
Abstract
Human papillomavirus-16 (HPV-16) is primary etiological agent of invasive cervical cancer development in the world population. There is no treatment for HPV infection. As a possible tool for prophylactic vaccination, the development of virus-like particles (VLPs) comprising the HPV-16 L1 capsid protein is highly preferred. The expression of HPV-16 L1 gene was carried out in Pichia pastoris. It was done as a promising vaccine candidate against HVP-16 infection for developing countries. The codon optimization for HPV-16 L1 gene was done and synthesized in pPICZA plasmid. The expression of HPV-16 L1 was evaluated in P. pastoris after induction with methanol. The purification of HPV-16 VLPs was accomplished by the ultra-centrifugation using the sucrose density gradient. The sera of 101 subjects including 16 patients positive for HPV-16 and 85 individuals negative for HPV-16 were tested by ELISA assay using anti-HPV-16 antibodies. The results of ELISA test have depicted no false positive and no false negative as compared with commercial ELISA kit. The results of HPV-16 L1 expression in P. pastoris showed a band of about 56 kDa by SDS-PAGE, and were confirmed by Western-blot assay. The formation of VLP and the self-assembly of HPV-16 L1 major capsid protein in VLP was observed by transmission electron and atomic force microscopies.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Cervical cancer is the major health problem in women worldwide. The cervical cancer is the fourth common cancer among women. It was estimated about 528 000 new cases of cervical cancer and 266 000 deaths each year worldwide [1]. The clinical, molecular and epidemiological studies of human papillomavirus (HPV) have been shown that the persistent infection with HPV is a prerequisite for cervical cancer [2]. More than 200 genotypes of HPV have been recognized and completely sequenced (pave. niaid.nih.gov; International Human Papillomavirus Reference Center; http://www.hpvcenter.se) [3]. Genital HPV is the most frequent sexually transmitted infection with an estimated prevalence of ~70% in both sexes [4]. Practically, all cervical cancers are caused by high risk group of HPV, primarily by HPV-16 (~50%) and HPV-18 (~20%). Less frequently of HPV infection was happen by other high risk HPV types (e.g. HPV-31, 33, 45, 52, 58 .…) [5, 6]. The HPV comprising a double-stranded DNA with approximately 8000 bp surrounded by two major (L1) and minor (L2) capsid proteins. The L1 protein (55–60 kD) builds up 72 pentameric capsomers arranged in an icosahedral array [7, 8]. HPV-16 L1 is the most significant target in production of HPV vaccines. The expression of the HPV-16 L1 capsid protein has been produced in different expression systems (e.g. insect cells or yeast) which resulted in formation of self-assembled virus-like particles (VLPs). Several studies have illustrated that HPV-16 VLPs resemble the native virions in size (55 nm) and shape, although, they do not carry the viral genome. They are highly immunogenic as they can induce high titer virus-neutralizing antibodies which could prevent persistent HPV infections [9–12]. Two prophylactic vaccines have been licensed based on VLPs, gardasil (Merck and Co., Inc., USA), and cervarix (GlaxoSmithKline, United Kingdom) [13–15]. Although these vaccines which are produced in eukaryotic cells (insect cells or yeast) are highly efficient, the major impediments of them are their high cost. Therefore, they are not cost effective for under developing countries. Since the number of cervical cancer due to HPV-16 increases in third world countries, the cost effective HPV-16 VLPs is the main purpose of the research. Thus, the aim of the study was to express HPV-16 L1 gene in Pichia pastoris to develop HPV-16 VLPs as a promising candidate vaccine for developing countries.
MATERIALS AND METHODS
Construction of the pPICZA L1 plasmid. HPV-16 L1 protein sequence was obtained from GenBank (accession no. KP161014). Codon optimization of L1 HPV-16 gene was done for P. pastoris using online integrated DNA technology program (https://eu.idtdna.com).
The 3D structure of the relevant protein was performed using the PyMOL molecular graphics system 1.1 and I-TASSER algorithm (http://zhanglab.ccmb.med. umich.edu) and presented in Fig. 1 [16–19].

In silico construction of HPV-16 L1 protein.
Following synthesizing the L1 gene in pPICZA L1 plasmid (Biomatik, Canada), primers were designed by OLIGO primer analysis software for amplifying the target gene which included forward 5'ACTTCGAAATGTCTCTTTGGCTGC3' comprising SfuI restriction site (shown as bold letters) and reverse primer 5'TACGAGCTCTTACAGCTTACGTTTTTTGC3' containing XhoI restriction site (shown as bold letters) and stop codon (shown as italic underlined letters).
HPV-16 L1 gene (amplicon) was purified by NucleoSpin gel and PCR clean-up kit (Macherey-Nagel, Germany). The amplicon of L1 HPV-16 gene was digested by XhoI and sfuI restriction enzymes (Thermo Fisher Scientific, USA). The digested amplicon was ligated to pPICZA plasmid using T4 DNA ligase (Invitrogen, USA) according to the manufacturer’s protocol. The ligated gene in plasmid was affirmed by PCR using mentioned above primers.
The expected size of plasmid containing target gene was examined using vector-NTI software. High copy amplification of the pPICZA-L1 plasmid was obtained by transformation of the plasmid into competent E. coli TOP 10 cells TOP 10 (Invitrogen, USA) using the heat shock method [20].
The plasmid was extracted from E. coli cells using easy-pure plasmid mini-prep kit (Beijing, China). A PCR assay was done to approve the inserted gene in plasmid. The linearization of plasmid of interest was carried out using SacI restriction enzyme (Thermo Fisher Scientific, USA) according to the manufacturer’s protocol.
Transformation of linearized pPICZA L1 plasmid into P. pastoris. The cold linearized plasmid (10 μg) was blended with P. pastoris competent cells strain KM71H (100 μL) provided by Razi Vaccine and Serum Research Institute, Iran. The relevant mixture was pulsed at 2500 V, 25 μF and 200 Ω by gene pulser X-cell electroporation equipment (Bio-Rad, USA). Negative control was done by pulsing the P. pastoris competent cells with no L1 gene. The transformed cells were cultured in YPDS media plate containing (g/L): dextrose—20.0, yeast extract—10.0, peptone—20.0 and agar—20.0, supplemented with 1 M sorbitol and 100 μg/mL zeocin. The plates were incubated at 28°C for 7 days.
Screening the multicopy transformed colonies was carried out by culturing the colonies in YPDS media containing high zeocin concentration (500, 1000 and 2000 μg/mL).
The presence of L1 gene in the transformed P. pastoris cells was done by PCR colony assay using the primers mentioned in easy-select Pichia kit, (Invitrogen, USA). The extraction of DNA from transformed cells was done using boiling extraction method [21].
Expression and purification of HPV-16 L1 protein. The transformed P. pastoris cells were cultured in 500 mL of BMGY medium containing (g/L): peptone—20.0, yeast nitrogen base—13.4, potassium phosphate—13.4, yeast extract—10.0, glycerol—10.0, biotin—0.02 supplemented with 100 μg/mL zeocin. Flasks containing cultured transformed P. pastoris in BMGY media were incubated at 180 rpm and 28°C for 24 h. The incubation was continued till the OD600 reached 6. The cells were centrifuged at 5000 g for 11 min. The supernatant was discarded, the precipitated cells were suspended in 100 mL of BMMY medium containing (g/L): peptone—20.0, yeast nitrogen base—13.4, potassium phosphate—13.4, yeast extract—10.0, biotin—0.02 supplemented with 0.5% methanol, and incubated at 180 rpm and 28°C. At every 24 h interval, 1 mL of the media containing P. pastoris cells of interest was centrifuged under conditions described above and the obtained cells kept at –80°C. Next, 0.5% of the final volume (1 mL) methanol was added to the flask to induce and enhance of the expression level of L1 protein in P. pastoris cells. After incubation for 4 days, the cells were centrifuged at 5000 g for 10 min. The cells were resuspended in the breaking 50 mM Na-phosphate buffer containing 5% glycerol, 1 mM EDTA and 1 mM PMSF (pH 7.4). An equivalent volume of the acid-washed glass beads (diameter of 0.5 mm, Sigma-Aldrich, USA) was added to the tubes containing transformed cells. The mixture was vortexed for 45 s and immediately placed on ice for 45 s. This cycle was repeated 10 times. Then the suspension was centrifuged at 13 000 g for 10 min (4°C). Subsequently, the supernatant was dialyzed against the same buffer containing 0.2 M PMSF and 0.5 M NaCl at 4°C overnight. After the ultra-centrifugation at 130 000 g for 4 h (4°C) in 10–40% sucrose density gradient, the fractions were collected [21]. Each fraction was tested by SDS-PAGE. Finally, purified protein was collected and dialyzed as described previously.
Qualification and quantification of protein. The expression of L1 HPV-16 protein was affirmed using SDS-PAGE and Western-blot. After electrophoresis in PAGE and transfer of proteins to PVDF-membrane, the protein of interest was blocked on membrane by 5% BSA-PBS solution for 2 h at 4°C. It was washed with 0.05% Tween 20-PBS and kept with the anti-HPV-16 L1 monoclonal antibody (Creative Diagnostic, USA) at 4°C overnight. The protein reacted with secondary horseradish peroxidase (HRP)-conjugated donkey anti-goat antibodies (Thermo Fisher Scientific, USA) at 4°C for 2 h. Finally, the reaction between antibodies and relevant protein was visualized using 4% diaminobenzidine, 1% H2O2 in 50 mM Tris-HCL buffer (pH 7.5). The quantification of protein of interest was examined by the Bradford protein assay [22].
Atomic-force microscopy (AFM). The highly purified L1 HPV-16 VLPs were placed on a piece of stone membrane (mica sheet), dried at room temperature, and visualized under atomic force microscope (C26, DME, Denmark). The analyzing the AFM data was carried out using SPM software (v. 2.0.0.9).
Electron microscopy. A drop of the relevant highly purified VLPs was placed on the carbon coated grid and stained by uranyl acetate [7]. Finally, the grid was examined under Zeiss EM900 transmission electron microscope (Carl Zeiss AG, Germany).
ELISA method. The optimized 55 ng HPV-16 L1 VLPs protein was coated on the polystyrene wells of ELISA plate (Nuck, Denmark) for detection using commercial HPV-16 L1 antibody (Thermo Fisher Scientific, USA). One hundred μL of antibody prepared in blocking buffer (skim milk) was added to the wells with HPV-16 L1 VLPs protein and incubated at 37°C for 2 h. After 4 times washing the tested wells with PBS containing 0.5% Tween-20, 100 μL of secondary HRP-conjugated donkey anti-goat antibodies was added to the wells and incubated for 2 h at 37°C. Wells were washed with washing buffer as described in the previous step, and 100 μL 0.1% 3,3',5,5'-tetramethylbenzidine (TMB) was added to the wells and placed in dark for 30 min. Subsequently, 50 μL 2 N sulfuric acid was added to wells and the results of the ELISA tests were read at OD450 [23].
In addition, 55 ng of purified VLPs were coated on ELISA plate wells (Nuck, Denmark) for detection of anti-L1 antibodies in the sera of 16 patients who had diagnosed HPV-16 infection lasted for more than 3 years. The sera of 85 individuals with no history of papilloma virus infections were used as negative control. One hundred µL of serum of each individual was added to the tested wells. After washing the wells, 100 µL of secondary HRP- conjugated rabbit anti-human antibodies (L1) (Thermo Fisher Scientific, USA) was added to the each tested well and incubated for 2 h at 37°C. Then, 100 μL 0.1% TMB was added to the wells and incubated in dark 30 min. Finally, after adding the sulfuric acid as stopping reagent the OD was read at 450 nm. The cut off value was determined and results of this ELISA test were compared with those for commercial ELISA kit (Diapro, Italy) according to the manufacturer instructions.
The ethic consent of this test was obtained from all the participating in this research project. This study was conducted in accordance with the Declaration of Helsinki (as revised in 2008) and according to local guidelines and laws. The ethical approval for this study was obtained with the registration number of OG94100 from the university ethic committee of Ahvaz Jundishapur University of Medical Sciences, Ahvaz, Iran.
RESULTS AND DISCUSSION
In silico analysis of the chimeric HPV-16 L1 protein. The analysis of 3D structure of HPV-16 L1 protein along with native control which was carried out by PyMOL molecular graphics system 1.1, I-TASSER algorithm and ExPASy (https://www.expasy.org/) online software, revealed that the formation of VLPs was designed properly as indicated in Fig. 1.
Codon adaptation index (CAI) was analyzed using online genscript software (www.genscript.com) before and after the optimization. CAI in this study was 0.9 which showed high expression level. The CAI greater than 0.8 indicates high level of expression.
The amplification of HPV-16 L1 gene (1534 bp) was successfully done using primers which were mentioned above (data not shown). The purified HPV-16 L1 gene with XhoI and SfuI restriction sites was inserted into pPICZA plasmid (data not shown). The PCR amplification of the relevant gene was achieved with primers mentioned in easy-select Pichia kit and affirmed that the relevant gene was properly inserted in correct position (1858 bp). Linearization of the plasmid containing HPV-16 L1 gene (1534 bp) was observed in the agarose gel electrophoresis as shown in Fig. 2.

Linearization of the plasmid containing HPV-16 L1 gene. 1—Ladder; 2—linearized pPICZA- HPV-16 L1 gene plasmid, 3—negative control.
Transformation and selection of multi-copy colonies. The colonies of P. pastoris comprising HPV-16 L1 pPICZA plasmid were appeared on YPDS agar plates containing zeocin after 7 days of incubation. The result of colony PCR illustrated that transformation of the plasmid into P. pastoris cells was done properly (1858 bp).
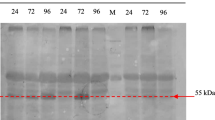
Expression analysis of HPV-16 L1 protein. The expression of HPV-16 L1 protein was primarily confirmed by SDS-PAGE method which revealed a band of 56 kDa (Fig. 3a). This band did not exist in the negative control. The yield of protein expression depended on expression phase (0, 1, 2, 3 and 4 days). The highest protein yield of 11 mg/L was revealed after 96 h induction which is indicated in Fig. 3a. SDS-PAGE analysis exhibited the highly purified HPV-16 L1 protein with no extra bands which was obtained by ultracentrifugation as it was shown in Fig. 3b. Reactivity of the highly purified VLPs of interest with the anti-HPV-16 antibody was approved using Western-blot analysis (Fig. 3c) and ELISA method.

Analysis of HPV-16 L1 protein. (a) SDS-PAGE for the expression of the chimeric protein in P. pastoris. 1, 2 and 3—proteins expressed in P. pastoris after 4, 3 and 2 days of induction, respectively; 4—negative control; 5—markers; (b) SDS-PAGE for the purified by ultracentrifugation expressed protein. 1—Purified protein; 2—markers; (c) Western-blot analysis using the anti-HPV-16 L1 monoclonal antibody. 1—Negative control; 2—purified expressed protein.
Verifying VLP formation. The VLP formation was tested using AFM and TEM as shown in Figs. 4 and 5, respectively. The both methods illustrated the presence of purified particles with approximately 60 nm in diameter. Turning the AFM images by SPM software showed the presence of true particles on the membrane (data not shown). No particles with the same size of HPV-16 L1 VLPs were observed in the negative control according to Fig. 4b.

AFM images of expressed HPV-16 L1 VLPs (a) and negative control (b).

TEM image of purified HPV-16 L1 VLPs.
The wells coated of 55 ng of purified VLPs were able to detect the lowest and highest titer of commercial antibody in the ELISA test. The wells coated HPV-16 L1 showed reactivity with positive samples and no false reaction with the negative control. The results showed that the mean OD450 of anti–HPV-16 L1 antibody was 2.405 while the OD450 of standard HPV-16 L1 VLPs was 2.500. The results obtained for wells coated with HPV-16 L1 protein for patients having sera positive for HPV-16 infection and sera of individuals negative for HPV infection found similar results gained by commercial test kit, with no observation of false positive and no false negative controls.
HPV-16 infection is the main risk factor for development of cervical cancer [24]. The expression of HPV16-L1 protein in yeast cells lead to formation of VLPs that mimic the natural virus structure and elicit high titers to virus neutralizing antibodies in animal models and humans [25]. HPV-16 L1-VLPs have been widely accepted as the best candidate of HPV-16 prophylactic vaccines [26]. However, the cost of current HPV vaccines is unaffordable in less-developed countries, where the incidence of HPV-associated disease is high, and the vaccine is the most required to prevent the spreading of HPV-16 infection. High prevalence of HPV-16 has been reported in Iran. Therefore, the designed vaccines should comprise high risk papillomaviruses specially type 16 [23, 27–29].
In the present study, we have described the construction, transcription and expression of HPV-16 L1 gene carried out in P. pastoris. We have displayed that HPV-16 L1 gene is correctly expressed in P. pastoris cells, as demonstrated by Western blotting, PCR and ELISA methods.
The highest level expression of the relevant protein was gained using 96 h induction which was in contrast with the research by Bazan et al. [20, 30] reported high level of exoression was observed following induction for 48 h. The single band of HPV-16 L1 expressed with approximately 56 kDa was observed by SDS-PAGE which was in a agreement with the study reported by Abdoli et al. [31] and Kirnbauer et al. [9]. The purified VLPs protein was recovered by ultracentrifugation which has been observed by SDS-PAGE analysis. Reactivity of the protein of interest was approved by Western-blot method,
The results of ELISA test depicted no false positive and no false negative as compared to commercial ELISA kit.
In this study, high expression of HPV-16 L1 gene leading to pure HPV-16 L1 VLP recombinant protein was successfully generated using P. pastoris cells. It was confirmed by SDS-PAGE (single band of 56 kDa) and Western-blot assay. The formation of VLP was visualized by TEM and AFM. High yield of 11 mg/L for purified HPV 16 L1 was obtained after induction of methanol 0.5% for 96 h. The expression of HPV-16 L1 VLPs in the P. pastoris system is promising for prophylactic vaccine development for less-developing countries. Further investigation is required on humoral response and formulation with appropriate adjuvant with HPV-16 L1 protein in animal model.
REFERENCES
Ferlay, J., Soerjomataram, I., Dikshit, R., Eser, S., Mathers, C., Rebelo, M., et al., Int. J. Cancer, 2015, vol. 136, no. 5, pp. E359–386.
Bosch, F.X., Lorincz, A., Muñoz, N., Meijer, C.J.L.M., and Shah, K.V., J. Clin. Pathol., 2002, vol. 55, no. 4, pp. 244–265.
Bernard, H-U., Burk, R.D., Chen, Z., van Doorslaer, K., zur Hausen, H., and de Villiers, E.M., Virology, 2010, vol. 401, no. 1, pp. 70–79.
Hu, D. and Goldie, S., Am..J. Obstet. Gynecol., 2008, vol. 198, no. 5, pp. 500e1–500e7.
Munoz, N., Bosch, F.X., Castellsagué, X., Díaz, M., de Sanjose, S., Hammouda, D. et al., Int. J. Cancer, 2004, vol. 111, no. 2, pp. 278–285.
de Sanjose, S., Quint, W.G., Alemany, L., Geraets, D.T., Klaustermeier J.E., Lloveras, B., et al., Lancet Oncol., 2010, vol. 11, no. 11, pp. 1048–1056.
Finnen, R.L., Erickson, K.D., Chen, X.S., and Garcea, R.L., J. Virology, 2003, vol. 77, no. 8, pp. 4818–4826.
Bishop, B., Dasgupta, J., Klein, M., and Garcea, R.L., J. Biol. Chem., 2007, vol. 282, no. 43, pp. 31803–31811.
Kirnbauer, R., Taub, J., Greenstone, H., Roden, R., Dürst, M., Gissmann, L., et al., J. Virology, 1993, vol. 67, no. 12, pp. 6929–6936.
Koutsky, L.A., Ault, K.A., Wheeler, C.M., Brown D.R., Barr, E., Alvarez, F.B., et al., N. Engl. J. Med., 2002, vol. 347, no. 21, pp. 1645–1651.
Harper, D.M., Franco, E.L., Wheeler, C., Ferris, D.G., Jenkins, D., Schuind, A., et al., Lancet, 2004, vol. 364, no. 9447, pp. 1757–1765.
Villa, L., Costa, R., Petta, C., Andrade, R.P., Paavonen, J., Iversen, O.E., et al., Br. J. Cancer, 2006, vol. 95, no. 11, p. 1459.
Munoz, N., Kjaer, S.K., Sigurdsson, K., Iversen, O.E., Hernandez-Avila, M., Wheeler, C.M., et al., J. Nat. Cancer Inst., 2010, vol. 102, no, 5, pp. 325–339.
Palefsky, J.M., Giuliano, A.R., Goldstone, S., Moreira, E.D., Aranda, C., Jessen, H., et al., N. Engl. J. Med., 2011, vol. 365, pp. 1576–1585.
Lehtinen, M., Paavonen, J., Wheeler, C.M., Jaisamrarn, U., Garland, S.M., Castellsagué, X., et al., Lancet Oncol., 2012, vol.13, no. 1, pp. 89–99.
Maniatis, T., Fritsch, E., and Sambrook, J., Molecular Cloning: A Laboratory Manual, New York: Cold Spring Harbor Laboratory, 1982.
Zhang, Y., BMC Bioinform., 2008, vol. 9, p. 40.
Roy, A., Kucukural, A., and Zhang, Y., Nat. Protoc., 2010, vol. 5, p. 725.
Yang, J., Yan, R., Roy, A., Xu D., Poisson J., and Zhang Y., Nat. Methods, 2015, vol. 12, no. 1, p. 7–8.
Rao, N.H., Babu, P.B., Rajendra, L., Sriraman R., Pang, Y.-Y.S., Schiller, J.T., et al., Vaccine, 2011, vol. 29, no. 43, pp. 7326–7334.
Kotzé, L., Smith, J., den Haan, van Zyl, R.W.H., and Görgens, J.F., Afr. J. Biotechnol., 2010, vol. 10, no. 2, pp. 214–219.
Ernst, O. and Zor, T., J. Vis. Exp., 2010, no. 38.
Hosseini, S.Z., Makvandi, M., Samarbafzade, A., Timori, A., Ranjbar, N., Saki, N., et al., Asian Pac. J. Cancer Prev., 2017, vol. 18, no. 4, pp. 889–893.
Zur Hausen, H., Biochim. Biophys. Acta—Rev.Cancer, 1996, vol. 1288, no. 2, f55–78.
Paavonen, J., Jenkins, D., Bosch, F.X., Naud, P., Salmerón, J., Wheeler, C.M., et al., Lancet, 2007, vol. 369, no. 9580, pp. 2161–2170.
Koutsky, L.A. and Harper, D.M., Vaccine, 2006, vol. 24, pp. S114–S121.
Jalilvand, S., Shoja, Z., and Hamkar, R., Asian Pac. J. Cancer Prev., 2014, vol. 15, no. 17, pp. 7029–7035.
Kiani, S.J., Malekshahi, S.S., Ghalejoogh, Y.Z., Ghavvami, N., Shafiei Jandaghi, N.Z., Shahsiah, R., et al., Jundishapur J. Microbiol., 2015, vol. 8, no. 12, e26441.
Yaghoobi, R., Makvandi, M., Afshar, N., Nader, P., Hamidifard, M. and Sharifpour, C., Jundishapur J. Microbiol., 2015, vol. 8. no. 11. e25882.
Bazan, S.B., Chaves, A., Aires, K.A., Cianciarullo, A.M., Garcea, R.L., and Ho, P.L., Arch. Virol., 2009, vol.154, no. 10, p. 1609.
Abdoli, A., Soleimanjahi, H., Fotouhi, F., Pour Beiranvand, S., and Kianmehr Z., Iran. J. Basic Med. Sci., 2013, vol. 16, p. 891.
ACKNOWLEDGMENTS
We appreciate the staff at the Department of Genomics and Genetic Engineering specially Mr. M. Aali and Mrs. S. Karimi Naghlani, Razi Vaccine and Serum Research Institute (RVSRI), Karaj, Alborz, Iran. We are very grateful to personnel of Razi Laboratory, Karaj, Iran for providing positive and negative anti-HPV-16 sera.
Funding
This study was as a part of research project conducted by Mr. A. Sanchooli Ph.D. student (virology), with the registration number 94 155, which was financially supported by Infectious and Tropical Diseases Research Center, Health Research Institute, Ahvaz Jundishapur University of Medical Sciences, Ahvaz Iran.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The authors declare that they have no conflict of interest. This article does not contain any studies involving animals or human participants performed by any of the authors.
Rights and permissions
About this article

Cite this article
Sanchooli, A., Aghayipour, K., Naghlani, S.K. et al. Production of Human Papillomavirus Type-16 L1 VLP in Pichia pastoris. Appl Biochem Microbiol 56, 51–57 (2020). https://doi.org/10.1134/S0003683820010147
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S0003683820010147