by E. Kats (Wiley, Chichester, 1987).
Experimental Methods of Chemical Kinetics, The School-Book, Ed. by N. M. Emanuel’ and M. G. Kuz’min (Mosk. Gos. Univ., Moscow, 1985), Chap. 7, p. 326 [in Russian].
K. I. Sakodynskii, V. V. Brazhnikov, S. A. Volkov, et al., Analytical Chromatography (Khimiya, Moscow, 1993) [in Russian].
B. V. Aivazov, Introduction to Chromatography (Vysshaya Shkola, Moscow, 1983) [in Russian].
Funding
This work was supported by the Russian Science Foundation as part of a joint project with the National Natural Science Foundation of China, project no. 21-43-00006 Polysulfide Ion-Solvent Complexes and Their Electrochemical Behavior in Lithium-Sulfur Batteries.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Translated by M. Drozdova
Rights and permissions
Open Access. This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sheina, L.V., Karaseva, E.V., Battalova, E.A. et al. Solvate Ionic Liquids: Assessing the Possibility of Determining the Composition by Means of Gas–Liquid Chromatography. Russ. J. Phys. Chem. 96, 1322–1327 (2022). https://doi.org/10.1134/S0036024422060231
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S0036024422060231