
Abstract
Cockayne syndrome is an autosomal recessive disorder principally characterized by postnatal growth failure and progressive neurological dysfunction, due primarily to mutations in ERCC6 and ERCC8. Here, we report our diagnostic experience for two patients in a Chinese family suspected on clinical grounds to have Cockayne syndrome. Using multiple molecular techniques, including whole exome sequencing, array comparative genomic hybridization and quantitative polymerase chain reaction, we identified compound heterozygosity for a maternal splicing variant (chr5:60195556, NM_000082:c.618-2A > G) and a paternal complex deletion/inversion/deletion rearrangement removing exon 4 of ERCC8, confirming the suspected pathogenesis in these two subjects. Microhomology (TAA and AGCT) at the breakpoints indicated that microhomology-mediated FoSTeS events were involved in this complex ERCC8 rearrangement. This diagnostic experience illustrates the value of high-throughput genomic technologies combined with detailed phenotypic assessment in clinical genetic diagnosis.
Similar content being viewed by others
Introduction
Cockayne syndrome (CS; MIM# 133540, 216400), a rare autosomal recessive disorder first described by Cockayne in 19361, involves multiple systems, and its major features include progressive growth failure, microcephaly, mental retardation, retinal pigmentary degeneration, sensorineural deafness, photosensitivity and premature death2. Different clinical subtypes of CS have been defined based upon severity and phenotypic heterogeneity2. For example, the classic form, CS type I, presents with developmental abnormalities in the first two years, and usually leads to death in the first or second decade. In contrast, CS type II is more severe with symptoms present at birth, while CS type III is a milder/later-onset form with normal growth and cognitive development. Due to its wide spectrum of symptoms, precise diagnosis of CS must rely on molecular genetic testing. CS cases may be caused by mutation in ERCC8 (Cockayne syndrome A or CSA; 35% of cases) or ERCC6 (Cockayne syndrome B or CSB; 65% of cases)2,3,4,5,6,7.
Here, we report our diagnostic experience for two clinically suspected CS patients in a Chinese family. Using multiple molecular techniques, including whole exome sequencing (WES), array comparative genomic hybridization (aCGH) and quantitative polymerase chain reaction (qPCR), we completed genetic diagnosis and counseling for this family, confirming CS due to compound ERCC8 variants. Both siblings were compound heterozygotes for variants not reported previously: a maternal splicing variant (chr5:60195556, NM_000082:c.618-2A > G) and a paternal complex rearrangement removing exon 4 of ERCC8 (chr5:60211534-60213756-60212086-60217114, hg19). Our report illustrates the diagnostic puzzle presented by some CS cases, and the advantage of approaching molecular diagnosis with high-throughput genomic techniques in the clinical genetics laboratory.
Results
Clinical report
The Capital Institute of Pediatrics Review Board approved this project. Written informed consent was obtained from the patients’ guardian/parent/next of kin for the publication of this report and any accompanying images.
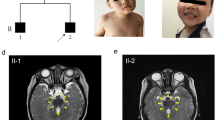
There are two affected boys in this large Chinese family. Their clinical phenotypes were evaluated by a neurologist, ophthalmologist, otolaryngologist, and a clinical geneticist. Their parents are nonconsanguineous and healthy. The mother had two marriages and ten pregnancies. During her first marriage she gave birth to one boy who died from pneumonia at one year of age and to two normal girls. Her second marriage produced three normal girls and the two affected boys in this study (Fig. 1a). Two abortions were also recorded during the second marriage.

(a) The family Pedigree. Black symbols denote affected individuals, and open symbols denote unaffected individuals. Genotypes are given for all family members who provided DNA. Del: the rearrangement removing exon 4 allele. -: the wild-type allele. (b) The facial features of the two patients. The top two photos show the proband (II:6); the bottom two photos show the affected sibling (II:10). (c) Fundus examination of the two patients. Figure PR and PL show optic nerve atrophy and retinitis pigmentosa in the right and left eye of the proband, respectively. Figure BR and BL show optic nerve atrophy in both eyes of the affected sibling. (d) The brain CT scan of the proband shows calcification of bilateral globus pallidus and subcortex of left frontal lobe, mild cerebral atrophy (widened sulcus and cleft, enlarged superatentorial ventricle) and cerebellar vermis dysplasia.
The proband (II:6) is a 13 year old boy. He was severely hypotonic at birth and could not sit or stand until age four. At five years of age, he could sit steadily and stand with aid, and began to murmur simple words such as “baba” and “mama”. He was first referred to Jiangxi Children’s Hospital at six years old due to significant developmental delay and facial photosensitivity which failed to respond to any medical treatment. Subsequently, his parents pursued a medical diagnosis at many children’s hospitals. He was transferred to the affiliated hospital of the Capital Institute of Pediatrics, and given a detailed physical checkup and genetic counseling. At 10 years old, his height and weight was 93 cm and 12 kg, and head circumference was 42.5 cm; all were 3 standard deviation (SD) or more below the mean. He had dysmorphic facial features (Fig. 1b). The brainstem auditory evoked potential (BAEP) confirmed hearing loss, and the fundus examination showed optic nerve atrophy and retinitis pigmentosa (Fig. 1c). In addition, the brain CT scan showed calcification of bilateral globus pallidus, mild cerebral atrophy and cerebellar vermis agenesis (Fig. 1d). His clinical information is summarized in Table 1.
The affected brother (II:10) is three years old, with milder clinical features. At age of three, he can stand with aid, but can’t walk alone; he can say “baba” and “mama”, and can count from 1 to 10. Unlike his brother, he has bilateral cryptorchidism. His dysmorphic facial features and clinical information are summarized in Table 1. Fundus examination showed optic nerve atrophy (Fig. 1c). The brain CT scan showed cerebellar vermis dysplasia and decreased white matter volume (data not shown).
Based upon their major clinical and facial features (severe postnatal growth failure, neurologic dysfunction, intracranial calcifications), the two patients were thought to display CS. However, sequencing for ERCC8/ERCC6 did not detect homozygous or compound heterozygous pathogenic variants although it did reveal a variant presumed to affect splicing in ERCC8. Karotyping and aCGH (Agilent SurePrint G3 4 × 180 K CGH+ SNP array) were performed for both patients, but no pathogenic genomic imbalance or uniparental disomy (UPD) was detected, so no precise genetic diagnosis could be given.
Pathogenic variant and family co-segregation analysis
To complete genetic diagnosis and counseling for this family, we pursued WES for the two affected siblings (II:6 and II:10) in an attempt to identify the cause of pathogenesis for this as yet uncertain disorder.
An average of 55.80 million reads of DNA sequence were generated with average 49.37 fold coverage, and above 97.76% of the reads were aligned to hg19, providing sufficient depth to call single nucleotide polymorphisms (SNPs) and deletion/insertion (del/ins). Common variants reported in dbSNP138 or in the 1000 Genomes Project with minor allele frequency (MAF) ≥0.005 were excluded. The Exome Aggregation Consortium (ExAC) database was used to confirm the novelty of single nucleotide variants (SNVs). Given the similar characteristics of the two siblings, shared rare variants were collected and uploaded to Ingenuity Pathways Analysis (IPA) software for genotype-phenotype analysis. A total of 826 rare variants was shared between the two patients. To narrow these to a manageable number, we set key phenotypes including “photosensitivity”, “intellectual and developmental disabilities”, “dwarfism”, “hearing loss” and “retinitis pigmentosa” in the IPA system. According to the pedigree, autosomal recessive and X-linked recessive genetic disease were both possible models for inheritance in this family. Recently, the American College of Medical Genetics and Genomics (ACMG) developed standards and guidelines for the interpretation of sequence variants: the ACMG recommends five specific standard terminologies (pathogenic, likely pathogenic, uncertain significance, likely benign and benign) to describe any individual variant identified in genes known to cause Mendelian disorders8. We strictly followed ACMG guidelines to classify pathogenic/likely pathogenic variants from WES, with null variants (nonsense, frameshift, canonical splice sites, initiation codon, single exon or multiexon deletion) and predicted deleterious missense variants receiving preferential consideration. With this strategy, 23 heterozygous variants on autosomes and one variant on the X chromosome were chosen for co-segregation analysis using all family members (Supplementary Table S1).
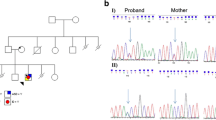
However, all but one of these candidate variants were excluded by co-segregation analysis: ERCC8 c.618-2A > G (NM_000082), a potential splicing variant, was present in both siblings (viewed by NextGENe software in Fig. 2a). Subsequent Sanger sequencing in the family members confirmed that these two siblings and one normal sister (II:4) inherited this heterozygous variant from their mother, while the father and two other unaffected sisters have the reference sequence at this site (Fig. 2b).

(a) NextGENe views of the c.618-2A > G variant of two patients. (b) Sanger traces for PCR products show the c.618-2A > G variant in the family members, confirming it as a maternally inherited splice site variant. (c) Sanger traces for RT-PCR products show 9 bp (underlined in red) deleted from exon 8 in the two patients (II:6 and II:10) and their mother (I:2) due to the c.618-2 A > G splicing variant.
Splicing variant leads to deletion of three amino acid residues
To test whether the identified c.618-2A > G (NM_000082) variant does affect pre-mRNA splicing, we generated cDNA from RNA isolated from peripheral blood of the two patients and their parents. ERCC8 reverse transcription polymerase chain reaction (RT-PCR) amplification products were cloned and sequenced. We found that this splicing variant resulted in absence in the mRNA of the first 9 bp (TGCTGACAG) of exon 8 (Fig. 2c), corresponding to loss of three amino acids (ADS, NP_000073) from position 207 to 209 within the fourth WD motif of the ERCC8 (CSA) protein.
Complex ERCC8 deletion/inversion/deletion rearrangement detected by qPCR and 1 M aCGH
We hypothesized that perhaps an undetected intragenic deletion in ERCC8 combined with the splicing variant to generate the CS phenotype. Accordingly, both exon-targeted qPCR and high density 1 M aCGH were performed to test this possibility. Using primers targeted on each exon of ERCC8, we detected a possible deletion covering exon 4 in the affected siblings (Supplementary Fig. S1). Meanwhile, 1 M aCGH conducted for the proband detected an abnormal log ratio for two probes (chr5:60212688-60219701, 7 kb, green dots in Fig. 3a) in ERCC8 suggesting the possibility of a small intragenic deletion. Long-range PCR with a series of deletion-specific primers (red arrowheads in Fig. 3a, Fm and Rm primers in Supplementary Table S2) was then performed in all family members to confirm and map this intragenic deletion. Fragment size analysis indicated deletion of 3.8 kb in the two affected patients (Fm5 and Rm4 primers in Supplementary Table S2), the father (I:3) and one sister (II:7), but not in the mother and the two other sisters (II:4 and II:8) (Fig. 3b).

(a) The location of probes in the 1 M aCGH and the deletion-specific primers. The black rectangles indicate the locations of non-deleted probes while the green rectangles indicate deleted probes. The red arrows below the map area represent the deletion-specific primers which were designed to amplify the deletion breakpoints. (b) Long-range PCR shows that the heterozygous deletions were detected in two patients (II:6 and II:10), father (I:3) and one sister (II:7). The 5.8 kb band is the wild-type allele (wt) and the 2.0 kb band is the deletion allele (del). (c) Two FoSTeS events are revealed by sequence analysis of the breakpoints. Two microhomologies (TAA and AGCT, respectively) are found at four breakpoint sites. Four base del/ins bases (tcat, yellow) were identified near one junction. (d) The putative rearrangement model of complex ERCC8 exonic deletion (not to scale) illustrates that two putative FoSTeS events occurred at four genomic rearrangement sites, resulting in deletion/inversion/deletion in the ERCC8 region. Fm and Rm were used to amplify the 2.0 kb joint product.
To confirm the breakpoints of this intragenic ERCC8 deletion, we purified the deletion-specific PCR product from an agarose gel and sequenced it (using the Fsq, Rsq, Fm5, Rm4, primers in Supplementary Table S2). These sequencing data revealed a complex deletion/inversion/deletion rearrangement (chr5:60211534-60217114) in which two distinct deletions (chr5:60211534-60213756 which removes exon 4 and chr5:60212086-60217114) flank a 1670 bp inverted segment (chr5:60212086-60213756). The sequence characteristics surrounding the four breakpoints revealed microhomologies (TAA on chr5:60211534/60213756; AGCT on chr5:60212086/60217114. see Fig. 3c,d) as potential factors contributing to the ERCC8 rearrangement. We also performed the cDNA sequencing for all available family members, and confirmed the skipping of exon 4 in the proband, sibling and father (Supplementary Fig. S2).
Discussion
ERCC8 (encoding CSA protein) is located on chromosome 5q12.1 and encodes a 44-kDa protein of 396 amino acids with seven predicted WD-40 repeat motifs5,9. CSA interacts with CSB (the product of ERCC6) to recruit other repair factors (XAB2, HMGN1, TFIIS) to the repair site during repair of UV-generated DNA damage10,11,12,13. Mutations in ERCC8 account for about 35% of the CS cases and comprise nonsense, missense, frameshift and splicing mutations, along with dosage imbalance due to small insertions and deletions7,14. In the current study, we investigated two boys characterized by growth failure, neurological impairment, microcephaly, short statue, hearing and insight loss, and photosensitivity, the characteristics of classical CS (CS type I). However, the genetic diagnosis was not given to the family until the proband was 13 years old. We performed multiple genetic testing modalities for the affected siblings to finally achieve a precise molecular diagnosis for this family. The affected siblings carry a splicing variant and a complex exonic deletion of ERCC8, which were inherited from the mother and the father, respectively. Both variants were interpreted as “pathogenic” in a recessive mode, according to the guidelines of ACMG based upon the following criteria: absent in population databases, predicted as null/deleterious variants, co-segregating with disease in multiple affected family members, presence in trans8. The maternal splicing variant (c.618-2A > G) leads to an in-frame deletion of 9 bp in exon 8, resulting in loss of three amino acids (ADS) within the fourth WD motif of CSA. At this same splice site, a different variant, c.618–1G > A, has been reported repeatedly in Caucasian patients, and the transcript analysis has confirmed that it also produces mis-splicing that removes the same amino acids (ADS) within the same WD motif7,15. We reviewed CS case reports and found that all causative missense mutations of ERCC8 identified in CS patients are located in the WD repeats7,16, implying the importance of WD motifs for building beta propeller structures and protein–protein interactions. In vitro functional analysis has also indicated that missense mutation (A205P) in the fourth WD motif affects its interaction with other proteins (DDB1)17. Beside SNVs, exonic deletions account for 16.7% (7/42) of ERCC8 mutations in CS7. Examples include a deletion containing exon 1 and upstream regulatory sequences in a CS patient reported by Ren et al.18 and double deletions in a CS patient composed of a paternally-inherited deletion covering exon 4 and a maternal whole gene deletion of ERCC814. In our patients, the deletion of exon 4 was so small that it was easily missed during routine aCGH testing due to few probes covering each exon of the ERCC8 region on the particular arrays used. This initiated a long diagnostic odyssey for this family.
With the dramatic improvements in genomic technologies and the significantly decreased price for sequencing, high-throughput whole genomic testing has been accepted as the most comprehensive and valuable testing approach for clinical diagnosis laboratory19,20,21. In general, high-throughput whole genomic testing includes whole genomic aCGH and WES. The former can detect genomic imbalances more effectively than FISH or karyotyping22,23, and the latter can detect any SNVs and del/ins in coding sequences across the genome. Compared with candidate gene or gene panel sequencing, WES provides an unbiased approach to affordably screen a patient’s entire exome to establish the genetic basis of disease. These high throughout genomic technologies have increased the diagnostic yield among individuals with unexplained developmental disorders, and rapidly revolutionized molecular diagnostics strategy, enabling physicians and patients to move to more accurate diagnostics and appropriate treatment (precision medicine). For patients with unknown unexplained autism spectrum disorder (ASD), the clinical utility of chromosomal microarray analysis (CMA) has dramatically increased the rate of diagnosis from 2.23% to 7%24. Recently, the Scherer group combined CMA and WES assays to study the genomic basis for a heterogeneous sample of ASD patients; they found that the combined molecular diagnostic yield was 15.8%, higher than the diagnostic yield of WES (8.4%) or CMA (9.3%) alone25. However, these high-throughput genomic testing approaches cannot handle all diagnostic puzzles if the geneticist does not fully integrate the information from these different techniques with accurate and complete clinical data. The identification of precise/unique symptoms is a key step in phenotype-genotype interpretation during WES. When we initially performed data analysis in the IPA system, we used the symptoms “intellectual and developmental disabilities”, “dwarfism”, and “hearing loss” but failed to identify the mode of pathogenesis because these symptoms are seen in many genetic/genomic disorders. In the second round of interpretation, we used two more specific symptoms (photosensitivity and retinitis pigmentosa) for data analysis, reducing the candidate variant pool sufficiently to permit testing of co-segregation in family members. Our results indicated the value of repeated and detailed review of the detailed phenotype of the patient to prioritize and ultimately identify causative pathogenic variants in clinical WES testing.
Notably, deletion of ERCC8 has been identified in the normal population, as evidenced by the Database of Genomic Variant track in the UCSC Genome Browser, indicating no evidence of a clinical phenotype caused by haploinsufficiency. We compared the frequency of intragenic ERCC8 deletions from one large control cohort (dgv9811n54, dgv9810n54)26 and one CS patient cohort (Laugel’s study)7. ERCC8 deletion was significantly more frequent in CS patients than in controls (7/42 vs. 12/8329, p < 0.001, two-tailed Fisher’s exact test). This emphasized that, while ERCC8 deletion is not recognized as a pathogenic CNV in the normal population, it is involved in the pathogenesis of CS when it co-occurs in trans with another pathogenic ERCC8 variant. While in the process of submitting this paper, we identified another unrelated CS patient carrying exact same exonic deletion of ERCC8 as the two patients in this study (data not shown), arguing that otherwise benign ERCC8 deletions in the general population represent a risk for causing CS in a recessive model of inheritance.
Copy number variants (CNVs) are generated by a variety of genomic rearrangements27,28,29,30,31,32,33,34. Non-allelic homologous recombination (NAHR) between low-copy repeats can cause recurrent rearrangements and lead to genomic disorders, such as DiGeorge syndrome and Williams-Beuren syndrome35. Non-homologous end-joining (NHEJ) is another mechanism contributing to chromosomal abnormality via the formation and repair of DNA double-strand breaks (DSBs), and is one of the key mechanisms underlying non-recurrent rearrangements36. DNA replication fork stalling and template switching (FoSTeS) is a recently identified mechanism for non-recurrent and especially complex rearrangements due to faulty DNA replication. A short stretch of microhomology is a key feature of the FoSTeS process. The FoSTeS molecular mechanistic details have been provided in the microhomology-mediated break-induced replication (MMBIR) model. The DNA replication FoSTeS/ MMBIR mechanism can generate complex genomic, genic and exonic rearrangements in humans37. FoSTeS had been implicated in many pathogenic genomic rearrangements, such as non-recurrent intragenic NRXN1 deletion38 and duplication/deletion in PLP1 associated with Pelizaeus-Merzbacher disease (PMD)39. In the current study, we mapped this complex ERCC8 intragenic rearrangement (deletion/inversion/deletion) at the nucleotide level and found microhomologies (TAA and AGCT) around the breakpoint sites supporting the view that two FoSTeS events were involved in generating the ERCC8 complex rearrangement.
In summary, we describe the clinical and genetic characterization of two affected boys with CS type I in a Chinese family. Following the guidelines for the interpretation of sequence variants of the ACMG, we identified compound heterozygosity, including a paternal intragenic rearrangement and a maternal splicing variant in ERCC8, as the cause of CS pathogenesis in these individuals. In the clinical diagnosis of many rare neurological diseases, combined utilization of careful clinical phenotyping with high-throughput genomic technologies, such as WES and aCGH, can increase the diagnostic yield and provide much-needed answers for families faced with these disorders.
Methods
Ethics statement
This study was performed in accordance with the Declaration of Helsinki and approved by the ethics committee of Capital Institute of Pediatrics (SHERLL 2015069). Written informed consent was obtained from the patients’ guardian/parent/next of kin for the publication of this clinical information and any accompanying images.
aCGH
Genomic DNAs of the proband and all available family members were extracted from peripheral blood using the QIAamp DNA Blood Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. Two aCGH (4 × 180 K and 1 M) (Agilent Technologies, Palo Alto, CA, USA) were used to detect genomic imbalance according to previously published methods40. aCGH data were analyzed via DNA CytoGenomics software (Agilent Technologies, Palo Alto, CA, USA).
Whole exome sequencing and variant analysis
Genomic DNAs of the two patients were fragmented by Covaris S2 (Covaris, Massachusetts, USA) to 200–300 bp. The paired-end libraries were prepared following the Illumina protocol. Whole exome sequences were captured from the genomic DNA using Agilent SureSelect V5 Enrichment kit (Agilent, Cedar Creek, TX). The exon-enriched DNA libraries were sequenced by 100 bp paired-end reads on a Hiseq2000 sequencer (Illumina, San Diego, California). Raw image files were processed by the Illumina Pipeline for base calling using default parameters.
After image analysis and base calling was conducted using the Illumina Pipeline, raw data were transfer into fastq form and filtered to generate “clean reads” by removing adapters and low quality reads (Q20). Sequencing reads were mapped to the reference human genome version hg19 (200902 release, http://genome.ucsc.edu/) and variants were visualized and analyzed by NextGENe software version 2.1.1.1 (SoftGenetics, StateCollege, PA) and Ingenuity Variant Analysis software (http://www.ingenuity.com).
qPCR
To test whether the affected patients (II:6, II:10) had the ERCC8 intragenic deletion, qPCR was performed using the 7500 Fast Real-Time PCR System with GAPDH as internal reference gene for each sample. Two unrelated testing samples without ERCC8 deletion (validated by 244k aCGH beforehand) were used as controls. The qPCR was carried out in the presence of SYBR Green, measuring the fluorescence signal produced by the binding of SYBR Green to the studied amplicons compared with the control. The qPCR primers for reference and target genes are in Supplementary Table S3.
Long-range PCR and breakpoint mapping on the junction sequence
Long-range PCR was carried out to amplify the truncated fragment covering exon 4 of ERCC8 and map out the characteristics of breakpoints (Platinum PCR SuperMix High Fidelity kit, Invitrogen, Carlsbad, CA). Using a series of primers around the approximate breakpoints (Supplementary Table S2), the junction fragments were amplified successfully and were visualized on a 1% agarose gel. The truncated fragment was purified from the gel (QIAquick gel extraction kit, Qiagen, Valencia, CA) and sequenced (ABI 3700).
RT-PCR, Cloning and Sequencing
Total RNA was isolated from fresh peripheral blood using the RNeasy mini kit (Qiagen, Hilden, Germany) and was reverse transcribed with ProtoScript® First Strand cDNASynthesis Kit (New England Biolabs Inc., MA, USA). PCR amplification of cDNA including spanning exon 3-12 was performed using the primer pair 5′-CACATGTAAAGCAGTGTGTTCC-3′and 5′-GCATTTCATGTTTAAGCCAGATT-3′, visualized on a 1% agarose gel, purified using QIAquick PCR Purification Kit (Qiagen, Hilden, Germany), and then cloned into a pUC19 vector for Sanger sequencing following the manufacturer’s protocol. A mixture of DNA from 10 normal children was used as the control.
Additional Information
How to cite this article: Xie, H. et al. A complex intragenic rearrangement of ERCC8 in Chinese siblings with Cockayne syndrome. Sci. Rep. 7, 44271; doi: 10.1038/srep44271 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Cockayne, E. A. Dwarfism with retinal atrophy and deafness. Archives of disease in childhood 11, 1–8 (1936).
Nance, M. A. & Berry, S. A. Cockayne syndrome: review of 140 cases. American journal of medical genetics 42, 68–84 (1992).
Troelstra, C. et al. ERCC6, a member of a subfamily of putative helicases, is involved in Cockayne’s syndrome and preferential repair of active genes. Cell 71, 939–953 (1992).
Bootsma, D. et al. Nucleotide excision repair syndromes: molecular basis and clinical symptoms. Philosophical transactions of the Royal Society of London. Series B, Biological sciences 347, 75–81 (1995).
Henning, K. A. et al. The Cockayne syndrome group A gene encodes a WD repeat protein that interacts with CSB protein and a subunit of RNA polymerase II TFIIH. Cell 82, 555–564 (1995).
Pasquier, L. et al. Wide clinical variability among 13 new Cockayne syndrome cases confirmed by biochemical assays. Archives of disease in childhood 91, 178–182 (2006).
Laugel, V. et al. Mutation update for the CSB/ERCC6 and CSA/ERCC8 genes involved in Cockayne syndrome. Human mutation 31, 113–126 (2010).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in medicine: official journal of the American College of Medical Genetics 17, 405–424 (2015).
Zhou, H. X. & Wang, G. Predicted structures of two proteins involved in human diseases. Cell biochemistry and biophysics 35, 35–47 (2001).
Fousteri, M., Vermeulen, W., van Zeeland, A. A. & Mullenders, L. H. Cockayne syndrome A and B proteins differentially regulate recruitment of chromatin remodeling and repair factors to stalled RNA polymerase II in vivo. Molecular cell 23, 471–482 (2006).
Groisman, R. et al. The ubiquitin ligase activity in the DDB2 and CSA complexes is differentially regulated by the COP9 signalosome in response to DNA damage. Cell 113, 357–367 (2003).
Saijo, M. et al. Functional TFIIH is required for UV-induced translocation of CSA to the nuclear matrix. Molecular and cellular biology 27, 2538–2547 (2007).
Groisman, R. et al. CSA-dependent degradation of CSB by the ubiquitin-proteasome pathway establishes a link between complementation factors of the Cockayne syndrome. Genes & development 20, 1429–1434 (2006).
Ting, T. W. et al. Cockayne Syndrome due to a maternally-inherited whole gene deletion of ERCC8 and a paternally-inherited ERCC8 exon 4 deletion. Gene 572, 274–278 (2015).
Bertola, D. R. et al. Cockayne syndrome type A: novel mutations in eight typical patients. Journal of human genetics 51, 701–705 (2006).
Saijo, M. The role of Cockayne syndrome group A (CSA) protein in transcription-coupled nucleotide excision repair. Mech Ageing Dev 134, 196–201.
Jin, J., Arias, E. E., Chen, J., Harper, J. W. & Walter, J. C. A family of diverse Cul4-Ddb1-interacting proteins includes Cdt2, which is required for S phase destruction of the replication factor Cdt1. Molecular cell 23, 709–721 (2006).
Ren, Y. et al. Three novel mutations responsible for Cockayne syndrome group A. Genes & genetic systems 78, 93–102 (2003).
Han, S. M. et al. Genetic testing of Korean familial hypercholesterolemia using whole-exome sequencing. PloS one 10, e0126706 (2015).
Johar, A. S. et al. Candidate gene discovery in autoimmunity by using extreme phenotypes, next generation sequencing and whole exome capture. Autoimmunity reviews 14, 204–209 (2015).
Ding, D. et al. Identification of a de novo DYNC1H1 mutation via WES according to published guidelines. Scientific reports 6, 20423 (2016).
Kan, A. S. et al. Whole-genome array CGH evaluation for replacing prenatal karyotyping in Hong Kong. PloS one 9, e87988 (2014).
Evangelidou, P. et al. Implementation of high resolution whole genome array CGH in the prenatal clinical setting: advantages, challenges, and review of the literature. BioMed research international 2013, 346762 (2013).
Shen, Y. et al. Clinical genetic testing for patients with autism spectrum disorders. Pediatrics 125, e727–735 (2010).
Tammimies, K. et al. Molecular Diagnostic Yield of Chromosomal Microarray Analysis and Whole-Exome Sequencing in Children With Autism Spectrum Disorder. Jama 314, 895–903 (2015).
Genet, N. et al. Home care in Europe: a systematic literature review. BMC health services research 11, 207 (2011).
Iafrate, A. J. et al. Detection of large-scale variation in the human genome. Nature genetics 36, 949–951 (2004).
Sebat, J. et al. Large-scale copy number polymorphism in the human genome. Science 305, 525–528 (2004).
Feuk, L., Carson, A. R. & Scherer, S. W. Structural variation in the human genome. Nature reviews. Genetics 7, 85–97 (2006).
Redon, R. et al. Global variation in copy number in the human genome. Nature 444, 444–454 (2006).
Flores, M. et al. Recurrent DNA inversion rearrangements in the human genome. Proceedings of the National Academy of Sciences of the United States of America 104, 6099–6106 (2007).
Korbel, J. O. et al. Paired-end mapping reveals extensive structural variation in the human genome. Science 318, 420–426 (2007).
Kidd, J. M. et al. Mapping and sequencing of structural variation from eight human genomes. Nature 453, 56–64 (2008).
Perry, G. H. et al. The fine-scale and complex architecture of human copy-number variation. American journal of human genetics 82, 685–695 (2008).
Edelmann, L., Pandita, R. K. & Morrow, B. E. Low-copy repeats mediate the common 3-Mb deletion in patients with velo-cardio-facial syndrome. American journal of human genetics 64, 1076–1086 (1999).
Chen, J. M., Cooper, D. N., Ferec, C., Kehrer-Sawatzki, H. & Patrinos, G. P. Genomic rearrangements in inherited disease and cancer. Seminars in cancer biology 20, 222–233 (2010).
Zhang, F. et al. The DNA replication FoSTeS/MMBIR mechanism can generate genomic, genic and exonic complex rearrangements in humans. Nature genetics 41, 849–853 (2009).
Chen, X. et al. Molecular analysis of a deletion hotspot in the NRXN1 region reveals the involvement of short inverted repeats in deletion CNVs. American journal of human genetics 92, 375–386 (2013).
Lee, J. A., Carvalho, C. M. & Lupski, J. R. A DNA replication mechanism for generating nonrecurrent rearrangements associated with genomic disorders. Cell 131, 1235–1247 (2007).
Shen, Y. & Wu, B. L. Microarray-based genomic DNA profiling technologies in clinical molecular diagnostics. Clinical chemistry 55, 659–669 (2009).
Acknowledgements
This work is partially funded by the Capital Health Research and Development of Special (2014-2-1131), the National Nature Science Fund [31671310 and 81370708 to XLC, 81401207 to HX], and Beijing Natural Science Foundation [7162029 to XLC] and the advanced Personnel Training Program of Beijing Municipal Health Bureau and Beijing nova program interdisciplinary collaborative project [xxjc201717] to XLC, and NIH grant [P01GM061354 to JFG] as well as CAMS Initiative for Innovative Medicine (CAMS-I2M).
Author information
Authors and Affiliations
Contributions
X.L.C. designed the study; H.X., X.Y.L., J.P.P., X.Z.S., X.L.C. and C.H.L. performed D.N.A. and R.N.A. extraction, 4 × 180 K aCGH experiment, WES experiment, Sanger sequencing, RT-PCR, qPCR, mapping and data analysis; W.Y.L. and JQX performed 1 M aCGH experiment; H.X., X.L.C. performed manuscript writing. Q.C., Z.J.G. and J.M.Z. helped in patient and family members’ recruitment and clinical checkup and obtaining a genetics questionnaire; J.F.G. revised the manuscript; T.Z. gave key suggestions for design and manuscript writing. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Xie, H., Li, X., Peng, J. et al. A complex intragenic rearrangement of ERCC8 in Chinese siblings with Cockayne syndrome. Sci Rep 7, 44271 (2017). https://doi.org/10.1038/srep44271
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep44271
- Springer Nature Limited