
Abstract
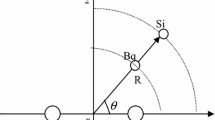
We present the results of a detailed study of the first accurate 3D ground state interaction potential energy surface (PES) of the Ne–Li2 system by quantum calculations using the coupled-cluster singles and doubles excitation approach with perturbative treatment of triple excitations [CCSD(T)]. The calculations were carried out for the frozen molecular equilibrium geometries and for an extensive range of the remaining two Jacobi coordinates, R and θ, for which a total of about 1976 points is generated for the surface. Mixed basis sets, aug-cc-pVTZ for the Ne atom and cc-pCVTZ for the Li atom, with an additional (3s3p2d2f1g) set of midbond functions are used. The ab initio points on the PES are fitted to a 96-parameter algebraic form with an average absolute error of 0.00000255% and a maximum error less than 0.00888%. The experimental results are compared with our ab initio potential surface calculations. Our PES gives more accurate results along with the experimental data.







Similar content being viewed by others


REFERENCES
D. A. Otoniel, Th. Stoecklin, and Ph. Halvick, J. Chem. Phys. 140, 084316 (2014).
T. Yuan, M. L. Yang, and H. Zhu, Computat. Theor. Chem. 1070, 88 (2015).
S. Lepp, P. C. Stancil, and A. Dalgarno, J. Phys. B: At. Mol. Opt. Phys. 35, R57 (2002).
D. H. Ross, D. Y. Sachin, K. Sabre, and S. F. Joseph, J. Chem. Phys. 144, 204121 (2016).
E. Bodo, F. A. Gianturco, and R. Martinazzo, Phys. Rep. 384, 85 (2003).
M. H. Alexander, D. E. Manolopoulos, and H. J. Werner, J. Chem. Phys. 95, 6524 (1991).
S. Brian, D. P. Magil, and E. P. David, J. Phys. Chem. A 104, 10566 (2000).
D. Zanuttini, J. Douady, E. Jacquet, E. Giglio, and B. Gervais, J. Chem. Phys. 134, 044308 (2011).
M. A. Rosenberry, R. Marhatta, and B. Stewart, Chem. Phys. Lett. 523, 15 (2012).
J. Wang, B. W. Yang, and X. D. Yang, J. Sichuan Univ. (Nat. Sci. Ed.) 43, 832 (2006).
K. A. Peterson and G. C. McBane, J. Chem. Phys. 123, 283 (2005).
E. Y. Feng, Z. Q. Wang, M. Y. Gong, and Z. F. Cui, J. Chem. Phys. 127, 284 (2007).
J. V. Lill, G. A. Parker, and J. C. Light, Chem. Phys. Lett. 89, 483 (1982).
D. T. Colbert and W. H. Miller, J. Chem. Phys. 96, 1982 (1992).
E. M. Goldfield and S. K. Gray, Comput. Phys. Commun. 98, 1 (1998).
H.-J. Werner and P. J. Knowles, MOLPRO, a Package of ab initio Programs. http://www.molpro.net.
S. F. Boys and F. Bernardi, Mol. Phys. 19, 553 (1970).
W. C. Kenneth and D. Power, J. Chem. Phys. 110, 860 (1999).
K. T. Tang and J. P. Toennies, J. Chem. Phys. 80, 3726 (1984).
G. Ihm, M. W. Cole, and F. Toigo, Phys. Rev. A 42, 5244 (1990).
M. Yasuhiko, I. Haruki, and M. Naohiko, J. Chem. Phys. 122, 154302 (2005).
ACKNOWLEDGMENTS
The author Wang thanks his parents for their encouragements. This work is supported by the Key projects of science research in University of Anhui Province (grant nos. KJ2018A0482, KJ2018A0476), the National College Students’ innovative training program (grant no. 201610383035), the College Students’ innovative training program (grant no. 2017tlxydxs082).
Author information
Authors and Affiliations
Corresponding author
Additional information
The article is published in the original.
Rights and permissions
About this article

Cite this article
Wang Yue, Gan, G., De Zhi, D. et al. Ab initio Potential Energy Surface for Ne–Li2 in Its Ground Electronic State. Russ. J. Phys. Chem. 93, 488–493 (2019). https://doi.org/10.1134/S0036024419030233
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S0036024419030233