
Abstract
Nonsyndromic oculocutaneous Albinism (nsOCA) is clinically characterized by the loss of pigmentation in the skin, hair, and iris. OCA is amongst the most common causes of vision impairment in children. To date, pathogenic variants in six genes have been identified in individuals with nsOCA. Here, we determined the identities, frequencies, and clinical consequences of OCA alleles in 94 previously unreported Pakistani families. Combination of Sanger and Exome sequencing revealed 38 alleles, including 22 novel variants, segregating with nsOCA phenotype in 80 families. Variants of TYR and OCA2 genes were the most common cause of nsOCA, occurring in 43 and 30 families, respectively. Twenty-two novel variants include nine missense, four splice site, two non-sense, one insertion and six gross deletions. In vitro studies revealed retention of OCA proteins harboring novel missense alleles in the endoplasmic reticulum (ER) of transfected cells. Exon-trapping assays with constructs containing splice site alleles revealed errors in splicing. As eight alleles account for approximately 56% (95% CI: 46.52–65.24%) of nsOCA cases, primarily enrolled from Punjab province of Pakistan, hierarchical strategies for variant detection would be feasible and cost-efficient genetic tests for OCA in families with similar origin. Thus, we developed Tetra-primer ARMS assays for rapid, reliable, reproducible and economical screening of most of these common alleles.
Similar content being viewed by others


Introduction
Autosomal recessive non-syndromic oculocutaneous albinism (nsOCA) is a genetic disorder characterized by the partial or complete loss of pigmentation in the skin, hair, and iris that is due to a decrease or absence of melanin production1,2. Other clinically substantial aberrations associated with OCA are foveal hypoplasia, photoreceptor rod cell deficit, misrouting of the optic nerves at the chiasm, photophobia, nystagmus, and vision impairment2,3. The prevalence of albinism worldwide has been estimated at 1 in 17,000, meaning that approximately 1 in 70 people are carriers of the OCA allele. To date, pathogenic variants in six genes, TYR, OCA2, TYRP1, SLC45A2, SLC24A5, and C10orf11, have been identified in individuals with nsOCA4. We previously mapped a new locus for nsOCA on chromosome 4q24 in a large consanguineous Pakistani family5, for which the gene is currently unknown. In published population studies, however, the detection rate of alleles causing albinism varies from 60% to 90%6,7,8,9,10,11.
The clinical range of OCA varies, with OCA1A being the most severe type, characterized by a complete lack of melanin production throughout life. The less severe forms (OCA1B, OCA2, OCA3, and OCA4) show some pigmentation over a lifetime. Although the diverse types of OCA are caused by variants in different genes, their clinical phenotypes are not always distinguishable, making molecular diagnosis an essential tool for genetic counseling12, and for emerging therapeutic interventions13. Our goals here were to determine identities, frequencies, and clinical consequences of OCA alleles in Pakistani cohort, predominantly enrolled from Punjab province, and to develop hierarchical strategies for rapid, feasible, and cost-efficient genetic diagnostic assays for improved carrier detection and genetic counseling.
Results
After institutional review board approval, we enrolled 94 Pakistani families segregating nsOCA (Supplementary Fig. S1). All the affected individuals of these families have congenital hypopigmentation phenotype. Inter-familial variation of hair color was noted among individuals, ranging from white to honey blonde or brown (Table 1). Similarly, variation in iris color was noted, with tones ranging from light grey to brown (Table 1). However, due to limited facilities available in the remote areas of Pakistan, detailed clinical evaluations in every affected person from every family was not possible, therefore, we refrained ourselves from commenting on genotype-phenotype correlation for every variant. Through the combination of Sanger and whole exome sequencing techniques, we identified 38 variants, including 22 novel variants, segregating with the phenotype in 80 families (Table 2). The 22 new variants include 9 missense, 4 splice site, 2 non-sense, 1 insertion, and 6 gross deletions (Table 2 and Fig. S2). None of these variants were found in ethnically matched control samples (Table 2 and Table S1). We also documented the frequencies of polymorphic alleles of nsOCA genes in our cohort (Table S2).
Missense variants altered the targeting of encoded OCA proteins
The nine novel missense variants include two alleles of TYR (OCA1), six of OCA2 (OCA2) and one in TYRP1 (OCA3). Three prediction programs, specifically, Polyphen-214, MutationTaster15 and SIFT16, suggested that each of these missense variants were deleterious (Table 2). To further evaluate the effects of these variants on the encoded proteins, we performed in silico molecular modeling and in vitro protein targeting studies.
TYR encodes tyrosinase in melanocytes, an essential enzyme for the biosynthesis of melanin17. Previously, it was shown that missense alleles of tyrosinase lead to ER retention of encoded protein due to misfolding18. To evaluate the targeting of p.Cys55Ser and p.Asp75Tyr harboring proteins, we introduced these variants in a GFP-tagged tyrosinase and transiently transfected human melanocytes. Wild type tyrosinase was localized throughout the cytoplasm of melanocytes (Fig. 1). Immunofluorescence studies with calregulin (an ER marker) demonstrated that the mutant proteins, however, predominantly co-localized with calregulin, indicating retention in the ER (Fig. 1). In contrast, p.(Asp86Tyr) missense variant did not affect the targeting of TYRP1 protein (Fig. 1).

eGFP-tagged TYR and TYRP1 wild type and mutant constructs (green) were transiently transfected in melanocytes grown at 37 °C. Calregulin (red) and DAPI (blue) were used as markers for the endoplasmic reticulum and nucleus, respectively. Merged images show the co-localization of only tyrosinase variants with calregulin, which indicated ER retention. The scale bar represents 20 μm for all panels.
OCA2 protein, with 12 putative transmembrane α-helices, belongs to the Na+/H+ antiporter family. Besides its presumed role in maintaining the pH of the melanosomes19,20,21,22, OCA2 also participates in the sorting and transport of tyrosinase and tyrosinase-related protein 1 (TYRP1) to the plasma membrane23,24,25. We performed comparative computational modeling of wild type and six novel missense alleles harboring OCA2 proteins using Phyre226 program. All the identified missense alleles were predicted to impact either protein folding, interaction with the lipid-bilayer, protein topology, or protein-protein interactions (Fig. S3). When transiently transfected in HEK293 cells, in contrast to wild type, disease-associated alleles of OCA2 proteins showed retention in the ER (Fig. 2). Collectively, these studies support the deleterious nature of novel missense variants identified in our OCA cohort.

eGFP-tagged OCA2 wild type and mutant constructs (green) were transiently transfected in HEK293 cells grown at 37 °C. Calregulin (red) and DAPI (blue) were used as markers for the endoplasmic reticulum and nucleus, respectively. Merged images show partial co-localization of OCA2 variants with calregulin, which indicated increased ER retention. The scale bar represents 20 μm for all panels.
Ex vivo splicing is defective due to splice site variants identified in Pakistani families
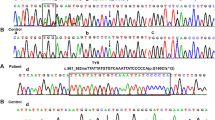
To evaluate the impact of four new splice site variants elucidated in our cohort, we examined the RNA splicing pattern of wild type and mutated exons by transfecting minigene constructs in COS7 cells. Results of our splicing assays are summarized in Fig. 3. The c.1037–18 T > G variant in exon 3 of TYR generated an upstream cryptic splice acceptor site, which resulted in insertion of 17 base intronic region in the spliced product (Fig. 3). Retention of 17 base intronic region in the spliced mRNA would result in the frameshift and premature truncation of the encoded tyrosinase. In contrast, the c.1184 + 2 T > C change in exon 3 of TYR revealed loss of canonical splice donor site. Splicing assay revealed utilization of a cryptic donor site within exon 3, resulting in loss of 55 bp from the coding region (Fig. 3), predicted to cause frameshift and premature truncation of the encoded protein. Both splice site variants of OCA2 (c.1182 + 2 T > TT and c.1951 + 4 A > G) revealed skipping of their respective exons in the minigene splicing assays (Fig. 3). The skipping of exon 11 (66 bp) due to c.1182 + 2 T > TT leads to in-frame deletion of 22 amino acids, while the loss of exon 18 (109 bp) of OCA2 due to c.1951 + 4 A > G variant would cause frameshift and premature truncation.

Schematic representation and Sanger sequencing chromatograms of minigene assays for TYR and OCA2 novel splice site variants revealed aberrant splicing products, supporting their pathogenic nature.
Exome sequencing revealed six gross deletions in OCA families
Approaches to detect indels using exome sequence data are an active area of research. As yet there is no single method that guarantees consistent success. We used the widely-evaluated methods XHMM27 and CoNIFER28 to identify gross insertions/deletions in our exome data. Our analyses revealed six novel gross deletions of TYR and OCA2 genes segregating with OCA phenotype in six families (Fig. 4). To investigate the mechanisms involved in these deletions, the intervals surrounding the breakpoints were analyzed through RepeatMasker (http://www.repeatmasker.org/). In addition, significantly over-represented motifs within ±15 bp of GRaBD translocations breakpoints were also sought29. Thorough bioinformatics analysis revealed that the breakpoint for these deletions lie in the repetitive element, and the highly similar Alu short interspersed nuclear elements (SINEs) may serve as the substrate for nonhomologous recombination.

(A) Compare the exome sequencing data (green-blue peaks) of control sample wild type sample, the exon 4 and 5 (indicated by red arrows) were deleted in DNA samples from PKAB64 and PKAB168 families. Breakpoints for genomic deletions are given according to the human genome build hg19/GRCh37. (B) Gross genomic deletions observed in OCA2 gene as compared to control samples are shown either by red arrows or red lines.
Next, to analyze the impact of deletions on protein 3D structure, we used Phyre2 modeling software. Removal of p.Ser395 to p.Leu529 amino acids due to deletion of exons 4 and 5 would eliminate the tyrosinase central domain that binds to its copper ligand for subsequent function (Fig. S4A). Similarly, deletions of OCA2 exons would result in the partial or complete loss of Na-Sulphur-symporter domain, which mediates the intake of several different molecules with the concomitant uptake of Na+ (Fig. S4B) and thus predicted to result in non-functional, truncated proteins.
Alleles of TYR and OCA2 are the common cause of nsOCA in our cohort
Variants of TYR (allele frequency: 15/38) and OCA2 (19/38) were the most common cause of nsOCA, occurring in 43 and 30 families, respectively (Fig. 5A). To further refine the prevalence estimates, we reviewed the alleles frequencies in known albinism genes among our larger cohort of 143 families, including 48 previously reported families (Fig. 5B)5,30,31,32,33 and in published literature (Table S1). TYR and OCA2 alleles are the frequent cause of OCA in Pakistanis (Table S1). In our cohort, variants in TYR and OCA2 collectively account for the majority [67.8% (97/143); 95% confidence interval (CI): 60.2–76.0%] of the genetic causes of nsOCA, which is comparable to prevalence in European population (Table S3). In approximately 14% of our OCA families, we did not find any pathogenic variant in the known OCA genes (Fig. 5B).

(A–C) Relative distribution of variants in albinism genes in Pakistani families. (A) The distribution of OCA1-4 genes in 94 families screened in the current study. Number of families and their percentage contribution are given in parentheses. (B) Total pool of albinism genes in our cohort. (C) Frequencies of recurrent alleles of nsOCA genes in Pakistanis. (D) Results of tetra-primer ARMS assays for detection of recurrent variants. The top band in each gel represents the positive control amplimer in all samples generated using the outer primers. Nested allele-specific primers were used to generate the wild type (Wild) or variant harboring (Mutant) PCR products. IC: inner control product.
Next, we investigated the frequency of alleles of nsOCA genes in our cohort. Overall, four alleles of TYR, three of OCA2, and one of SLC45A2 together account for ~56% (95% CI: 46.52–65.24%) of the variants responsible for nsOCA in our cohort (Fig. 5C). Therefore, we developed rapid and inexpensive assays for detecting carriers and homozygotes (Fig. 5D). For most of these alleles, we were able to develop tetra-primer ARMS assays. The sensitivity and specificity of these assays were confirmed on multiple DNA samples with different genotypes followed by Sanger sequencing.
Discussion
Our study illustrates the relative genetic contribution of four major OCA genes in the prevalence of albinism, primarily in families (69) currently residing in the Punjab province of Pakistan. However, our study also includes 19 families from Sindh and 6 families from Khyber Pakhtunkhwa (KPK) provinces. Pakistanis have a rich anthropogenic background owing to successive waves of invasions and adaptations of haplogroups. Most did not intermingle with the original local population and practiced endogamy, giving rise to genetic isolates that persists even today. Parental consanguinity is an important risk factor (0.25–20% higher chances) for recessive genetic defects34. In Pakistan, 62.7% of marriages are consanguineous, ~80% of which are between first cousins35. Specific clans and high consanguinity in Pakistan are the root causes of increased incidences of recessive disorders, including OCA. In our cohort, OCA phenotype was observed in families of different linguistic/ethnic origins (Table 1). We did not observe any apparent enrichment of a particular OCA allele within families of certain clans (Table 1). For instance, c.832 C > T in TYR and c.1045–15 T > G in OCA2 are the most frequent alleles observed in our samples (Fig. 5C). However, both these alleles were observed in families of various ethnical and geographical origins (Table 1). Similarly, other common variants (e.g. c.649 C > T, c.1255 G > A, c.1456 G > T) were also found in families enrolled from Punjab, Sindh and KPK (Table 1). Therefore, with the current samples size for each of the identified allele, it would be inapplicable to comment on ethnic/linguistic origins of different alleles observed in our cohort.
Our overall estimated prevalence of TYR and OCA2 alleles is quite similar in certain cases from Europe but fairly different from other studies in different populations (Table S3). For instance, TYR and OCA2 variants account for 70% and 10%, respectively, of OCA in a study of 127 patients from a Chinese population (Table S3). In a few eastern and central regions of China, TYR and OCA2 contribution varies; however, TYR alleles remain the most common cause of OCA. In India, a study of 82 OCA patients revealed approximately 60% and 11% prevalence for TYR and OCA2, respectively36. Similarly, in the US, Europe, Italy, Japan, and Korea, the alleles of TYR are the most common cause of nsOCA7,8,9,11,37 (Table S3). In contrast, variants in OCA2 account for ~80% of the OCA cases in an African population38.
Usually the alleles of TYR result in the retention of encoded tyrosinase in the ER18,30. Here, we also observed retention of OCA2 proteins harboring missense and truncating alleles in the ER (Fig. 2). A portion of the known human and mouse TYR variants have shown temperature-sensitive behavior30,39,40,41,42,43,44. Cultivating mammalian cells at permissive temperature (31 °C) resulted in increased cytoplasmic expression of tyrosinase harboring missense alleles, especially for those present in the copper-binding region30,39,41,42,44. However, the new OCA2 alleles were imaged at 31 °C and did not reveal an apparent increased cytoplasmic expression (data not shown).
Besides single nucleotide variants, our study, for the first time in Pakistani population, revealed six novel gross deletions in TYR and OCA2 genes (Fig. 4). These genetic aberrations span from a single exon deletion up to 12 exons with the encompassed introns (Fig. 4). These deletions confer pigmentation disorders either because of almost entire protein would be missing or non-functional truncated forms would be encoded. Considering their relative contribution simplified methods and bioinformatics tools will be needed for rapid detection of gross deletions for clinical genetic diagnosis of OCA.
Clinical presentation of OCA is often not very helpful in genetic diagnosis due to significantly overlapping features (Table 1). For economic and geographical reasons, it is not feasible to routinely perform Sanger sequencing of all the known OCA genes to detect underlying genetic defects. However, PCR-based assays that are developed to detect common alleles are quick and affordable. Twenty of the variants identified in this study were private (found in single families). However, eight variants (Fig. 5C) account for more than half of the alleles found in our families (Table 2). Therefore, we developed tetra-primer ARMS assays for these common alleles for rapid detection and genetic screening in large cohorts before embarking for exome/genome wide studies. We are cognizant of the fact that the majority of families in our cohort are from the Punjab province of Pakistan. Hence, the details of a hierarchical nonsyndromic OCA genes mutational screening strategy may need to be refined for other geographical regions and lingo-ethnic groups within Pakistan. Our results will be helpful for future diagnosis, genetic counseling, molecular epidemiology, and functional studies of nsOCA genes associated pigmentation disorders.
Intriguingly, fourteen families in our cohort did not reveal any pathogenic variants in common nsOCA genes. There are several possibilities for our failure to detect disease-associated alleles in these fourteen families. First, potential pathogenic variants may alter sequence of cis-acting regulatory or deep intronic splicing elements that are necessary for expression of these genes in melanocytes. Presently, we have very limited knowledge of the location of the regulatory elements of nsOCA genes. Secondly, the hypopigmentation segregating in these fourteen families may be syndromic and requires comprehensive clinical phenotyping, which would help in filtering the candidate genetic variants for further assessments. Third, there may be additional genes responsible for OCA in humans. For instance, in the US population the detection rate of alleles in known nsOCA genes is around 75%10. Thus, there exists both a need for further genetic understanding of albinism and an opportunity to improve the molecular diagnosis of albinism, and quite possibly its prevention.
Methods
Patients
This study followed the tenets of the Declaration of Helsinki. This study was approved by the IRB Committees at the University of Maryland School of Medicine (UMSOM), USA, the Institute of Molecular Biology & Biotechnology, Multan, Liaquat University of Medical & Health Sciences, Jamshoro, and Gomal Centre of Biochemistry and Biotechnology, Gomal University, D.I. Khan, Pakistan. All the methods were performed in accordance with the UMSOM relevant guidelines and regulations. Pedigrees were drawn after interviewing multiple individuals to confirm the relationships. Informed written consents were obtained from the adult subjects and the parents of minor subjects. Two to five ml of peripheral blood samples were collected from each participating individual. Human genomic DNA was isolated from peripheral blood by using inorganic method45. Detailed medical histories were taken for all of the participating individuals of the enrolled families, including the hypopigmentation phenotype of the hair, skin, eye, disease onset, segregation, presence of eye abnormalities (nystagmus, strabismus, photophobia, and poor vision) and information about immunological, neurological or bleeding time.
Sanger sequencing and segregation analysis
Primers (Table S4) covering the coding regions of the TYR, OCA2, TYRP1 and SLC45A2 genes were designed in primer3 software (http://bioinfo.ut.ee/primer3-0.4.0/primer3/input.htm) for Sanger sequencing and segregation analysis. PCR amplification, cleaning, and sequencing reactions were performed as previously described30,32.
Exome Sequencing
DNA samples from some of the participating family members were submitted for exome sequencing (WES) at the Center for Mendelian Genomics (CMG), University of Washington. WES on PKAB137 was performed at the Genetic core facility of Cincinnati Children’s Hospital. Genomic libraries were recovered for exome enrichment using the NimbleGen Exome kits (Roche Diagnostics; San Francisco, CA). Paired-end sequencing was performed using the Illumina Hi-Seq 2000 system (Illumina, San Diego, CA). The obtained sequencing data were analyzed following the guidelines outlined in the Broad Institute’s Genome Analysis Toolkit46. The raw data were mapped using the Burrows Wheeler Aligner47. Variants were called using Unified Genotyper software, and the data were then subjected to further processing and quality control46,47. Golden Helix software was used to analyze the variants found through exome sequencing. The coverage of exome was checked through Golden Helix Genome Browser.
Bioinformatics Analysis
Pathogenicity of novel missense variants was assessed by different bioinformatics prediction programs; Polyphen-2, Sift, and Mutation Taster. Allele frequencies of identified variants were checked in 1000 Genome browser (http://browser.1000genomes.org/), NHLBI-ESP variant database (http://evs.gs.washington.edu/EVS/) and ExAC database (http://exac.broadinstitute.org/). Multiple sequence alignments of orthologous OCA proteins were performed by using Clustal Omega multiple alignment program (http://www.ebi.ac.uk/Tools/msa/clustalo/). The 3D protein structures were predicted by Phyre2 server (http://www.sbg.bio.ic.ac.uk/phyre2/html/page.cgi?id=index) with “Intensive Mode” option which is a combination of template-based modeling and ab initio methods48.
Tetra Primer Amplification Refractory Mutation System (ARMS) assay
PCR primers (Table S5) used for Tetra-primers ARMS assay were designed by adjusting maximum (1:8) and minimum (1:3) relative size difference of two inner products and keeping others by default settings. Reaction mixture contains 10 ul 2X ECONOTAQ, 0.5 ul of 10 Um outer primers and 1.0 ul of 10 Um inner primers each, 50 ng (2 ul) genomic DNA and 5 ul H2O. The thermo cycling conditions are: initial denaturation of 4 min at 95 °C and followed by 35 cycles of 95 °C for 30 sec, 60 °C for 45 sec, 72 °C for 45 sec, and a final extension at 72 °C for 8 min. The PCR product was visualized in 2.0% agarose gel stained with ethidium bromide.
Exon-trapping assay
To ascertain the consequences of novel splice site variants found in our OCA families, the wild and mutant exons along with flanking intronic region (200 bp) were PCR amplified, cloned in pSPL3 vector (Invitrogen, Carlsbad, CA) and sequence verified as described49. Purified cloned constructs were transfected into COS7 cells using PEI (Polyethylenimine). After 48 hours of transfection, RNA was extracted using TRIzol reagent (Invitrogen, Carlsbad, CA) and single stranded cDNA (Clontech, Mountain View, CA) was synthesized. Primary PCR amplification of cDNA was performed using SD6 and SA2 vector primers and amplified products were cloned in TA-cloning vector (Invitrogen). At least ten bacterial clones for each construct were Sanger sequenced.
Expression constructs, transfection and immunofluorescence
All expression constructs used for immunofluorescence study were in eGFP-tagged vector (Clontech). For generating mutant constructs, mutagenesis was performed by using QuikChange kit (Stratagene, La Jolla, CA) and used specific wild type eGFP construct as a template. Each construct was transiently transfected in melanocytes or HEK293 cells seeded on cover slips with Lipofectamine 3000 (Invitrogen). After 48 hours of transfection either at 37 °C or 31 °C, cells were fixed and permeabilized in 4% paraformaldehyde and 0.1% Triton X-100 in PBS, respectively. For endoplasmic reticulum and nucleus visualization anti-Calregulin antibodody (Santa Cruz Biotechnology, Santa Cruz, CA) and DAPI staining was performed. A Zeiss LSMDUO confocal microscope was used for imaging.
Additional Information
How to cite this article: Shahzad, M. et al. Molecular outcomes, clinical consequences, and genetic diagnosis of Oculocutaneous Albinism in Pakistani population. Sci. Rep. 7, 44185; doi: 10.1038/srep44185 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Spritz, R. A., Chiang, P. W., Oiso, N. & Alkhateeb, A. Human and mouse disorders of pigmentation. Curr Opin Genet Dev 13, 284–289 (2003).
Tomita, Y. & Suzuki, T. Genetics of pigmentary disorders. Am J Med Genet C Semin Med Genet 131C, 75–81, doi: 10.1002/ajmg.c.30036 (2004).
Oetting, W. S. & King, R. A. Molecular basis of albinism: mutations and polymorphisms of pigmentation genes associated with albinism. Hum Mutat 13, 99–115, doi: 10.1002/(SICI)1098-1004(1999)13:2<99::AID-HUMU2>3.0.CO;2-C (1999).
Montoliu, L. et al. Increasing the complexity: new genes and new types of albinism. Pigment Cell Melanoma Res 27, 11–18, doi: 10.1111/pcmr.12167 (2014).
Kausar, T., Bhatti, M. A., Ali, M., Shaikh, R. S. & Ahmed, Z. M. OCA5, a novel locus for non-syndromic oculocutaneous albinism, maps to chromosome 4q24. Clin Genet 84, 91–93, doi: 10.1111/cge.12019 (2013).
Hutton, S. M. & Spritz, R. A. A comprehensive genetic study of autosomal recessive ocular albinism in Caucasian patients. Invest Ophthalmol Vis Sci 49, 868–872, doi: 10.1167/iovs.07-0791 (2008).
Mauri, L. et al. Clinical evaluation and molecular screening of a large consecutive series of albino patients. J Hum Genet, doi: 10.1038/jhg.2016.123 (2016).
Park, S. H., Chae, H., Kim, Y. & Kim, M. Molecular analysis of Korean patients with oculocutaneous albinism. Jpn J Ophthalmol 56, 98–103, doi: 10.1007/s10384-011-0098-z (2012).
Rooryck, C. et al. Molecular diagnosis of oculocutaneous albinism: new mutations in the OCA1-4 genes and practical aspects. Pigment Cell Melanoma Res 21, 583–587, doi: 10.1111/j.1755-148X.2008.00496.x (2008).
Simeonov, D. R. et al. DNA variations in oculocutaneous albinism: an updated mutation list and current outstanding issues in molecular diagnostics. Hum Mutat 34, 827–835, doi: 10.1002/humu.22315 (2013).
Tomita, Y., Miyamura, Y., Kono, M., Nakamura, R. & Matsunaga, J. Molecular bases of congenital hypopigmentary disorders in humans and oculocutaneous albinism 1 in Japan. Pigment Cell Res 13 Suppl 8, 130–134 (2000).
Oetting, W. S., Fryer, J. P., Shriram, S. & King, R. A. Oculocutaneous albinism type 1: the last 100 years. Pigment Cell Res 16, 307–311 (2003).
Onojafe, I. F. et al. Nitisinone improves eye and skin pigmentation defects in a mouse model of oculocutaneous albinism. J Clin Invest 121, 3914–3923, doi: 10.1172/JCI59372 (2011).
Adzhubei, I. A. et al. A method and server for predicting damaging missense mutations. Nat Methods 7, 248–249, doi: 10.1038/nmeth0410-248 (2010).
Schwarz, J. M., Rodelsperger, C., Schuelke, M. & Seelow, D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods 7, 575–576, doi: 10.1038/nmeth0810-575 (2010).
Kumar, P., Henikoff, S. & Ng, P. C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc 4, 1073–1081, doi: 10.1038/nprot.2009.86 (2009).
Tripathi, R. K., Hearing, V. J., Urabe, K., Aroca, P. & Spritz, R. A. Mutational mapping of the catalytic activities of human tyrosinase. J Biol Chem 267, 23707–23712 (1992).
Halaban, R. et al. Endoplasmic reticulum retention is a common defect associated with tyrosinase-negative albinism. Proc Natl Acad Sci USA 97, 5889–5894 (2000).
Puri, N., Gardner, J. M. & Brilliant, M. H. Aberrant pH of melanosomes in pink-eyed dilution (p) mutant melanocytes. J Invest Dermatol 115, 607–613, doi: 10.1046/j.1523-1747.2000.00108.x (2000).
Preising, M. N. et al. Mutation analysis in a family with oculocutaneous albinism manifesting in the same generation of three branches. Mol Vis 13, 1851–1855 (2007).
Matsunaga, J. et al. Sequence-based diagnosis of tyrosinase-related oculocutaneous albinism: successful sequence analysis of the tyrosinase gene from blood spots dried on filter paper. Dermatology 196, 189–193 (1998).
Gershoni-Baruch, R. et al. Mutations of the tyrosinase gene in patients with oculocutaneous albinism from various ethnic groups in Israel. Am J Hum Genet 54, 586–594 (1994).
Rosemblat, S. et al. Melanosomal defects in melanocytes from mice lacking expression of the pink-eyed dilution gene: correction by culture in the presence of excess tyrosine. Exp Cell Res 239, 344–352, doi: 10.1006/excr.1997.3901 (1998).
Orlow, S. J. & Brilliant, M. H. The pink-eyed dilution locus controls the biogenesis of melanosomes and levels of melanosomal proteins in the eye. Exp Eye Res 68, 147–154, doi: 10.1006/exer.1998.0599 (1999).
Chen, K., Manga, P. & Orlow, S. J. Pink-eyed dilution protein controls the processing of tyrosinase. Mol Biol Cell 13, 1953–1964, doi: 10.1091/mbc.02-02-0022. (2002).
Kelley, L. A., Mezulis, S., Yates, C. M., Wass, M. N. & Sternberg, M. J. The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protoc 10, 845–858, doi: 10.1038/nprot.2015.053 (2015).
Plagnol, V. et al. A robust model for read count data in exome sequencing experiments and implications for copy number variant calling. Bioinformatics 28, 2747–2754, doi: 10.1093/bioinformatics/bts526 (2012).
Krumm, N. et al. Copy number variation detection and genotyping from exome sequence data. Genome Res 22, 1525–1532, doi: 10.1101/gr.138115.112 (2012).
Chuzhanova, N., Abeysinghe, S. S., Krawczak, M. & Cooper, D. N. Translocation and gross deletion breakpoints in human inherited disease and cancer II: Potential involvement of repetitive sequence elements in secondary structure formation between DNA ends. Hum Mutat 22, 245–251, doi: 10.1002/humu.10253 (2003).
Jaworek, T. J. et al. Molecular genetic studies and delineation of the oculocutaneous albinism phenotype in the Pakistani population. Orphanet J Rare Dis 7, 44, doi: 10.1186/1750-1172-7-44 (2012).
Kausar, T. et al. Genetic studies of TYRP1 and SLC45A2 in Pakistani patients with nonsyndromic oculocutaneous albinism. J Invest Dermatol 133, 1099–1102, doi: 10.1038/jid.2012.432 (2013).
Shahzad, M. et al. Identification and functional characterization of natural human melanocortin 1 receptor mutant alleles in Pakistani population. Pigment Cell Melanoma Res 28, 730–735, doi: 10.1111/pcmr.12400 (2015).
Yousaf, S. et al. Identification and clinical characterization of Hermansky-Pudlak syndrome alleles in the Pakistani population. Pigment Cell Melanoma Res, doi: 10.1111/pcmr.12438 (2015).
Bittles, A. Consanguinity and its relevance to clinical genetics. Clin Genet 60, 89–98 (2001).
Hussain, R. & Bittles, A. H. The prevalence and demographic characteristics of consanguineous marriages in Pakistan. J Biosoc Sci 30, 261–275 (1998).
Sengupta, M. et al. Comprehensive analysis of the molecular basis of oculocutaneous albinism in Indian patients lacking a mutation in the tyrosinase gene. Br J Dermatol 163, 487–494, doi: 10.1111/j.1365-2133.2010.09830.x (2010).
Hutton, S. M. & Spritz, R. A. Comprehensive analysis of oculocutaneous albinism among non-Hispanic caucasians shows that OCA1 is the most prevalent OCA type. J Invest Dermatol 128, 2442–2450, doi: 10.1038/jid.2008.109 (2008).
Puri, N. et al. Type 2 oculocutaneous albinism (OCA2) in Zimbabwe and Cameroon: distribution of the 2.7-kb deletion allele of the P gene. Hum Genet 100, 651–656 (1997).
Giebel, L. B., Tripathi, R. K., King, R. A. & Spritz, R. A. A tyrosinase gene missense mutation in temperature-sensitive type I oculocutaneous albinism. A human homologue to the Siamese cat and the Himalayan mouse. J Clin Invest 87, 1119–1122, doi: 10.1172/JCI115075 (1991).
Kidson, S. H. & Fabian, B. C. The effect of temperature on tyrosinase activity in Himalayan mouse skin. J Exp Zool 215, 91–97, doi: 10.1002/jez.1402150111 (1981).
King, R. A., Mentink, M. M. & Oetting, W. S. Non-random distribution of missense mutations within the human tyrosinase gene in type I (tyrosinase-related) oculocutaneous albinism. Mol Biol Med 8, 19–29 (1991).
King, R. A. et al. Temperature-sensitive tyrosinase associated with peripheral pigmentation in oculocutaneous albinism. J Clin Invest 87, 1046–1053, doi: 10.1172/JCI115064 (1991).
Kwon, B. S., Halaban, R. & Chintamaneni, C. Molecular basis of mouse Himalayan mutation. Biochem Biophys Res Commun 161, 252–260 (1989).
Toyofuku, K., Wada, I., Spritz, R. A. & Hearing, V. J. The molecular basis of oculocutaneous albinism type 1 (OCA1): sorting failure and degradation of mutant tyrosinases results in a lack of pigmentation. Biochem J 355, 259–269 (2001).
Grimberg, J. et al. A simple and efficient non-organic procedure for the isolation of genomic DNA from blood. Nucleic Acids Res 17, 8390 (1989).
McKenna, A. et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20, 1297–1303, doi: 10.1101/gr.107524.110 (2010).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760, doi: 10.1093/bioinformatics/btp324 (2009).
Kelley, L. A. & Sternberg, M. J. Protein structure prediction on the Web: a case study using the Phyre server. Nat Protoc 4, 363–371, doi: 10.1038/nprot.2009.2 (2009).
Ahmed, Z. M. et al. Nonsyndromic recessive deafness DFNB18 and Usher syndrome type IC are allelic mutations of USHIC. Hum Genet 110, 527–531, doi: 10.1007/s00439-002-0732-4 (2002).
Acknowledgements
We thank the participants for their cooperation. We thank Shakeel Sheikh, Fareeha Zulfiqar, Suruchi Shrestha and Thomas Jaworek for their technical assistance. We thank Drs. Saumil Sethna, Elodie Richard and Arnaud Giese for their valuable critiques of the work and helpful discussions. The authors acknowledge support of the National Human Genome Research Institute and the National Heart, Lung, and Blood Institute. Funding was provided by HG006504 (Yale Center for Mendelian Disorders), HG006542 (Baylor-Hopkins Center for Mendelian Genomics) and HG006493 (University of Washington Center for Mendelian Genomics). This study was supported by National Institute of Arthritis and Musculoskeletal and Skin Diseases research grant R56AR065483 to ZMA.
Author information
Authors and Affiliations
Consortia
Contributions
Conception and design of the work: M.A.K., A.M.W., R.S.S., S.R., Z.M.A. Acquisition of data: M.S., S.Y., T.K., U.W.C.M.G., Y.M.W., H.G., N.T., U.M. Analysis and interpretation of data: all authors. Drafting of the manuscript: M.S., S.Y., S.R., Z.M.A. Critical revision of the manuscript: S.R., Z.M.A. Final approval of the version to be published: all authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Shahzad, M., Yousaf, S., Waryah, Y. et al. Molecular outcomes, clinical consequences, and genetic diagnosis of Oculocutaneous Albinism in Pakistani population. Sci Rep 7, 44185 (2017). https://doi.org/10.1038/srep44185
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep44185
- Springer Nature Limited
This article is cited by
-
PMEL is mutated in oculocutaneous albinism
Human Genetics (2023)
-
Novel deletion of exon 3 in TYR gene causing Oculocutaneous albinism 1B in an Indian family along with intellectual disability associated with chromosomal copy number variations
BMC Medical Genomics (2022)
-
Novel compound heterozygous mutations in OCA2 gene associated with non-syndromic oculocutaneous albinism in a Chinese Han patient: a case report
BMC Medical Genetics (2019)
-
Tyrosinase (TYR) gene sequencing and literature review reveals recurrent mutations and multiple population founder gene mutations as causative of oculocutaneous albinism (OCA) in Pakistani families
Eye (2019)