
Abstract
Human cytomegalovirus (HCMV) DNA and proteins are often detected in malignant tumors, warranting studies of the role that HCMV plays in carcinogenesis and tumor progression. HCMV proteins were shown to regulate the key processes involved in tumorigenesis. While HCMV as an oncogenic factor just came into focus, its ability to promote tumor progression is generally recognized. The review discusses the viral factors and cell molecular pathways that affect the resistance of cancer cells to therapy. CMV inhibits apoptosis of tumor cells, that not only promotes tumor progression, but also reduces the sensitivity of cells to antitumor therapy. Autophagy was found to facilitate either cell survival or cell death in different tumor cells. In leukemia cells, HCMV induces a “protective” autophagy that suppresses apoptosis. Viral factors that mediate drug resistance and their interactions with key cell death pathways are necessary to further investigate in order to develop agents that can restore the tumor sensitivity to anticancer drugs.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
Human cytomegalovirus (HCMV), like other members of the family Herpesviridae, becomes latent after primary infection and persists life-long in approximately 90% of the adult population. Only a minor part of infection carriers develop cancer, and the role of HCMV in carcinogenesis is therefore difficult to assess epidemiologically. However, HCMV DNA and proteins were often found in various tumors, including malignant glioma and prostate, breast, colorectal, and other cancers, in the past 20 years [1–7]. For example, the nonstructural IE1/IE2 and structural pp65 proteins of HCMV were detected in approximately 75% of primary tumors or lymph node metastases in breast cancer [8]. Because data on the HCMV presence in glioblastomas are discrepant, a recent study analyzed the results from 645 articles, which described 9444 clinical samples [9]. Immunohistochemistry was observed to reliably detect the HCMV proteins in tumors (84.2%), while viral nucleic acids were often undetectable by PCR.
The role of HCMV infection in cancer is most important to understand. Is the virus merely a passenger in tumor cells, or does it play a certain role? If the latter is true, what its role is? Various mechanisms whereby viruses lead to tumorigenesis were studied, including oncogene expression, mutations, epigenetic processes, chronic inflammation, and distorted metabolism of infected cells [10]. More than 200 proteins are produced by HCMV, and only a minor part of them is essential for virus replication. The majority of viral proteins act to change the cell behavior [11]. HCMV gene products and especially those expressed early in the virus life cycle are capable of regulating the processes related to hallmarks of cancer, as many virology studies showed [12]. While HCMV as an oncogenic factor that facilitates cell malignant transformation just came into focus [10, 13, 14], its oncomodulatory properties, that is, the ability to promote tumor progression, are generally recognized [15–17].
Hanahan and Weinberg [18, 19] formulated the hallmarks of cancer, which are expressed at various stages of tumor development. The set includes maintenance of a cell proliferative potential, lack of contact growth inhibition, resistance to cell death, unlimited proliferation, activation of angiogenesis and metastasis, and resistance to or inhibition of the host immune system. The inflammatory processes in tissues that form the tumor microenvironment was recently identified as a crucial element in tumor progression and metastasis [20]. The changes are possibly based not only on genetic mutations, but also on regression (loss of specialized functions by cells), epigenetic alterations that affect gene expression, and roles played by microbiota and neuronal signaling [21, 22].
Mutations resulting from certain processes of DNA damage and repair were long believed to be a main cause of cancer because they are capable of activating cell oncogenes and eventually triggering cell malignant transformation. Substantial changes are now introduced into the concept. The main new idea is that cancer is not only a genetic disorder, but also a metabolic one. The following findings are a matter of intense discussion. Seyfried et al. [23, 24] showed that dramatic metabolic changes occur in early cell transformation to facilitate higher energy supply, to change the type of energy production, and to switch metabolic pathways to syntheses of the macromolecules that are necessary for cell growth and division. Otto Warburg hypothesized in the 1920s that mitochondrial dysfunction underlies the majority of cancers and that oxidative phosphorylation changes to aerobic glycolysis in the course of cell malignant transformation. The hypothesis is further developed now [25–27]. HCMV was shown to reprogram infected cells towards Warburg-like metabolism [28–30]. However, it should be noted that the Warburg effect is currently considered as a decoupling of mitochondrial respiratory activity and glycolysis rather than a distortion of this activity [31].
Mass mutations of many genes, rather than activation of single critical oncogenes, are observed in the majority of tumors. Random mutations caused by reactive oxygen species and nitrogen radicals should activate not only the genes that promote tumor growth, but also those that prevent it. A series of strictly directional steps should be involved in the conversion of a precancer cell to a cancer cell [32] to trigger the cell survival mechanisms and to suppress the cell death mechanisms. Random mutations seem unlikely to ensure the process. At the same time, viruses are programmed to facilitate similar processes in infected cells to ensure long-term persistence of the latent virus as well as virus replication [10]. A latent virus is inactive in terms of reproduction of virus particles, but still produces oncomodulatory proteins. Moreover, mutant HCMV strains, which are incapable of efficient replication in transformed cells, are predominantly found in tumors. This circumstance can explain why viral DNA is not always detectable by PCR. However, viral proteins can be involved in oncomodulatory and carcinogenic processes.
Cancer stem cells and the tumor microenvironment are currently recognized as important factors in tumor behavior [33, 34]. It was demonstrated that stem cells are especially sensitive to HCMV and provide a reservoir for its persistence and reactivation. Expression of viral genes in stem cells increases the likelihood of mutations [35], activates virtually all signaling pathways important for carcinogenesis, and causes critical metabolic alterations [36, 37], thus converting stem cells to tumor-initiating cells [38]. Although only a minor part of cells in a tumor are actually infected with the virus, viral proteins released from stem and microenvironmental stromal cells affect tumor behavior and aggressiveness. The fact explains why infection of all cells of a tumor is not essential for oncomodulation. Exosomes are now thought to play an important role in cell-to-cell communication. Secretion of viral proteins and genetic material as exosome components by infected cells is a possible mechanism whereby HCMV exerts a systemic effect on uninfected cells [39].
Thus, cell oncomodulation is based on the fact that various signal transduction pathways are altered by HCMV to accelerate cell proliferation, to block cell death, to trigger angiogenesis, to increase cell motility and adhesion, and to produce a proinflammatory microenvironment. A combination of these properties increases the malignant properties of the tumor. HCMV binding to cell receptors initiates the first wave of signal transduction modulation, which is followed by the effects of virion components and effects of viral gene products [14].
One of the most important unsolved problems is that infected cells survive and overcome the death programs induced by anticancer agents. The problems are considered here with the example of apoptosis, which is the cell death program best understood now. The inhibitory effect of HCMV on apoptosis in cancer cells promotes further tumor progression and decreases the sensitivity to anticancer therapy. The resistance to apoptotic stimuli facilitates uncontrolled survival and expansion of cancer cells, and accumulation of mutations, and further malignant progression. Loss of sensitivity to anticancer drugs and immune-mediated destruction in cancer cells is thought to be a main cause of unfavorable outcomes in cancer.
CYTOMEGALOVIRUS SUPPRESSES APOPTOSIS IN CANCER CELLS
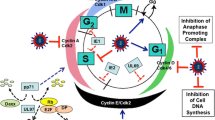
Extrinsic and intrinsic pathways are recognized in the activation of apoptosis. The extrinsic pathway depends on the interaction of external signaling molecules with cell receptors, which either triggers or blocks apoptosis. The intrinsic apoptosis pathway is mediated by intracellular stress signals, including those due to anticancer therapy. The final step of either pathway is activation of specific effector proteases: caspases 3, 6, and 7. Caspases recognize the critical cell substrates, and destruction of the substrates leads to morphological and functional changes associated with apoptosis. In fact, each of the pathways can be utilized by HCMV to suppress not only apoptosis, but also other cell death mechanisms. Mimicry is a strategy used by HCMV, which codes for false ligands and false receptors to evade immune mechanisms [40]. HCMV activates caspase inhibitors, can oppositely affect activities of cell proteins of the BCL-2 family, synthesizes homologs of these proteins, influences the cell microRNA repertoire, and produces its own microRNAs to control the cell defense mechanisms [17, 41, 42].
The viral inhibitor of caspase activation (vICA) is encoded by the HCMV gene UL36 and protects cells from apoptosis triggered through certain death receptors, such as TNFR1, FAS/CD95, and the Trail receptors. A direct interaction of vICA with the caspase prodomain inhibits the activation of caspase 8, which acts as an intermediate in the activation cascade of effector caspases 3, 6, and 7 [43]. Thus, vICA is functionally similar to cell protease inhibitors, although there are no homologous sequences and structural similarity between them. A cell caspase-8 inhibitor (FLIP) is additionally activated in infected cells, and the immediate-early 2 (IE2) protein of HCMV is involved in this activation [44].
Various strategies are used by HCMV to prevent the cell response to proapoptotic signals and are changed in the course of infection. Apart from a direct effect on caspase, an indirect effect on apoptosis is possible to occur upon ligand binding with respective receptors. For example, the interaction of TNFα with TNFRI activates the signal transduction pathways that eventually induce the nuclear factor κB (NF-κb) and c-JUN kinase, which are two main regulators of cell proliferation, apoptosis, and differentiation. HCMV was shown to induce TNFα expression and to simultaneously change the localization of a TNFα receptor (TNFRI), thus differentially affecting the surrounding uninfected cells and protecting the infected cells from apoptosis [45]. The ligand regulation aimed at increasing activity is possible to consider as a tactics that allows the virus to evade apoptosis induced by infiltrating immune cells with proper surface receptors. Alterations of receptor exposure on the cell membrane excludes TNFα-induced Jun kinase activity in infected cells.
Another mechanism of the HCMV effect on the receptor-mediated signaling pathway of apoptosis activation is possibly associated with the viral protein encoded by UL-144. The product of the gene is an ortholog of TNFR and acts as its competitor. While TNFR binds various ligands (LIGHT, LTα, BTLA, CD160, and HCMV gD) and can activate or inhibit the immune response, pUL144 binds only BTLA to inhibit B- and T-cell activation [46]. In addition, pUL144 was shown to act as a potential activator of NF-kB-induced transcription of the gene for chemokine CCL22, which binds to its receptor on suppressor T cells and blocks the immune response [47].
HCMV has a long reproduction period and broad cell tropism and, accordingly, possesses many mechanisms to regulate the antivirus response, to inhibit apoptosis in various infected cells [48], and to help infected cells to evade the immune system [49]. Products of certain viral genes inhibit apoptosis induced by cytotoxic T lymphocytes (CTLs) and natural killer (NK) cells. For example, US3, which is in the unique short region of the viral genome, codes for a protein that binds main histocompatibility complex (MHC) class I molecules and retains them in the endoplasmic reticulum. The products of US2 and US11 cause translocation of the molecules into the cytosol, where the molecules are degraded. The US6 viral protein blocks antigenic peptide transport into the endoplasmic reticulum [50]. Antigen presentation with MHC class II molecules can also be altered by HCMV [51, 50]. Glycoproteins encoded by UL16, UL18, and UL40 of the unique long region of the viral genome help infected cells to avoid recognition by the NK cells, which perform nonspecific protective functions in early infection [52–54]. HCMV stimulates the cytokines IL-10 and TGF-β in infected cancer cells [55] and produces a functional analog of IL-10. The UL111A HCMV gene codes for cmvIL-10, which is an ortholog of human IL-10; binds with the cell IL-10 receptor; activates the STAT3 transcription factor; and exerts a potent immunosuppressive effect, in particular, by inhibiting expression of MCH class I and class II proteins. The latency-associated isoform LAcmvIL-10 inhibits the miR-92a cell microRNA, thus activating the CCL8 chemokine and eventually inhibiting CD4+ T cells [56].
Thus, the antiapoptotic mechanisms of HCMV that involve the ligand–receptor signal transduction pathways may be part of the strategy of virus evasion of immune clearance.
Apoptosis induction in response to various cytotoxic agents usually proceeds through the intrinsic signaling pathway, which is associated with altered permeability of the mitochondrial membrane and release of cytochrome C and other proapoptotic factors from the intermembrane space into the cytoplasm. Cytochrome C acts together with APAF1 and procaspase 9 to form an apoptosome in the cytoplasm. This reaction cascade activates caspase 3 [57]. The apoptosis-inducing factor (AIF) is another proapoptotic factor released from mitochondria and acts as a main effector of its own apoptosis pathway. AIF triggers the apoptosis-related reactions in a caspase-independent manner, and, consequently, its apoptosis pathway is also known as the caspase-independent pathway [58].
Proteins of the BCL-2 family play a central role in controlling the mitochondrial apoptosis pathway. Antiapoptotic family members (BCL-2, BCL-XL, BCL-W, MCL-1, and A1) stabilize the mitochondrial membrane and thus prevent cytochrome C release, while proapoptotic family members (BAX, BAD, BAK, BIK, PUMA, NOXA, and BID) destabilize the membrane and facilitate cytochrome C release. The tumor suppressor protein p53 is involved in regulating the activities of BAX and other key apoptosis proteins at the levels of transcription and direct interactions in the cytoplasm [59, 60]. The exact mechanism whereby the BCL-2 family proteins play their roles in the process remains unclear, but certain experimental findings indicate that some of the proteins directly affect the permeability of the mitochondrial membrane through the formation of megapores or their inhibition by other proteins of the family [61]. It was demonstrated that BCL-XL directly binds with APAF1 and blocks its ability to activate procaspase 9 [57]. On the other hand, cytosolic cytochrome C interacts with BCL-XL to impair the apoptosome function [62]. Whichever the mechanism, when the balance is shifted towards antiapoptotic proteins, cells acquire a higher resistance to apoptosis induced by cytotoxic agents or various physiological stimuli, such as endoplasmic reticulum stress, distorted calcium homeostasis, DNA damage, and lysosomal or oxidative stress [63]. Note additionally that mitochondria do not directly mediate the extrinsic apoptosis pathway in the majority of cells, but may regulate the extrinsic pathway via a feedback loop and, in particular, activation of the Bid proapoptotic protein by initiator caspase 8 [64].
Apart from the proapoptotic and antiapoptotic proteins of the BCL-2 family, the PI3K/AKT/mTOR signaling pathway modulates the mitochondrial apoptosis pathway triggered by various stimuli. Overexpression or activation of protein kinase B (also known as AKT) is observed in many malignancies, where AKT serves as a main mediator of cell survival [65]. HCMV may affect AKT to modulate the intrinsic apoptosis pathway. The IE1 and EI2 HCMV proteins were shown to directly activate phosphatidylinositol 3-kinase (PI3K), which acts as the first component in the AKT activation cascade [66, 67]. AKT activation leads to phosphorylation and subsequent repression of several proteins, such as BAD, caspase 9, and transcription factors targeting the proapoptotic protein-coding genes [68, 69]. In addition, AKT can regulate the cell survival by phosphorylating the inhibitor of NF-κB (IκB), and its phosphorylation leads to translocation of NF-κB into the nucleus and activation of the promoters of antiapoptotic genes [70]. For example, BCL-XL expression is upregulated in HCMV-infected endothelial cells, and higher BCL-2 expression is observed in infected rectal cancer cells [68]. Neuroblastoma cells with persistent HCMV infection display higher BCL-2 activity and a lower sensitivity to the cytotoxic drugs etoposide and cisplatin as compared with uninfected cells [71]. Constitutively active kinase AKT protects the cell from PTEN-mediated apoptosis [72]. The HCMV immediate early protein IE1 activates NF-kB and AKT to increase expression of A20, which is a regulatory gene and codes for a protein that protects cells from apoptosis induced by various stimuli in a cell-specific manner [73].
To enter the cell, HCMV utilizes its glycoproteins to bind to integrins, the epidermal growth factor receptor (EGRF), and the platelet-derived growth factor receptor (PDGRF) [74, 75]. Upregulated EGFR expression was observed in a number of malignancies [76]. The HCMV proteins pUL135 and pUL138 were found to finely regulate the EGFR level on the infected cell surface [77]. UL138-mediated stimulation of EGFR expression in latently infected cells indicates that the EGFR regulation contributes to the oncomodulatory properties of HCMV [78]. PDGFR is expressed to a low level in normal cells and overexpressed in many tumors. Virus binding to the receptors leads to their phosphorylation, which activates the PI3K signaling pathway and induces mitogen-activated protein kinase (MAPK). Viral glycoprotein B (gB) acts together with gH to cause atypical AKT activation and eventually suppresses apoptosis. In particular, HCMV gB-induced AKT activation was shown to promote monocyte survival [79, 80]. Thus, the inhibition of apoptosis starts as early as the virus comes to interact with the cell.
To prevent apoptosis, HCMV may activate the RAS/RAF/MEK/ERK signaling pathway, which is the best-known MAPK cascade and plays a crucial role in cell proliferation, differentiation, and survival [81, 82]. The pathway may be affected by the HCMV-encoded microRNA miR-US5-2, which inhibits the EGF-mediated pathways that include MEK/ERK and PI3K. Both PI3K and MEK/ERK pathways are important for cell survival and proliferation. It was observed that miR-US5-2 regulates UL138 expression by decreasing EGFR signal transduction during infection [83]. Many mechanisms are utilized by miR-US5-2 to block proliferation of various cells. HCMV-encoded microRNAs play an important role in modulating the cell signaling pathways not only to change the cell environment, but also to control expression of viral proteins [83].
The viral mitochondrial inhibitor of apoptosis (vMIA) is encoded by UL37x1 and can also be used by HCMV to affect the intrinsic apoptosis pathway. This transmembrane protein is localized in mitochondria and inhibits activation of mitochondrial megapores, like the antiapoptotic proteins of the BCL-2 family. HeLa cells that express vMIA are resistant to doxorubicin-induced apoptosis. Although vMIA is structurally similar to BCL-XL and exerts a similar effect on apoptosis, there is no sequence homology between the two proteins. However, like BCL-XL, vMIA binds and sequesters the proapoptotic BAX protein on the outer mitochondrial membrane [84]; vMIA acts as a broad-range apoptosis inhibitor and suppresses not only the intrinsic apoptosis pathway, but also the pathway induced by the death domains. In addition, the inhibitor of BAK oligomerization (vIBO) is encoded by the HCMV open reading frame m41.1 and acts together with vMIA to completely suppress mitochondrial apoptosis [85], be it caspase dependent or independent.
A unique mechanism is associated with the untranslated HCMV RNA β2.7, which interacts with the inner mitochondrial membrane and protects the cell from apoptosis [86]. It is of interest that HCMV additionally upregulates the HSATII RNA, which is expressed to a high level in certain cancers and cancer cell lines. Its induction was shown to involve simultaneously two immediate-early viral proteins, IE1 and IE2 [87]. The HSATII induction is observed in both infected and cancer cells, indicating that both of the processes depend on HSATII-activated regulatory mechanisms. High-level transcription of the HSATII satellite is therefore possible to consider as an oncomodulatory viral factor. The HSATII RNA was additionally found to affect innate immunity by inducing IL-6 and TNFα [88].
The regulation of cell processes with microRNAs has certain advantages over the regulation that involves viral proteins. In contrast to viral proteins, microRNAs lack immunogenicity, take up less space in the genome, and can rapidly start acting in various cells. To help the virus to survive, viral microRNAs regulate the cell genes involved in immune defense, block apoptosis, and cooperate with viral and cell proteins involved in the same processes [89]. HCMV microRNAs were found in astrocytic tumors [90], glioblastomas [91], and extracellular vesicles from serum samples of infected children [92]. It was observed that HCMV miR UL-112 downregulates expression of MICB, a ligand of NK cell receptor, and thus protects the infected cell from NK cell-induced apoptosis [93]. The function of miR UL-112 was studied experimentally. To study the functions of other HCMV microRNAs that promote the survival of infected cells, genes for microRNAs involved in apoptosis were searched for by bioinformatics methods and the microRNAs were compared with homologous cell microRNAs with known functions [94]. Because latent or low-level infection is important for expression of carcionogenic and oncomodulatory properties, the microRNAs that help HCMV to establish and maintain latency are of particular interest in the total viral microRNA repertoire. The latent virus genome is still broadly expressed, although to a very low level, according to transcription profiling [95]. HCMV microRNAs presumably act as key regulators of protein expression in the latent period [95].
Of the 26 known HMCV microRNAs, only miR-UL70-3p and miR-UL148D efficiently bind to the 3′-UTRs of MOAP1, PHAP, and ERN1. All of the three genes play a role in apoptosis. MOAP1 (modulator of apoptosis 1) is involved in mitochondrial and receptor-mediated apoptosis by facilitating BAX activation [96, 97] and transmission of apoptotic signals induced by TNFα and TRAIL [98]. PHAP1 controls the formation of the CytC + Casp9 + Apaf-1 apoptosome [99]. ERN1 (endoplasmic reticulum-to-nucleus signaling 1) helps to induce apoptosis via endoplasmic reticulum stress [100]. Thus, the intrinsic apoptosis pathway is regulated mostly by viral microRNAs.
Many proapoptotic proteins of the BCL-2 family are controlled by p53-related transcription factors, which are activated in response to conventional anticancer drugs. As is known, p53 arrests the cell cycle and can trigger apoptosis due to accumulating DNA lesions. The response depends on the cell type. The product of the ATM (ataxia telangiectasia) gene is involved in the process that relates detection of DNA damage with p53 upregulation. A p53 tetramer functions as a transcription factor and binds to consensus sequences in 5′-UTRs of target genes, which include the above genes ВAX, PUMA, and NOXA. Upregulation of p53 increases expression of its target genes and consequently leads to apoptosis. However, the respective proteins are not the only effectors of p53-mediated apoptosis.
Activation of p53 in non-small cell lung cancer cells by anticancer agents inhibits signal transduction through EGFR and stimulates generation of reactive oxygen species (ROS). ROS induce cytochrome C release from mitochondria and possibly act by distorting ion transfer into mitochondria, suggesting an additional mechanism for p53-dependent apoptosis induction [101].
HCMV IE2 was shown to bind with p53 and to suppress its transactivation function, which is important for the induction of apoptosis [70]. Expression of IE1 alone activates the PI3K/AKT signaling pathway and simultaneously decreases the levels of major suppressor proteins of the Rb and p53 families in cultured glioblastoma cells [12, 102]. However, there are grounds to believe that other members of the p53 family, such as p63 and p73, should act together with p53 to induce apoptosis [103, 104].
CYTOMEGALOVIRUS CAN DECREASE THE CHEMOTHERAPY SENSITIVITY BY SHIFTING THE ISOFORM BALANCE OF THE p73 TRANSCRIPTION FACTOR
The p73 gene codes for a p53-related protein. It was demonstrated in the past years that p73 acts as a main determinant of cell sensitivity to chemotherapy and that mutant p53 forms inhibitory complexes with p73 to confer resistance to anticancer agents [105, 106].
Proteins of the p53 family are similar in structural organization and consequently activate the same target genes in experiments in vitro, the set including BAX, PUMA, NOXA, BAD, BIK, and the gene for the p53AIP1 mitochondrial membrane protein [107]. Each of the proteins certainly performs its specific function in the cell. In spite of the structural similarity of the p53-related proteins and their genes, there is a substantial functional difference between the proteins. For example, mutations of the p73 gene are extremely rare (0.5%) in human tumors, while the p53 gene is mutated in more than 50% of tumors. Moreover, p73 overexpression is observed in many tumors and can hardly agree with a p73 role as a tumor suppressor.
The structural organization of the p73 gene explains the phenomenon and allows expression of both tumor suppressor and carcinogenic p73 isoforms. Several different protein products result from alternative splicing of the 5′ and 3′ mRNA ends and the use of alternative promoters. N-terminally truncated isoforms (DNp73) lack a functional transactivation (TA) domain and inhibit transcriptional activities of both p73 with the full-size TA domain and its homolog p53 by forming heterotetramers with them. In addition, the isoform transcribed from the second (internal) promoter possesses its own, although weak, transcriptional activity towards the genes whose products have antiapoptotic potential, including caspase 2S, heat shock protein 70 (HSP70), etc. [108, 109]. It was found also that the DNp73 isoform occurs directly in sites of DNA damage, interacts with the 53BP1 sensory protein, and inhibits ATM activation and subsequent p53 phosphorylation, thus affecting p53-dependent apoptosis [110]. Thus, certain p73 isoforms possess tumor suppressor proteins, while other isoforms act as carcinogenic proteins, and the isoform balance determines the cell fate [111–113].
In some cases, HCMV can change the balance between the tumor suppressor and carcinogenic p73 isoforms, and a predominance of the truncated isoforms facilitates the anticancer drug resistance [113–115]. HCMV infection was observed to activate the molecular pathways that induce expression of the E2F1 transcription factor [115], which is capable of both activating and inhibiting p73 [116]. The truncated p73 isoforms prevent TAp73-dependent transcription of miR-205, which controls the E2F1 accumulation and acts as a negative regulator of the BCL2 antiapoptotic protein and the ATP-binding cassette transporters ABCA-2 and ABCA-5, which are responsible for elimination of cytotoxic anticancer agents from the cell [117, 118]. Thus, lack of the inhibitory effect of miR-205 can mediate the resistance that is observed in HCMV-infected cells and is associated with higher levels of DNp73 and E2F1.
The DNp73 protein downregulates miR-205 expression and leads to an accumulation of E2F1, which contributes to the induced apoptosis resistance by directly interacting with TAp73 and inhibiting its transcriptional activity towards the MDM2 and BAX gene promoters. Higher E2F1 amounts were even found to induce TAp73 degradation [116]. Another mechanism that determines the resistance of HCMV-infected cells to cytotoxic drugs is related to the fact that the N-terminally truncated p73 isoforms upregulate expression of the ABCB1/MDR1 (multidrug resistance 1) transmembrane protein by blocking the inhibitory effect of p53 on the MDR1 promoter [119].
Transcriptional activity of p73 sensitizes cells to death receptor-mediated apoptosis in a caspase-dependent manner. Both FAS gene transcription and FAS expression on the cell membrane are induced when p73 is activated via cell treatment with cisplatin or ectopic overexpression. HCMV infection inhibits p73-dependent sensitization to apoptosis. The mechanism of this inhibition is associated with upregulation of the N-terminally truncated DNp73 isoform [120]. Because FAS-dependent apoptosis is possibly involved in the mechanism whereby cytotoxic lymphocytes eliminate tumor cells in vivo, the HCMV contribution to tumor progression may be substantial.
Normally, TAp73 increases in amount and activity in cell stress and the increase eventually activates the processes that suppress tumor growth, for example, by arresting the growth, inducing apoptosis, maintaining the genome integrity, and preventing anchorage-independent cell growth [111, 112, 121]. In addition to acting as a transcription factor, TAp73 can affect apoptosis by directly interacting with mitochondria, like p53 [122]. The truncated isoforms transcribed from the second internal promoter control p73 and p53 activities and are controlled by p73 and p53. A highly efficient p53/p73-binding element is found in the promoter and acts to increase transcription from the promoter, thus creating a feedback loop. A p53/p73-dependent activation of the internal promoter would contradict the proapoptotic role that p53 and p73 play in the response to DNA damage, if rapid and selective degradation of the truncated isoform were not induced simultaneously [123]. It is therefore a pressing problem to identify the key virus-induced factors that affect the balance of the suppressor and carcinogenic p73 isoforms and to study their association with cell molecular pathways involved in the response of cancer cells to therapy.
It is of interest that the observed shift in TA/DN isoforms is not associated with changes in isoform mRNA levels in HCMV-infected cancer cell cultures, possibly because transcription of both isoforms in cancer cells is substantially higher than in normal tissues [113]. Transcription of the DNp73 isoform is possibly maximal, which is consistent with hypomethylation of the internal promoter in lung cancer [124] and neuroblastoma [125] cells. Promoter methylation was found to redirect the NRF2 transcription factor, which regulates expression of the p73 gene, from the first to the second promoter in breast cancer cells [126]. The virus is therefore most likely to act at the protein level to ensure a predominance of the DNp73 isoform in cancer cells.
Various mechanisms can be responsible for selective degradation of the truncated isoform in genotoxic stress. A key role in regulating the process is possibly played by the ring finger domain of PIR2 ubiquitin ligase, which is a transcriptional target of TAp73 [127, 128]. PIR2 binds with both of the p73 isoforms to selectively stabilize TAp73 and degrade DNp73, thus shifting their balance. Two other HECT-type ubiquitin ligases, WWP2 and WWP1, may also affect the balance of the p73 isoforms. WWP2 ubiquitinates and targets the TAp73 isoform for proteasomal degradation. On the other hand, WWP2 can form a heterodimer with WWP1, and their heteromerization leads to degradation of the DNp73 isoform. In genotoxic stress, phosphatase PPM1G acts as a switch to change the balance between the monomeric and heteromeric states of WWP1 and WWP2 and, eventually, the balance between the p73 isoforms [129]. It remains unclear whether HCMV utilizes these pathways to improve infected cell survival.
A ubiquitin-independent mechanism, which is regulated by the polyamine metabolism system, can also mediate DNp73 degradation. Its activation involves independently three transcription factors: c-JUN, JUNB, and FOSB, which belong to the family of the activator protein 1 (AP-1) family and function in genotoxic stress. The factors inhibit transcription of the gene for acetylpolyamine oxidase (APAO), which is a catabolic enzyme of the polyamine cycle, and thereby change the balance of individual polyamines and eventually lead to the formation of functionally active antizyme 1 (Az1), which inhibits synthesis of compounds of the class. Az1 is a small inhibitory protein and binds with DNp73 to target it for proteasomal degradation [130]. HCMV was found to utilize this pathway to establish the doxorubicin resistance by increasing transcription of spermidine/spermine N1‑acyltransferase (SSAT), which is another catabolic enzyme. There is data also that direct inhibition of polyamine oxidases with their specific inhibitor MDL725.27 increases the sensitivity of virus-infected monocytic leukemia cells to doxorubicin-induced apoptosis. The TAp73/DNp73 balance shifts towards the full-size isoform in this case, and the effect of HCMV on SSAT is insufficient for protecting the truncated isoform from degradation [113].
CYTOMEGALOVIRUS IS INVOLVED IN REGULATING AUTOPHAGY IN CANCER CELLS
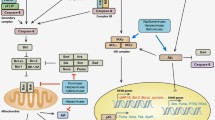
Little is still known as to how HCMV affects autophagy in infected cells and whether HCMV plays a role in cancer cell resistance to autophagic cell death. The term “autophagy” was first coined in 1963. Two Nobel Prizes were awarded for the discovery of autophagy and its mechanisms, to C.R. de Duve, G.E. Palade, and A. Claude in 1974 and Y. Ohsumi in 2016. Autophagy serves to eliminate and process structurally or functionally impaired organelles and intracellular components and is activated in critical periods of cell life, such as exposure to adverse changes in environmental or internal conditions or pathogen invasion. The opinion that autophagy is an exclusively protective response seems questionable. Available data indicate that autophagic cell death is necessary to occur during individual growth and development [131]. Moreover, autophagy is considered a mechanism of programmed cell death along with apoptosis and other cell death programs [132, 133]. There is a substantial overlap between the signaling pathways that regulate apoptosis and autophagy, leading to their competition or unidirectional interaction, but the relationship between autophagy and apoptosis is still poorly understood [134]. Comparing the two phenomena, apoptosis is possible to characterize as cell suicide and autophagy, as self-digestion [135]. Autophagy is hereafter understood as macrophagy, which is an autophagy type that is the most common; involves the formation of autophagosomes; is regulated by a broad range of special proteins (ATGs) and signaling pathways; and plays a role in cell development, differentiation, senescence, and death [136].
Several consecutive steps occur in the process of autophagy and produce double-membrane vesicles known as the autophagosomes and then, after fusion with lysosomes, autolysosomes, where the contents are degraded. More than 40 autophagy-related (ATG) genes were identified to date, and 16–18 of them are highly conserved among eukaryotes and consequently associated with a main autophagy mechanism [137]. An induction is the earliest step and requires expression of Unc-51-like kinase 1 (ULK1) [138] and activation of AMP-activated protein kinase (AMPK), which regulates the energy balance at the cell and organismic levels to maintain energy homeostasis [139]. ULK1 phosphorylates many targets important for the initiation of autophagy, including Beclin1, which is encoded by BECN1. Beclin1 plays an important role in regulating the formation of the autophagosome membrane, autophagosome maturation, and material transport [140]. The microtubule-associated protein light chain 3 (LC3) cycle is activated somewhat later to produce two forms, cytosolic LC3-I and membrane-associated LC3-II. Detection and testing of LC3-II is widely used to measure the autophagy activity because the LC3-II amount directly correlates with the autophagosome number and is indicative of the extent of mature autophagosome generation [141].
Viruses that take advantage of autophagy developed means to activate or to suppress autophagy during evolution [142, 143]. The term “virophagy” was introduced to describe how autophagy affects the viral components and selectively degrades virus particles, and virophagy was observed to play an important role in replication of certain viruses (coronaviruses, poliovirus, hepatitis C virus, Dengue virus, etc.) [144]. On the other hand, the induction of virophagy is thought to counteract infection at various levels and may provide a useful approach to fighting viruses, including the new coronavirus SARS-CoV-2 [145]. There is still no common opinion about the proviral or antiviral role of autophagy, possibly because the interaction of viruses and their target ce lls during the infection process is still poorly understood. (See reviews by Leonardi et al. [146] and Liang et al. [147] for data on the association of autophagy with viruses of various taxonomic groups.)
Discrepant results were obtained in studies of the HCMV effect on autophagy. Ambiguous data were similarly obtained in attempts to understand whether autophagy is a proviral or antiviral process [148–150]. Zimmermann et al. [151] studied the effect of HCMV on human fibroblasts and observed that the SQSTM1/p62 autophagy receptor colocalizes with HCMV capsids in infected cell nuclei. The observation indicates that autophagy already takes place at early (nuclear) stages of HCMV virion production. In addition, membrane-associated LC3-II and several autophagy receptors were detected in extracellular HCMV virions, indicating that autophagosome membranes are involved in the formation of the secondary envelope of a virion. The effect of autophagy on HCMV replication was studied using a mutant virus that expressed a dominant negative variant of cell protease ATG4B, which is necessary for LC3 cleavage and subsequent conjugation with the membrane [151]. Replication of the virus genome and virus release in cells infected with the mutant virus were more intense than in cells infected with control HCMV strains. Autophagy was concluded to act as an antiviral process in HCMV infection. On the other hand, a block of autophagy inhibits IE2 expression and virus replication [152]. The finding agrees with the HCMV strategy that is aimed at sustaining host cell survival to ensure successful virus replication. Thus, competing autophagy processes may be activated in cells with lytic HCMV infection. Virus-induced autophagy can prevent HCMV DNA replication and the release of virus progeny in early infection. At later stages, the virus utilizes the autophagosome membranes to produce virions, and autophagy may facilitate an increase in virus progeny and the spreading of infection.
It should be noted that certain data indicate that autophagy is suppressed by viral proteins. For example, the HCMV proteins TRS1 and IRS1 interact with the cell protein Beclin1 and inhibit autophagy, and the involvement of both of the viral proteins is necessary for complete autophagy inhibition [148]. There is also evidence that HCMV IE2 is involved in regulating autophagy because an increase in IE2 expression after infection is associated with an increase in autophagy markers, such as LC3II and ATG3, while a decrease in IE2 expression is accompanied by inhibition of autophagy [152].
Cells used to obtain the above data were sensitive to HCMV and capable of maintaining lytic infection. It is of interest to study autophagy in latently HCMV-infected cells, which are the most abundant in the body after primary or acute infection, and cancer cells. HCMV-infected THP-1 human leukemia cells were used as a model to study the problem [153]. It was found that latent infection is established 5 days after infecting THP-1 cells. Autophagosome counting by electron microscopy showed that the number of vesicles per cell was two or three in uninfected (control) cells; increased to 10–13 one day after infection; and reached 80 and 30 on days 5 and 9 after infection, respectively. The mRNA levels of autophagy-related genes (LC3, Beclin1, and Atg5) slightly differed during infection, but similarly increased on day 1, reached their maximum on day 5, and decreased by days 7–9 to a higher-than-normal level. The results showed that HCMV is capable of inducing autophagy in infected THP-1 leukemia cells, but its potential to induce autophagy decreases once latency is established. A comparison of data on autophagy inhibition in lytically infected cells [154] and autophagy induction in THP-1 cells gave grounds to assume that autophagy is involved in the establishment of latency. Autophagy induction in U251 glioma cells utilized the AMPK/Akt/mTOR pathway, led to the establishment of latency in cells infected with herpes simplex virus 1 (HSV-1), and ensured their survival [155]. It remains unclear why data on the effect of viruses on autophagy are so discrepant. Discrepancies may arise because the effect of infection differs between cells of different origins and different stages of the virus life cycle, such as active infection, the establishment of latency, and virus reactivation.
AUTOPHAGY AND CANCER CELL RESISTANCE TO CHEMOTHERAPY
A dual role in carcinogenesis is possible for autophagy: autophagy may protect cancer cells from damage and thus facilitate tumor progression, or autophagic cell death may be induced to suppress the tumor growth [156, 157].
Studies of the role that autophagy may play as a mechanism that affects anticancer drug resistance led to the conclusion that two autophagy types are necessary to recognize: lethal (toxic) autophagy causes increased cell death, while protective autophagy is presumably a main cause of cancer cell survival, metastasis, and resistance to chemotherapy [158]. Lethal autophagy was demonstrated with glioblastoma and hepatocellular carcinoma cells [159, 160]; protective autophagy, with pancreatic, colorectal, and lung cancer cells [161–163]. Autophagy was found to increase the resistance of cancer cells, including stem (tumor-initiating) cells, to conventional chemotherapies in the majority of cases [164–166].
To restore the cancer cell sensitivity to chemotherapy, it is necessary to understand the mechanisms that regulate autophagy and apoptosis, but the mechanisms are still unclear. However, a regulation of autophagy and apoptosis via the PI3K/AKT/mTOR pathway was described to restore the sensitivity in hepatocellular carcinoma cells [167]. Inhibition of autophagy restored the sensitivity of osteosarcoma cells and induced apoptosis via inactivation of the VEGFR2/STAT3/BCL-2 signaling pathway [168].
ROLES OF AUTOPHAGY AND APOPTOSIS IN CHEMOTHERAPY RESISTANCE OF CYTOMEGALOVIRUS-INFECTED CANCER CELLS
Acute and latent virus infections are capable of regulating autophagy and thus affecting the cancer cell resistance to anticancer therapy [169]. Certain viruses increase the infected cell survival by suppressing apoptosis via an autophagy-dependent mechanism. Various mechanisms are utilized by viruses: inhibition of ATG5 or Beclin1 and induced phosphorylation of STAT3, which is an important transcription factor involved in tumor cell survival [146].
The THP-1 leukemia cell line is sensitive to the anticancer drug doxorubicin, but acquires antibiotic resistance as a result of HCMV infection. Cell survival correlates with suppression of apoptosis pathways, including a decrease in activities of initiator and effector caspases 8, 9, and 3, and prevention of DNA breaks [113]. Given that autophagy intensifies in HMCV-infected THP-1 cells [153], it is possible to assume that autophagy is involved in the development of doxorubicin resistance in leukemia cells and that induction of autophagy prevents apoptotic cell death. The development of the resistance was observed to involve the enzymes that catabolize biogenic polyamines [113] and the PI3K/AKT/mTOR molecular pathway [170]. Doxorubicin resistance restores in response to mTOR inhibitors possibly because mTOR binds with ULK1 and suppresses its interaction with AMPK, thus inactivating ULK1 and inhibiting autophagy [171].
Data on the possibility to overcome the resistance of cancer cells supported more than 50 preclinical and clinical studies of autophagy-targeting agents. Galluzzi et al. [172] reviewed in detail many pharmacological approaches designed to modulate autophagy in various pathological conditions at various steps of the process. In spite of their great potential, treatments aimed at modulating autophagy have still not been approved for clinical use as of yet. Several approved drugs (rapamycin, chloroquine, and hydroxychloroquine) are known to activate or inhibit autophagy in fact, but were not tested comprehensively enough to be used for the purpose. A recent computer analysis of 1565 FDA-approved drugs identified three drugs (Ponatinib, Simeprevir, and Nilotinib) as agents potentially suitable for inhibiting autophagy and stimulating apoptosis in the tumor microenvironment [173]. However, uninfected cell lines were used to obtain the respective data.
Based on the above data that HCMV is present in the latent state in many tumors, autophagy markers are expressed in latently infected cells (THP-1), and viral proteins are involved in regulating autophagy, it is of interest to further study the role that viral factors may play in cancer cell resistance in the cases where HCMV infection is latent or HCMV is reactivated in cancer cells. Such data are essential for designing agents that can restore the cancer cell sensitivity to anticancer drugs.
Further research should be performed with due regard to the fact that autophagy and apoptosis can act synergistically to cause programmed cell death or antagonistically to ensure cell survival [174]. The effect of autophagy on the cell sensitivity may therefore depend on the pathways that come to interact with each other. Taking advantage of the therapeutic potential of autophagy modulators, new anticancer drugs will be developed and cancer cell sensitivity to chemotherapeutics will be increased.
CONCLUSIONS
The above data clearly demonstrate that HCMV intervenes in the processes that regulate apoptosis and autophagy, which control cell survival and death. Even latent, HCMV infection increases the anticancer drug resistance of cancer cells. Metabolic and redox-dependent processes play a crucial role in the resistance. Several examples are known where virus-associated resistance was overcome using low-molecular-weight inhibitors of enzymes involved in metabolic and signaling pathways. More ample data were obtained using uninfected cells. Thus, this research field has a potential for intense development and will help to design means to improve the efficacy of anticancer therapy.
REFERENCES
Cobbs C.S., Harkins L., Samanta M., Gillespie G.Y., Bharara S., King P.H., Nabors L.B., Cobbs C.G., Britt W.J. 2002. Human cytomegalovirus infection and expression in human malignant glioma. Cancer Res. 62, 3347–3350.
Samanta M., Harkins L., Klemm K., Britt W.J., Cobbs C.S. 2003. High prevalence of human cytomegalovirus in prostatic intraepithelial neoplasia and prostatic carcinoma. J. Urol. 170, 998–1002. https://doi.org/10.1097/01.ju.0000080263.46164.97
Harkins L.E., Matlaf L.A., Soroceanu L., Klemm K., Britt W.J., Wang W., Bland K.I.,Cobbs C.S. 2010. Detection of human cytomegalovirus in normal and neoplastic breast epithelium. Herpesviridae. 1, 8. https://doi.org/10.1186/2042-4280-1-8
Taher C., de Boniface J., Mohammad A.A., Religa P., Hartman J., Yaiw K.C., Frisell J., Rahbar A., Söderberg-Naucler C. 2013. High prevalence of human cytomegalovirus proteins and nucleic acids in primary breast cancer and metastatic sentinel lymph nodes. PLoS One. 8, e56795. https://doi.org/10.1371/journal.pone.0056795
Chen H.P., Chan Y.J. 2014. The oncomodulatory role of human cytomegalovirus in colorectal cancer: implications for clinical trials. Front. Oncol. 4, 314.https://doi.org/10.3389/fonc.2014.00314
Paradowska E., Jabłońska A., Studzińska M., Wilczyński M., Wilczyński J.R. 2019. Detection and genotyping of CMV and HPV in tumors and fallopian tubes from epithelial ovarian cancer patients. Sci. Rep. 9, 19935. https://doi.org/10.1038/s41598-019-56448-1
Athanasiou E., Gargalionis A.N., Boufidou F., Tsakris A. 2021. The association of human herpesviruses with malignant brain tumor pathology and therapy: two sides of a coin. Int. J. Mol. Sci. 22, 2250. https://doi.org/10.3390/ijms22052250
Touma J., Liu Y., Rahbar A., Pantalone M.R., Almazan N.M., Vetvik K., Söderberg-Naucler C., Geisler J., Sauer T. 2021. Detection of human cytomegalovirus proteins in paraffin-embedded breast cancer tissue specimens—a novel, automated immunohistochemical staining protocol. Microorganisms. 9, 1059. https://doi.org/10.3390/microorganisms905105
Peredo-Harvey I., Rahbar A., Söderberg-Nauclér C. 2021. Presence of the human cytomegalovirus in glioblastomas-a systematic review. Cancers (Basel). 13, 5051.https://doi.org/10.3390/cancers13205051
Soliman S.H.A., Orlacchio A., Verginelli F. 2021. Viral manipulation of the host epigenome as a driver of virus-induced oncogenes. Microorganisms. 9, 1179. https://doi.org/10.3390/microorganisms9061179
Söderberg-Nauclér C. 2008. HCMV microinfections in inflammatory diseases and cancer. J. Clin. Virol. 41, 218–223.
Cobbs C.S., Soroceanu L., Denham S., Zhang W., Kraus M.H. 2008. Modulation of oncogenic phenotype in human glioma cells by cytomegalovirus IE1-mediated mitogenicity. Cancer Res. 68, 724–730.
Cobbs C.S. 2011. Evolving evidence implicates cytomegalovirus as a promoter of malignant glioma pathogenesis. Herpesviridae. 2, 10.
Herbein G. 2018. The human cytomegalovirus, from oncomodulation to oncogenesis. Viruses. 10, E408. https://doi.org/10.3390/v10080408
Söderberg-Nauclér C., Geisler J., Vetvik K. 2019. The emerging role of human cytomegalovirus infection in human carcinogenesis: a review of current evidence and potential therapeutic implications. Oncotarget. 10, 4333–4347.
Blaylock R.I. 2019. Accelerated cancer aggressiveness by viral oncomodulation: new targets and newer natural treatments for cancer control and treatment. Surg. Neurol. Int. 10, 199.https://doi.org/10.25259/SNI_361_2019
Baba R.E., Herbein G. 2021. Immune landscape of CMV infection in cancer patients: from “canonical” diseases toward virus-elicited oncomodulation. Front. Immunol. 12, 730765.https://doi.org/10.3389/fimmu.2021.730765
Hanahan D., Weinberg R.A. 2000. The hallmarks of cancer. Cell. 100, 57–70. https://doi.org/ (00)81683-9https://doi.org/10.1016/S0092-8674
Hanahan D., Weinberg R.A. 2011. Hallmarks of cancer: the next generation. Cell. 144, 646–674. https://doi.org/10.1016/j.cell.2011.02.013
Colotta F., Allavena P., Sica A., Garlanda C., Mantovani A. 2009. Cancer-related inflammation, the seventh hallmark of cancer: links to genetic instability. Carcinogenesis. 30, 1073–1081. https://doi.org/10.1093/carcin/bgp127
Flavahan W.A., Gaskell E., Bernstein B.E. 2017. Epigenetic plasticity and the hallmarks of cancer. Science. 357 (6348), eaal2380.https://doi.org/10.1126/science.aal2380
Senga S.S., Grose R.P. 2021. Hallmarks of cancer–the new testament. Open Biol. 11, 200358. https://doi.org/10.1098/rsob.20.035
Seyfried T.N., Flores R.E., Poff A.M., D’Agostino D.P. 2014. Cancer as a metabolic disease: Implications for novel therapeutics. Carcinogenesis. 35, 515‒527.
Seyfried T.N., Chinopoulos C. 2021. Can the mitochondrial metabolic theory explain better the origin and management of cancer than can the somatic mutation theory? Metabolites. 11, 572. https://doi.org/10.3390/metabo11090572
Durah T., García-Romero N., Carrión-Navarro J., Madurga R., Mendivil A.O., Prat-Acin R., Garcia-Cañamaque L., Ayuso-Sacido A. 2021. Beyond the Warburg effect: oxidative and glycolytic phenotypes coexist within the metabolic heterogeneity of glioblastoma. Cells. 10, 202.https://doi.org/10.3390/cells10020202
Chen X., Yi C., Yang M.J., Sun X., Liu X., Ma H., Li Y., Li H., Wang C., He Y., Chen G., Chen S., Yu L., Yu D. 2021. Metabolomics study reveals the potential evidence of metabolic reprogramming towards the Warburg effect in precancerous lesions. J. Cancer. 12, 1563–1574. https://doi.org/10.7150/jca.54252.
Vaupel P., Multhoff G. 2021. Revisiting the Warburg effect: historical dogma versus current understanding. J. Physiol. 599, 1745–1757.https://doi.org/10.1113/JP278810
Munger J., Bajad S.U., Coller H.A., Shenk T., Rabinowitz J.D. 2006. Dynamics of the cellular metabolome during human cytomegalovirus infection. PLoS Pathog. 2, e132.
Yu Y., Clippinger A.J., Alwine J.C. 2011. Viral effects on metabolism: changes in glucose and glutamine utilization during human cytomegalovirus infection. Trends Microbiol. 19, 360–367. https://doi.org/10.1016/j.tim.2011.04.002
Williamson C.D., DeBiasi R.L., Colberg-Poley A.M. 2012. Viral product trafficking to mitochondria, mechanisms and roles in pathogenesis. Infect. Disord. Drug Targets. 12, 18–37.https://doi.org/10.2174/187152612798994948
DeBerardinis R.J., Chandel N.S. 2020. We need to talk about the Warburg effect. Nat. Metabolism. 2, 127–129.
Vogelstein B., Papadopoulos N., Velculescu V.E., Zhou S., Diaz L.A. J., Kinzler K.W. 2013. Cancer genome landscapes. Science. 339 (6127), 1546–1558. https://doi.org/10.1126/science.1235122
Hui L., Chen Y. 2015. Tumor microenvironment: sanctuary of the devil. Cancer Lett. 368, 7–13. https://doi.org/10.1016/j.canlet.2015.07.039
Bajaj J., Diaz E., Reya T.J. 2020. Stem cells in cancer initiation and progression. Cell Biol. 219, e201911053. https://doi.org/10.1083/jcb.201911053
Alonso-Álvarez S., Colado E., Moro-García M.A., Alonso-Arias R. 2021. Cytomegalovirus in haematological tumours. Front. Immunol. 12, 703256. https://doi.org/10.3389/fimmu.2021.703256
Soroceanu L., Matlaf L., Khan S., Akhavan A., Singer E., Bezrookove V., Decker S., Ghanny S., Hadaczek P., Bengtsson H., Ohlfestb J., Luciani-Torresa M.G., Harkinsf L., Perryg A., Guoc H., Soteropoulosc P., Charles S., Cobbs C.S. 2015. Cytomegalovirus immediate-early proteins promote stemness properties in glioblastoma. Cancer Res. 75, 3065–3076.https://doi.org/10.1158/0008-5472.CAN-14-3307
Teo W.H., Chen H.P., Huang J.C., Chan Y.J. 2017. Human cytomegalovirus infection enhances cell proliferation, migration and upregulation of EMT markers in colorectal cancer-derived stem cell-like cells. Int. J. Oncol. 51, 1415–1426. https://doi.org/10.3892/ijo.2017.4135
Li J.-W., Yang D. Yang D., Chen Z. Miao J, Liu W., Wang X., Qiu Z., Jin M., Shen Z. 2017. Tumors arise from the excessive repair of damaged stem cells. Med. Hypotheses. 102, 112–122. https://doi.org/10.1016/j.mehy.2017.03.005
Zakaria S., Arakelyan A., Palomino R.A.Ñ., Fitzgerald W., Vanpouille C., Lebedeva A., Schmitt A., Bomsel M., Brittg W., Margolis L. 2018. Human cytomegalovirus-infected cells release extracellular vesicles that carry viral surface proteins. Virology. 524, 97–105.
McSharry B.P., Avdic S., Slobedman B. 2012. Human cytomegalovirus encoded homologs of cytokines, chemokines and their receptors: roles in immunomodulation. Viruses. 4, 2448–2470.https://doi.org/10.3390/v4112448
Fu M., Gao Y., Zhou Q., Zhang Q., Peng Y., Tian K., Wang J., Zheng X. 2014. Human cytomegalovirus latent infection alters the expression of cellular and viral microRNA. Gene. 536 (2), 272–278.
Buzdin A.A., Artcibasova A.V., Fedorova N.E., Suntsova M.V., Garazha A.V., Sorokin M.I., Allina D., Shalatonin M., Borisov N.M., Zhavoronkov A.A., Kovalchuk I., Kovalchuk O., Kushch A.A. 2016. Early stage of cytomegalovirus infection suppresses host microRNA expression regulation in human fibroblasts. Cell Cycle. 15, 3378–3389. https://doi.org/10.1080/15384101.2016.1241928
Skaletskaya A., Bartle L.M., Chittenden T., A. Louise McCormick A.L., Mocarski E.S., Victor S. Goldmacher V.S. 2001. A cytomegalovirus-encoded inhibitor of apoptosis that suppresses caspase-8 activation. Proc. Nat. Acad. Sci. U. S. A. 98, 7829–7834.
Chiou S.H., Yang Y.P., Lin J.C., Hsu C.H., Jhang H.C., Yang Y.T., Lee C.H., Ho L.L., Hsu W.M., Ku H.H., Chen S.J., Chen S.S., Chang M.D., Wu C.W., Juan L.J. 2006. The immediate early 2 protein of human cytomegalovirus (HCMV) mediates the apoptotic control in HCMV retinitis through up-regulation of the cellular FLICE-inhibitory protein expression. J. Immunol. 177, 6199–6206. https://doi.org/10.4049/jimmunol.177.9.6199
Baillie J., Sahlender D.A., Sinclair J.H. 2003. Human cytomegalovirus infection inhibits tumor necrosis factor alpha (TNF-α) signaling by targeting the 55-kilodalton TNF-α receptor. J. Virol. 77, 7007–7716.
Bitra A., Nemcovicová I., Picarda G., Doukov T., Wang J., Chris A., Benedict C.A., Zajonc D.M. 2019. Structure of human cytomegalovirus UL144, an HVEM orthologue, bound to the B and T cell lymphocyte attenuator. J. Biol. Chem. 294, 10519–10529.
Poole E., King C.A., Sinclair J.H., Alcami A. 2006. The UL144 gene product of human cytomegalovirus activates NFkB via a TRAF6-dependent mechanism. EMBO J. 25, 4390–4399.
Andoniou C.E., Degli-Esposti M.A. 2006. Insights into the mechanisms of CMV-mediated interference with cellular apoptosis. Immun. Cell Biol. 84, 99–106.
Cox M., Kartikasari A.E.R., Gorry P.R., Flanagan K.L., Plebanski M. 2021. Potential impact of human cytomegalovirus infection on immunity to ovarian tumours and cancer progression. Biomedicines. 9, 351. https://doi.org/10.3390/biomedicines9040351
Craig R.R., Salcedo S.P., Gorvel J.-P.E. 2006. Pathogen–endoplasmic-reticulum interactions: in through the out door. Nat. Rev. Immunol. 6, 137–147.
Johnson D.C., Hegde N.R. 2002. Inhibition of the MHC class II antigen presentation pathway by human cytomegalovirus. Curr. Top. Microbiol. Immunol. 269, 101–115.
Johnsen J.I., Baryawno N., Söderberg-Nauclér C. 2011. Is human cytomegalovirus a target in cancer therapy? Oncotarget. 2, 1329–1338.
Wilkinson G.W., Tomasec P., Stanton R.J., Armstrong M., Prod’homme V., Aicheler R., McSharry B.P., Rickardsa C.R., Cochrane D., Llewellyn-Lacey S., Wang E.C., Griffin C.A., Davison A.J. 2008. Modulation of natural killer cells by human cytomegalovirus. J. Clin. Virol. 41, 206–212.
Berry R., Watson G.M., Jonjic S., Degli-Esposti M.A., Rossjohn J. 2020. Modulation of innate and adaptive immunity by cytomegaloviruses. Nat. Rev. Immunol. 20, 113–127.https://doi.org/10.1038/s41577-019-0225-5
Dziurzynski K., Wei J., Qiao W., Hatiboglu M.A., Kong L.Y., Wu A., Wang Y., Cahill D., Levine N., Prabhu S., Rao G., Sawaya R., Heimberger A.B. 2011. Glioma-associated cytomegalovirus mediates subversion of the monocyte lineage to a tumor propagating phenotype. Clin. Cancer Res. 17, 4642–4649. https://doi.org/10.1158/1078-0432.CCR-11-0414
Chinta P., Garcia E.C., Tajuddin K.H., Akhidenor N., Davis A., Faure L., Spencer J.V. 2020. Control of cytokines in latent cytomegalovirus infection. Pathogens. 9, 858.https://doi.org/10.3390/pathogens9100858
Würstle M.L., Rehm M.A. 2014. Systems biology analysis of apoptosome formation and apoptosis execution supports allosteric procaspase-9 activation. J. Biol. Chem. 289, 26277–26289. https://doi.org/10.1074/jbc.M114.590034
Hevlera J.F., Chiozzia R.Z., Cabrera-Oreficec A., Brandtc U., Arnoldc S., Hecka A.J.R. 2021. Molecular characterization of a complex of apoptosis-inducing factor 1 with cytochrome c oxidase of the mitochondrial respiratory chain. Proc. Natl. Acad. Sci. U. S. A. 118, e2106950118.https://doi.org/10.1073/pnas.2106950118
Laptenko O., Prives C. 2006. Transcriptional regulation by p53: one protein, many possibilities. Cell Death Differ. 13, 951–961. https://doi.org/10.1038/sj.cdd.4401916
Geng Y., Walls K.C., Ghosh AP., Akhtar R.S., Klocke B.J., Roth K.A. 2010. Cytoplasmic p53 and activated Bax regulate p53-dependent, transcription-independent neural precursor cell apoptosis. J. Histochem. Cytochem. 58, 265–275. https://doi.org/10.1369/jhc.2009.954024
Dadsena S., King L.E., García-Sáez A.J. 2021. Apoptosis regulation at the mitochondria membrane level. Biochim. Biophys. Acta Biomembranes. 1863, 183716.https://doi.org/10.1016/j.bbamem.2021.183716
Bertini I., Chevance S., Del Conte R., Lalli D., Turano P. 2011. The anti-apoptotic Bcl-xL protein, a new piece in the puzzle of pytochrome C interactome. PLoS One. 6 (4), e18329. https://doi.org/10.1371/journal.pone.0018329
Singh R., Letai A., Sarosiek K. 2019. Regulation of apoptosis in health and disease: the balancing act of BCL-2 family proteins. Nat. Rev. Mol. Cell. Biol. 20 (3), 175–193. https://doi.org/10.1038/s41580-018-0089-8
Kantari C., Walczak H. 2011. Caspase-8 and Bid: caught in the act between death receptors and mitochondria. Biochim. Biophys. Acta. 1813, 558–563.https://doi.org/10.1016/j.bbamcr.2011.01.026
Song M., Bode A.M., Dong Z., Lee M.H. 2019. AKT as a therapeutic target for cancer. Cancer Res. 79, 1019–1031. https://doi.org/10.1158/0008-5472.CAN-18-2738
Johnson R.A., Wang X., Ma X.L., Huong S.M., Huang E.S. 2001. Human cytomegalovirus upregulates the phosphatidylinositol 3-kinase (PI3-K) pathway: inhibition of PI3-K activity inhibits viral replication and virus-induced signaling. J. Virol. 75, 6022–6032. https://doi.org/10.1128/JVI.75.13.6022-6032.2001
Yu Y., Alwine J.C. 2002. Human cytomegalovirus major immediate-early proteins and simian virus 40 large T antigen can inhibit apoptosis through activation of the phosphatidylinositide 3'-OH kinase pathway and the cellular kinase Akt. J. Virol. 76, 3731–3738. https://doi.org/10.1128/jvi.76.8.3731-3738.2002
Cinatl J., Jr., Vogel J.U., Kotchetkov R., Doerr H.W. 2004. Oncomodulatory signals by regulatory proteins encoded by human cytomegalovirus: A novel role for viral infection in tumor progression. FEMS Microbiol. Rev. 28, 59–77.https://doi.org/10.1016/j.femsre.2003.07.005
Kamada H., Nito C., Endo H., Chan P.H. 2007. Bad as a converging signaling molecule between survival PI3-K/Akt and death JNK in neurons after transient focal cerebral ischemia in rats. J. Cereb. Blood Flow Metab. 27, 521–533.https://doi.org/10.1038/sj.jcbfm.9600367
Paulus C., Nevels M. 2009. The human cytomegalovirus major immediate-early proteins as antagonists of intrinsic and innate antiviral host responses. Viruses. 1, 760–779.https://doi.org/10.3390/v1030760
Cinatl J.Jr., Cinatl J., Vogel J.U., Kotchetkov R., Driever P.H., Kabickova H., Kornhuber B., Schwabe D., Doerr H.W. 1998. Persistent human cytomegalovirus infection induces drug resistance and alteration of programmed cell death in human neuroblastoma cells. Cancer Res. 58, 367‒372.
Porta C., Paglino C., Mosca A. 2014. Targeting PI3K/Akt/mTOR signaling in cancer. Front. Oncol. 4, 64. https://doi.org/10.3389/fonc.2014.00064
Abbasi A., Forsberg K., Bischof F. 2015. The role of the ubiquitin-editing enzyme A20 in diseases of the central nervous system and other pathological processes. Front. Mol. Neurosci. 8, 21.https://doi.org/10.3389/fnmol.2015.00021
Soroceanu L., Akhavan A., Cobbs C.S. 2008. Platelet-derived growth factor-alpha receptor activation is required for human cytomegalovirus infection. Nature. 455, 391–395. https://doi.org/10.1038/nature07209
Kabanova A., Marcandalli J., Zhou T., Bianchi S., Baxa U., Tsybovsky Y., Lilleri D., Silacci-Fregni C., Foglierini M., Fernandez-Rodriguez B.M., Druz A., Zhang B., Geiger R., Pagani M., Sallusto F., Kwong P.D., Corti D., Antonio Lanzavecchia A., Perez L. 2016. Platelet-derived growth factor-alpha receptor is the cellular receptor for human cytomegalovirus gHgLgO trimer. Nat. Microbiol. 8, 16082. https://doi.org/10.1038/nmicrobiol.2016.82
Lindsey S., Langhans S.A. 2015. Epidermal growth factor signaling in transformed cells. Int. Rev. Cell Mol. Biol. 314, 1–41. https://doi.org/10.1016/bs.ircmb.2014.10.001
Buehler J., Zeltzer S., Reitsma J., Petrucelli A., Umashankar M., Rak M., Zagallo P., Schroeder J., Terhune S., Goodrum F. 2016. Opposing regulation of the EGF receptor: a molecular switch controlling cytomegalovirus latency and replication. PLoS Pathog. 12 (5), e1005655.https://doi.org/10.1371/journal. ppat.1005655
Goodrum F., Reeves M., Sinclair J., High K., Shenk T. 2007. Human cytomegalovirus sequences expressed in latently infected individuals promote a latent infection in vitro. Blood. 110, 937–945. https://doi.org/10.1182/blood-2007-01-070078
Cojohari O., Peppenelli M.A., Chan G.C. 2016. Human cytomegalovirus induces an atypical activation of Akt to stimulate the survival of short-lived monocytes. J. Virol. 90, 6443–6452. https://doi.org/10.1128/JVI.00214-16
Mahmud J., Miller M.J., Altman A.M., Chan G.C. 2020. Human cytomegalovirus glycoprotein-initiated signaling mediates the aberrant activation of Akt. J. Virol. 94 (16), e00167-20.https://doi.org/10.1128/JVI.00167-20
Filippakis H., Spandidos D.A., Sourvinos G. 2010. Herpesviruses: hijacking the Ras signaling pathway. Biochim. Biophys. Acta. 1803, 777–785. https://doi.org/10.1016/j.bbamcr.2010.03.007
Barbosa R., Acevedo L.A., Marmorstein R. 2021. The MEK/ERK network as a therapeutic target in human cancer. Mol. Cancer Res. 19, 361–374. https://doi.org/10.1158/1541-7786.MCR-20-0687
Hancock M.H., Mitchell J., Goodrum F.D., Nelson J.A. 2020. Human cytomegalovirus miR-US5-2 downregulation of GAB1 regulates cellular proliferation and UL138 expression through modulation of epidermal growth factor receptor signaling pathways. mSphere. 5 (4), e00582-20. https://doi.org/10.1128/mSphere.00582-20
Maa J., Edlichb F., Bermejoa G.A., Norrisb K.L., Youleb R.J., Tjandraa N. 2012. Structural mechanism of Bax inhibition by cytomegalovirus protein vMIA. Proc. Natl. Acad. Sci. U. S. A. 109, 20901–20906. www.pnas.org/cgi/doi/10.1073/pnas.1217094110.
Pauleau A.-L., Larochette N., Giordanetto F., Scholz S.R., Poncet D., Zamzami N., Goldmacher V.S., Kroemer G. 2007. Structure–function analysis of the interaction between Bax and the cytomegalovirus-encoded protein vMIA. Oncogene. 26, 7067–7080. https://doi.org/10.1038/sj.onc.1210511
Reeves M.B., Davies A.A., McSharry B.P., Wilkinson G.W., Sinclair J.H. 2007. Complex I binding by a virally encoded RNA regulates mitochondria-induced cell death. Science. 316, 1345–1348. https://doi.org/10.1126/science.1142984
Nogalski M.T., Solovyov A., Kulkarni A.S., Desai N., Oberstein A., Levine A.J., Ting D.T., Shenk T., Greenbaum B.D. 2019. A tumor-specific endogenous repetitive element is induced by herpesviruses. Nat. Commun. 10, 90. https://doi.org/10.1038/s41467-018-07944-x
Tanne A., Muniz L.R., Puzio-Kuter A., Leonova K.I., Gudkov A.V., Ting D.T., Monasson R., Cocco S., Levine A.J., Bhardwaj N., Greenbaum B.D. 2015. Distinguishing the immunostimulatory properties of noncoding RNAs expressed in cancer cells. Proc. Natl. Acad. Sci. U. S. A. 112, 15154–15159. https://doi.org/10.1073/pnas.1517584112
Gottwein E., Cullen B.R. 2008. Viral and cellular micro RNAs as determinants of viral pathogenesis and immunity. Cell Host Microbe. 3, 375–387. https://doi.org/10.1016/j.chom.2008.05.002
Deshpande R.P., Panigrahi M., Chandrasekhar Y.B.V.K., Babu P.P. 2018. Profiling of microRNAs modulating cytomegalovirus infection in astrocytoma patients. Neurol. Sci. 39, 1895–1902. https://doi.org/10.1007/s10072-018-3518-8
Liang Q., Wang K., Wang B., Cai Q. 2017. HCMV-encoded miR-UL112-3p promotes glioblastoma progression via tumour suppressor candidate 3. Sci. Rep. 7, 44705.
Zhang J., Huang Y., Wang Q., Ma Y., Qi Y., Liu Z., Deng J., Ruan Q. 2020. Levels of human cytomegalovirus miR-US25-1-5p and miR-UL112-3p in serum extracellular vesicles from infants with HCMV active infection are significantly correlated with liver damage. Eur. J. Clin. Microbiol. Infect. Dis. 39, 471–481.https://doi.org/10.1007/s10096-019-03747-0
Stern-Ginossar N., Elefant N., Zimmermann A., Wolf D.G., Saleh N., Biton M., Horwitz E., Prokocimer Z., Prichard M., Hahn G., Goldman-Wohl D., Greenfield C., Yagel S., Hengel H., Altuvia Y., Marqalit H., Mandelboim O. 2007. Host immune system gene targeting by a viral miRNA. Science. 317, 376–381.https://doi.org/10.1126/science.1140956
Babu S.G., Pandeya A., Verma N., Shukla N., Kumar R.V., Saxena S. 2014. Role of HCMV miR-UL70-3p and miR-UL148D in overcoming the cellular apoptosis. Mol. Cell. Biochem. 393, 89–98. https://doi.org/10.1007/s11010-014-2049-8
Diggins N.L., Skalsky R.L., Hancock M.H. 2021. Regulation of latency and reactivation by human cytomegalovirus miRNAs. Pathogens. 10, 200.https://doi.org/10.3390/pathogens10020200
Fu N.Y., Sukumaran S.K., Yu V.C. 2007. Inhibition of ubiquitin-mediated degradation of MOAP-1 by apoptotic stimuli promotes Bax function in mitochondria. Proc. Natl. Acad. Sci. U. S. A. 104, 10051–10056. www.pnas.orgcgidoi10.1073pnas.0700007104.
Tan C.T. Zhou Q.-L., Su Y.-C., Fu N.Y. Chang H.-C., Tao R.N., Sukumaran S.K., Baksh S., Tan Y.-J., Sabapathy K., Yu C.-D., Yu V.C. 2016. MOAP-1 mediates Fas-induced apoptosis in liver by facilitating tBid recruitment to mitochondria. Cell Rept. 16, 174–185.
Tan K.O., Fu N.Y., Sukumaran S.K., Chan S.L., Kang J.H., Chen B.S., Yu V.C. 2005. Map-1, is a mitochondrial effector of bax. Proc. Natl. Acad. Sci. U. S. A. 50, 14623–14628.
Monian P., Jiang X. 2012. Clearing the final hurdles to mitochondrial apoptosis: regulation post cytochrome C release. Exp. Oncol. 34, 185–191.
Tabas I., Ron D. 2011. Integrating the mechanism of apoptosis induced by endoplasmic reticulum stress. Nat. Cell. Biol. 13, 184–190. https://doi.org/10.1038/ncb0311-184
Zhang Y., Han C.Y., Duan F.G., Fan X.-X., Yao X.-J., Parks R.J., Tang Y.-J., Wang M.-F., Liu L., Tsang B.K., Leung E.L.-H. 2019. p53 sensitizes chemoresistant non-small cell lung cancer via elevation of reactive oxygen species and suppression of EGFR/PI3K/AKT signaling. Cancer Cell Int. 19, 188. https://doi.org/10.1186/s12935-019-0910-2
Hwang F.S., Zhang Z., Cai H., Huang D.Y., Huong S.M., Cha C.Y., Huang E.S. 2009. Human cytomegalovirus IE1-72 protein interacts with p53 and inhibits p53-dependent transactivation by a mechanism different from that of IE2-86 protein. J. Virol. 83, 12388–12398.
Alexandrova E.M., Moll U.M. 2012. Role of p53 family members p73 and p63 in human hematological malignancies. Leuk. Lymphoma. 53, 2116–2129.https://doi.org/10.3109/10428194.2012.684348
Rozenberg J.M., Zvereva S., Dalina A., Blatov I., Zubarev I., Luppov D., Bessmertnyi A., Romanishin A., Alsoulaiman L., Kumeiko V., Kagansky A., Melino G., Ganini C., Barlev N.A. 2021. The p53 family member p73 in the regulation of cell stress response. Biol. Direct. 16, 23.
Lunghi P., Costanzo A., Mazzera L., Rizzoli V., Massimo Levrero M., Bonati A. 2009. The p53 family protein p73 provides new insights into cancer chemosensitivity and targeting. Clin. Cancer Res. 15, 6495–6502.
Hong B., Prabhu V.V., Zhang S., van den Heuvel A.P.J., Dicker D.T., Kopelovich L, El-Deiry W.S. 2014. Prodigiosin rescues deficient p53 signaling and anti-tumor effects via up-regulating p73 and disrupting its interaction with mutant p53. Cancer Res. 74, 1153–1165.https://doi.org/10.1158/0008-5472.CAN-13-0955
Pietsch E.C., Sykes S.M., McMahon S.B., Murphy M.E. 2008. The p53 family and programmed cell death. Oncogene. 27, 6507–6521.
Toh W.H., Logette E., Corcos L., Sabapathy K. 2008. TAp73b and DNp73b activate the expression of the pro-survival caspase-2S. Nucl. Acids Res. 36, 4498–4509.
Tanaka Y., Kameoka M., Itaya A., Ota K., Yoshihara K. 2004. Regulation of HSF1-responsive gene expression by N-terminal truncated form of p73. Biochem. Biophys. Res. Commun. 317, 865–872.https://doi.org/10.1016/j.bbrc.2004.03.124
Wilhelm M.T., Rufini A., Wetzel M.K., Tsuchihara K., Inoue S., Tomasini R., Itie-Youten A., Wakeham A., Arsenian-Henriksson M., Melino G., Kaplan D.R., Miller F.D., Mak T.W. 2010. Isoform specific p73 knockout mice reveal a novel role for delta Np73 in the DNA damage response pathway. Genes Dev. 24, 549–560.
Vinogradskaya G.R. 2013. The p73 protein in carcinogenesis and in response to anticancer therapy. Vopr. Onkol. 59 (2), 42–48.
Engelmann D., Meier C., Alla V., Putzer B.M. 2015. A balancing act: orchestrating amino-truncated and full-length p73 variants as decisive factors in cancer progression. Oncogene. 34, 4287–4299.
Fedorova N.E., Chernoryzh Y.Y., Vinogradskaya G.R., Emelianova S.S., Zavalyshina L.E., Yurlov K.I., Zakirova N.F., Verbenko V.N., Kochetkov S.N., Kushch A.A., Ivanov A.V. 2019. Inhibitor of polyamine catabolism MDL72.527 restores the sensitivity to doxorubicin of monocytic leukemia THP-1 cells infected with human cytomegalovirus. Biochimie. 158, 82–89. https://doi.org/10.1016/j.biochi.2018.12.012
Allart S., Martin H. Detraves C., Terrasson J., Caput D., Davrinche C. 2002. Human cytomegalovirus induces drug resistance and alteration of programmed cell death by accumulation of DN-p73. J. Biol. Chem. 277, 29063–29068.
Emel’yanova S.S., Chernoryzh Ya.Yu., Yurlov K.I., Fedorova N.E., Ivanov A.V., Kochetkov S.N., Verbenko V.N., Kushch A.A., Vinogradskaya G.R. 2018. Involvement of transcription factors E2F1 and P73 in the formation of resistance to doxorubicin in THP-1 tumor cells infected with human cytomegalovirus, Tsitologiya. 60, 527–530.
Ozaki T., Okoshi R., Ono S., Kubo N., Nakagawara A. 2009. Deregulated expression of E2F1 promotes proteolytic degradation of tumor suppressor p73 and inhibits its transcriptional activity. Biochem. Biophys. Res. Commun. 387, 143–148.
Alla V., Kowtharapu B.S., Engelmann D., Emmrich S., Schmitz U., Steder M., Pulzer B.M. 2012. E2F1 confers anticancer drug resistance by targeting ABC transporter family members and Bcl-2 via the p73/DNp73-miR205 circuitry. Cell Cycle. 11, 3067–3078.
Ferrari E., Gandellini P. 2020. Unveiling the ups and downs of miR-205 in physiology and cancer: transcriptional and post-transcriptional mechanisms. Cell Death Dis. 11, 980. https://doi.org/10.1038/s41419-020-03192-4
Vilgelm A., Wei J.X., Piazuelo M.B., Washington M.K., Prassolov V., El-Rifai W., Zaika A. 2008. ΔNp73α regulates MDR1 expression by inhibiting p53 function. Oncogene. 27, 2170–2176.https://doi.org/10.1038/sj.onc.1210862
Terrasson J., Allart S., Martin H., Lulé J., Haddada H., Caput D., Davrinche C. 2005. P73-Dependent apoptosis through death receptor: impairment by human cytomegalovirus infection. Cancer Res. 65, 2787–2794.
Logotheti S., Richter C., Murr N., Spitschak A., Marquardt S., Pützer B.M. 2021. Mechanisms of functional pleiotropy of p73 in cancer and beyond. Front. Cell. Dev. Biol. 9, 737735. https://doi.org/10.3389/fcell.2021.737735.34650986
Liu T., Roh S.E., Woo J.A., Ryu H., Kang D.E. 2013. Cooperative role of RanBP9 and P73 in mitochondria-mediated apoptosis. Cell Death Dis. 4, e476.
Maisse C., Munarriz E., Barcaroli D., Melino G., De Laurenzi V. 2004. DNA damage induces the rapid and selective degradation of the DNp73 isoform, allowing apoptosis to occur. Cell Death Differ. 11, 685–687.
Daskalos A., Logotheti S., Markopoulou S., Xinarianos G., Gosney J.R., Kastania A.N., Zoumpourlis V., Field J.K., Liloglou T. 2011. Global DNA hypomethylation-induced DeltaNp73 transcriptional activation in non-small cell lung cancer. Cancer Lett. 300, 79–86.
Casciano I., Banelli B., Croce M., Allemanni G., Ferrini S., Tonini G.P., Ponzoni M., Romani M. 2002. Role of methylation in the control of DeltaNp73 expression in neuroblastoma. Cell Death Differ. 9, 343–345.
Lai J., Nie W., Zhang W., Wang Y., Xie R., Wang Y., Gu J., Xu J., Song W., Yang F., Huang G., Cao P., Guan X. 2014. Transcriptional regulation of the p73 gene by Nrf-2 and promoter CpG methylation in human breast cancer. Oncotarget. 5, 6909–6922.
Sayan B.S., Yang A.L., Conforti F., Tucci P., Piro M.C., Browne G.J. 2010. Differential control of TAp73 and DNp73 protein stability by the ring finger ubiquitin ligase PIR2. Proc. Natl. Acad. Sci. U. S. A. 107, 12877–12882.
Taebunpakul P., Sayan B.S., Flinterman M., Klanrit P., Gäken J., Odell E.W., Melino G., Tavassoli M. 2012. Apoptin induces apoptosis by changing the equilibrium between the stability of TAp73 and ΔNp73 isoforms through ubiquitin ligase PIR2. Apoptosis. 17, 762–776. https://doi.org/10.1007/s10495-012-0720-7
Chaudhary N., Maddika S. 2014. WWP2-WWP1 ubiquitin ligase complex coordinated by PPM1G maintains the balance between cellular p73 and DNp73 levels. Mol. Cell. Biol. 34, 3754–3764.
Bunjobpol W., Dulloo I., Igarashi K., Concin N., Matsuo K., Sabapathy K. 2014. Suppression of acetylpolyamine oxidase by selected AP-1 members regulates DNp73 abundance: mechanistic insights for overcoming DNp73-mediated resistance to chemotherapeutic drugs. Cell Death Differ. 21, 1240–1249.
Cao W., Li J., Yang K., Cao D. 2021. An overview of autophagy: mechanism, regulation and research progress. Bull Cancer. 108, 304–322.https://doi.org/10.1016/j.bulcan.2020.11.004
Yu L., Wan F., Dutta S., Welsh S., Liu Z., Freundt E., Baehrecke E.H., Lenardo M. 2006. Autophagic programmed cell death by selective catalase degradation. Proc. Natl. Acad. Sci. U. S. A. 103, 4952–4957.https://doi.org/10.1073/pnas.0511288103
Cui J., Zhao S., Li Y., Zhang D., Wang B., Xie J., Wang J. 2021. Regulated cell death: discovery, features and implications for neurodegenerative diseases. Cell Commun. Signal. 19, 120. https://doi.org/10.1186/s12964-021-00799-8
Shlyapina V.L., Yurtaeva S.V., Rubtsova M.P., Dontsova O.A. 2021. At the crossroads: mechanisms of apoptosis and autophagy in cell life and death. Acta Naturae. 13, № 2 (49). 106‒115.
Babaei G., Aziz S.G., Jaghi N.Z.Z. 2021. EMT, cancer stem cells and autophagy; the three main axes of metastasis. Biomed. Pharmacother. 133, 110909.https://doi.org/10.1016/j.biopha.2020.110909
Urbańska K., Orzechowski A. 2021. The secrets of alternative autophagy. Cells. 10, 3241. https://doi.org/10.3390/cells10113241
Gómez-Sánchez R., Rose J., Guimarães R., Mari M., Papinski D., Rieter E., Geerts W.J., Hardenberg R., Kraft C., Ungermann C., Reggiori F. 2018. Atg9 establishes Atg2-dependent contact sites between the endoplasmic reticulum and phagophores. J. Cell. Biol. 217, 2743–2763. https://doi.org/10.1083/jcb.201710116
Li X., He S., Ma B. 2020. Autophagy and autophagy-related proteins in cancer. Mol. Cancer. 19 (1), 12.https://doi.org/10.1186/s12943-020-1138-4
Bujak A.L., Crane J.D., Lally J.S., Ford R.J., Kang S.J., Rebalka I.A., Green A.E., Kemp B.E., Hawke T.J., Schertzer J.D., Steinberg G.R. 2015. AMPK activation of muscle autophagy prevents fasting-induced hypoglycemia and myopathy during aging. Cell. Metab. 21, 883–890.https://doi.org/10.1016/j.cmet.2015.05.016
Hill S.M., Wrobel L., Rubinsztein D.C. 2019. Post-translational modifications of Beclin 1 provide multiple strategies for autophagy regulation. Cell Death Differ. 26, 617–629. https://doi.org/10.1038/s41418-018-0254-9
Klionsky D.J., Abdelmohsen K., Abe A., et al. The consortium. 2016. Guidelines for the use and interpretation of assays for monitoring autophagy. 3rd ed. Autophagy. 12, 1–222. https://doi.org/10.1080/15548627.2015.1100356.26799652
Pradel B., Robert-Hebmann V., Espert L. 2020. Regulation of innate immune responses by autophagy: a goldmine for viruses. Front. Immunol. 11, 578038.https://doi.org/10.3389/fimmu.2020.578038
Sharma V., Verma S., Seranova E., Sarkar S., Kumar D. 2018. Selective autophagy and xenophagy in infection and disease. Front. Cell. Dev. Biol. 6, 147.https://doi.org/10.3389/fcell.2018.00147
Choi Y., Bowman J.W., Jung J.U. 2018. Autophagy during viral infection—a double-edged sword. Nat. Rev. Microbiol. 16, 341–354. https://doi.org/10.1038/s41579-018-0003-6
Mijaljica D., Klionsky D.J. 2020. Autophagy/virophagy: a “disposal strategy” to combat COVID-19. Autophagy. 16, 2271–2272. https://doi.org/10.1080/15548627.2020.1782022
Leonardi L., Sibéril S., Alifano M., Cremer I., Joubert P.E. 2021. Autophagy modulation by viral infections influences tumor development. Front. Oncol. 11, 743780. https://doi.org/10.3389/fonc.2021.743780
Liang W., Liu H., He J., Ai L., Meng Q., Zhang W., Yu C., Wang H., Liu H. 2021. Studies progression on the function of autophagy in viral infection. Front. Cell. Dev. Biol. 9, 772965. https://doi.org/10.3389/fcell.2021.772965
Mouna L., Hernandez E., Bonte D., Brost R., Amazit L., Delgui L.R., Brune W., Geballe A.P., Beau I., Esclatinea A. 2016. Analysis of the role of autophagy inhibition by two complementary human cytomegalovirus BECN1/Beclin 1-binding proteins. Autophagy. 12, 327–342.
Belzile J.P., Sabalza M., Craig M., Clark A.E., Morello C.S., Spector D.H. 2016. Trehalose, an mTOR-independent inducer of autophagy, inhibits human cytomegalovirus infection in multiple cell types. J. Virol. 90, 1259–1277.
Zhang X., Zhang L., Bi Y., Xi T., Zhang Z., Huang Y., Lu Y.Y., Liu X., Shu S., Fang F. 2021. Inhibition of autophagy by 3-methyladenine restricts murine cytomegalovirus replication. J. Med. Virol. 93, 5001–5016.https://doi.org/10.1002/jmv.26787
Zimmermann C., Krämer N., Krauter S., Strand D., Sehn E., Wolfrum U., Freiwald A., Butter F., Plachter B. 2021. Autophagy interferes with human cytomegalovirus genome replication, morphogenesis, and progeny release. Autophagy. 17, 779–795.https://doi.org/10.1080/15548627.2020.1732686
Zhang X., Xi T., Zhang L., Bi Y., Huang Y., Lu Y., Liu X., Fang F. 2021. The role of autophagy in human cytomegalovirus IE2 expression. J. Med. Virol. 93, 3795–3803. https://doi.org/10.1002/jmv.26357
Liu Y., Pan J., Liu L., Li W., Tao R., Chen Y., Li H., Shang S. 2017. The influence of HCMV infection on autophagy in THP-1 cells. Medicine (Baltimore). 96, e8298.https://doi.org/10.1097/MD.0000000000008298
Chaumorcel M., Souquère S., Pierron G., Codogno P., Esclatine A. 2008. Human cytomegalovirus controls a new autophagy-dependent cellular antiviral defense mechanism. Autophagy. 4 (1), 46–53. https://doi.org/10.4161/auto.5184
Tovilovic G., Ristic B., Siljic M., Nikolic V., Kravic-Stevovic T., Dulovic M., Milenkovic M., Knezevic A., Bosnjak M., Bumbasirevic V., Stanojevic M., Trajko-vic V. 2013. mTOR-independent autophagy counteracts apoptosis in herpes simplex virus type 1-infected U251 glioma cells. Microbes Infect. 15 (8–9), 615–624. https://doi.org/10.1016/j.micinf.2013.04.012
Usman R.M., Razzaq F., Akbar A., Farooqui A.A., Iftikhar A., Latif A., Hassan H., Zhao J., Carew J.S., Nawrocki S.T., Anwer F. 2021. Role and mechanism of autophagy-regulating factors in tumorigenesis and drug resistance. Asia Pac. J. Clin. Oncol. 17, 193–208.https://doi.org/10.1111/ajco.13449
Chiou J.T., Huang C.H., Lee Y.C., Wang L.J., Shi Y.J., Chen Y.J., Chang L.-S. 2020. Compound C induces autophagy and apoptosis in parental and hydroquinone-selected malignant leukemia cells through the ROS/p38 MAPK/AMPK/TET2/FOXP3 axis. Cell Biol. Toxicol. 36, 315–331.
Linder B., Kögel D. 2019. Autophagy in cancer cell death. Biology. 8 (4), 82. https://doi.org/10.3390/biology8040082
Tao Z., Li T., Ma H., Yang Y., Zhang C., Hai L., Liu P., Yuan F., Li J., Yi L., Tong L., Wang Y., Xie Y., Ming H., Yu S., Yang X. 2018. Autophagy suppresses self-renewal ability and tumorigenicity of glioma-initiating cells and promotes Notch1 degradation. Cell Death Dis. 9, 1063. https://doi.org/10.1038/s41419-018-0957-3
Barthet V.J.A., Brucoli M., Ladds M., Nössing C., Kiourtis C., Baudot A.D., O’Prey J., Zunino B., Müller M., May S., Nixon C., Long J.S., Bird T.G., Ryan K.M. 2021. Autophagy suppresses the formation of hepatocyte-derived cancer-initiating ductular progenitor cells in the liver. Sci. Adv. 7, eabf9141.https://doi.org/10.1126/sciadv.abf9141
Zhu H., Wang D., Zhang L., Xie X., Wu Y., Liu Y., Shao G., Su Z. 2014. Upregulation of autophagy by hypoxia-inducible factor-1α promotes EMT and metastatic ability of CD133+ pancreatic cancer stem-like cells during intermittent hypoxia. Oncol. Rep. 32, 935–942.https://doi.org/10.3892/or.2014.3298
Zhu Y., Huang S., Chen S., Chen J., Wang Z., Wang Y., Zheng H. 2021. SOX2 promotes chemoresistance, cancer stem cells properties, and epithelial-mesenchymal transition by β-catenin and Beclin1/autophagy signaling in colorectal cancer. Cell Death Dis. 12, 449.https://doi.org/10.1038/s41419-021-03733-5
Zhou Q., Cui F., Lei C., Ma S., Huang J., Wang X., Qian H., Zhang D., Yang Y. 2021. ATG7-mediated autophagy involves in miR-138-5p regulated self-renewal and invasion of lung cancer stem-like cells derived from A549 cells. Anti-Cancer Drugs. 32, 376–385.https://doi.org/10.1097/CAD.0000000000000979
Wang X., Lee J., Xie C. 2022. Autophagy regulation on cancer stem cell maintenance, metastasis, and therapy resistance. Cancers (Basel). 14, 381.https://doi.org/10.3390/cancers14020381
Bao L., Jaramillo M.C., Zhang Z., Zheng Y., Yao M., Zhang D.D., Yi X. 2015. Induction of autophagy contributes to cisplatin resistance in human ovarian cancer cells. Mol. Med. Rep. 11, 91–98.https://doi.org/10.3892/mmr.2014.2671
Cheng C.Y., Liu J.C., Wang J.J., Li Y.H., Pan J., Zhang Y.R. 2017. Autophagy inhibition increased the anti-tumor effect of cisplatin on drug-resistant esophageal cancer cells. J. Biol. Regul. Homeost. Agents. 31, 645–652.
Yang J., Pi C., Wang G. 2018. Inhibition of PI3K/Akt/mTOR pathway by apigenin induces apoptosis and autophagy in hepatocellular carcinoma cells. Biomed. Pharmacother. 103, 699–707.https://doi.org/10.1016/j.biopha.2018.04.072
Liu K., Ren T., Huang Y., Sun K., Bao X., Wang S., Zheng B., Guo W. 2017. Apatinib promotes autophagy and apoptosis through VEGFR2/STAT3/BCL-2 signaling in osteosarcoma. Cell Death Dis. 8, e3015.https://doi.org/10.1038/cddis.2017.422
Antonioli M., Pagni B., Vescovo T., Ellis R., Cosway B., Rollo F., Bordonia V., Agratia C., Labus M., Covelloe R., Benevoloe M., Ippolitoa G., Robinson M., Piacentini M., Lovatc P., Fimia G.M. 2021. HPV sensitizes OPSCC cells to cisplatin-induced apoptosis by inhibiting autophagy through E7-mediated degradation of AMBRA1. Autophagy. 17, 2842–2855.https://doi.org/10.1080/15548627.2020.1847444
Chernoryzh Yu.Yu., Fedorova N.E., Yurlov K.I., Simonov R.A., Kornev A.V., Karpov D.S., Zakirova N.F., Ivanov A.V., Kusch A.A., Gintsburg A.L. 2019. Resistance of THP-1 leukemia cells infected with cytome-galovirus to anti-tumor antibiotic doxorubicin and restoration of the sensitivity by inhibitors of the PI3K/AKT/mTOR molecular pathway. Dokl. Biochem. Biophys. 489, 388–391.
Chang P.H., Graham J., Hao J., Ni J., Bucci N.J., Cozzi P.J., Kearsley J.H., Li Y. 2013. Acquisition of epithelial-mesenchymal cancer stem cell phenotypes is associated with activation of the PI3K/Akt/mTOR pathway in prostate cancer radioresistance. Cell Death Dis. 4, e875.https://doi.org/10.1038/cddis.2013.407
Galluzzi L., Pedro J.M.B.-S., Levine B., Green D.R., Kroemer G. 2017. Pharmacological modulation of autophagy: therapeutic potential and persisting obstacles. Nat. Rev. Drug Discov. 16, 487–511.https://doi.org/10.1038/nrd.2017.22
Prerna K., Dubey V.K. 2021. Repurposing of FDA-approved drugs as autophagy inhibitors in tumor cells. J. Biomol. Struct. Dyn. 20, 1–12. https://doi.org/10.1080/07391102.2021.1873862
Xie Q., Liu Y., Li X. 2020. The interaction mechanism between autophagy and apoptosis in colon cancer. Transl. Oncol. 13, 100871. https://doi.org/10.1016/j.tranon.2020.100871
Funding
This work was supported by the Russian Science Foundation (project no. 19-14-00197).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The authors declare that they have no conflict of interest. This article does not contain any studies involving animals or human subjects performed by any of the authors.
Additional information
Translated by T. Tkacheva
Rights and permissions
About this article

Cite this article
Vinogradskaya, G.R., Ivanov, A.V. & Kushch, A.A. Mechanisms of Survival of Cytomegalovirus-Infected Tumor Cells. Mol Biol 56, 668–683 (2022). https://doi.org/10.1134/S0026893322050132
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S0026893322050132