
Abstract
In many bacteria, OxyR is the major regulator controlling cellular response to H2O2. A common phenotype resulting from OxyR loss is reduced growth rate, but the underlying mechanism is unknown. We demonstrated in Shewanella oneidensis, an important research model for applied and environmental microbes, that the defect is primarily due to an electron shortage to major terminal oxidase cytochrome cbb3. The loss of OxyR leads to enhanced production of electron carriers that compete for electrons against cytochrome cbb3, cytochrome bd in particular. We further showed that the oxyR mutation also results in increased production of menaquinone, an additional means to lessen electrons to cytochrome cbb3. Although regulation of OxyR on these biological processes appears to be indirect, these data indicate that the regulator plays a previously underappreciated role in mediating respiration.
Similar content being viewed by others


Introduction
Oxidative stress, caused by reactive oxygen species (ROS), including superoxide (O2−), hydrogen peroxide (H2O2), and hydroxyl radical (OH), is one of the unavoidable crises for the vast majority of aerobic organisms by damaging biomolecules such as lipids, proteins and DNA1. ROS (H2O2 in particular) are ubiquitous because they are metabolic by-products of cellular oxygen respiration, mostly through the accidental auto-oxidation of non-respiratory flavoproteins and the turnover of committed oxidases, in addition to those coming from environments2,3,4. To cope with oxidative stress, bacteria have evolved multiple protection systems, such as ROS detoxification enzymes (alkylhydroperoxide reductase (Ahp), catalases, and various peroxidases), iron-sequestering proteins (Dps in particular), and oxidative damage repairing macromolecules5. When ROS levels exceed safe limits, in response these systems are induced coordinately by regulators to ensure that the ROS concentrations are restrained at an acceptable level and damages are promptly repaired. In Escherichia coli, the model organism in which the oxidative stress response is best understood, and many other bacteria, SoxRS and OxyR, are the predominant regulatory systems mediating cellular response to O2− (redox-cycling compounds) and H2O2, respectively1. OxyR, a LysR family transcriptional regulator, directly senses H2O2 via its two conserved cysteine residues that react rapidly with H2O2. In the presence of H2O2 that is sufficient to mount the response, these residues are oxidized and an intramolecular disulfide bond is formed, leading to OxyR activation, which subsequently turns on expression of the defending systems. However, exceptions to the E. coli OxyR model are found. In Neisseria, OxyR acts as both a repressor of catalase expression and an activator for other members of its regulon6.
Shewanella, a group of Gram-negative facultative γ-proteobacteria, inhabit redox-stratified environments and are renowned for their versatile respiratory abilities, which underlies great potential for bioremediation and microbial fuel cells7. These bacteria are substantially more sensitive to H2O2 than E. coli, in concert with the high susceptibility to UV and ionizing radiation, understandings primarily derived from studies on the model species S. oneidensis8,9,10,11. S. oneidensis OxyR appears to be the only possible regulator mediating the cellular response to both O2− and H2O2 because it lacks a homologue of E. coli SoxRS12,13. S. oneidensis OxyR acts as both an activator and a repressor as Neisseria OxyR proteins6; upon OxyR activation, S. oneidensis mainly relies on derepression of katB (encoding the major catalase) and dps genes to alleviate oxidative damage by degrading H2O2 and by sequestering the intracellular free iron, respectively12. As a result, an S. oneidensis oxyR mutant degrades H2O2 more rapidly than the wild-type. Despite this, the oxyR mutant bears defects in both viability (plating defect) and growth, which are commonly observed in bacteria whose OxyR proteins act as a positive regulator for catalase genes, such as E. coli12,13,14,15.
Oxygen respiration is carried out by respiratory oxygen reductases (traditionally dubbed as terminal oxidases)16. Among them, heme-copper oxidases (HCOs) and cytochrome bd are found in a wide variety of prokaryotes. While HCOs generate a proton motive force (PMF) via a highly efficient “proton pumping” mechanism, cytochrome bd does not and therefore is characterized by a lower energetic efficiency17. The S. oneidensis genome encodes three terminal oxidases: a bd-type quinol oxidase (cydABX) and two heme-copper oxidases (HCOs), a caa3-type (SO4606-9) and a cbb3-type (ccoNOQP)18,19,20. Cytochrome cbb3 is abundant in membranes under both aerobic and microaerobic conditions whereas cytochrome caa3 is not present; thus cytochrome cbb3 is the predominant force for oxygen respiration18,21. Cytochrome bd, like its E. coli counterpart, plays a minor role in oxygen respiration, and more importantly, confers S. oneidensis tolerance to various stresses, especially nitrite22,23,24. Consequently, the loss of cytochrome cbb3 but not cytochrome bd leads to a significant defect in growth under aerobic conditions18. All terminal oxidases, either directly or via soluble cytochrome c, accept electrons from quinones (UQs), which are the most important compounds in the membrane of most prokaryotes, working as lipophilic electron and proton shuttles16,25. In S. oneidensis, quinones include ubiquinones (UQs) and menaquinones (MKs); MKs, such as MK-7 (−80 mV), have low midpoint redox potentials compared with those of UQs, such as UQ-8 (+110 mV)26,27,28. While cytochrome cbb3 (via cytochrome bc1) and cytochrome bd predominantly use UQs as the electron donor MKs supply electrons to terminal reductases for many non-oxygen EAs with CymA working as quinone oxidase29,30,31. Interestingly, in batch cultures under aerobic conditions, MKs are still substantial in abundance, accounting for slightly less 50% of the quinone composition, one of features underlying its respiratory diversity27,29,32.
Although most, if not all, of oxyR mutants studied to date carry growing defect, the underlying mechanism remains elusive. Previously we have shown that the plating defect of the S. oneidensis oxyR mutant is primarily due to H2O2 that is generated abiotically, providing intriguing insights into current understandings of OxyR physiology10,13. The goal of this study was to address biological factors that are responsible for the growing defect of bacteria, with S. oneidensis as research model. We demonstrated that this defect is independent of the plating defect. By using a random mutational analysis, we identified cytochrome bd to be associated with the phenotype. We then showed evidence to suggest that cytochrome bd, as well as CymA, hampers growth by diminishing the quantity of electrons to cytochrome cbb3. Furthermore, we found that the oxyR mutation also results in increased production of menaquinone-7, which acts as additional means to lessen electrons to cytochrome cbb3.
Methods
Bacterial strains, plasmids and culture conditions
All bacterial strains and plasmids used in this study can be found in Table 1. Information about all of the primers used in this study was available upon request. All chemicals were obtained from Sigma-Aldrich unless otherwise noted. E. coli and S. oneidensis were grown in Lysogeny broth (LB, Difco, Detroit, MI) under aerobic conditions at 37 and 30 °C for genetic manipulation. When necessary, the growth medium was supplemented with chemicals at the following concentrations: 2,6-diaminopimelic acid (DAP), 0.3 mM; ampicillin, 50 μg/ml; kanamycin, 50 μg/ml; and gentamycin, 15 μg/ml; streptomycin, 100 μg/ml; and catalase on plates, 2000 U/ml.
Growth in liquid media was monitored by recording values of optical density at 600 nM (OD600) as all strains used in this study were morphologically similar. Both LB and defined medium MS13 were used for phenotypic assays in this study and comparable results with respect to growth were obtained.
In-frame mutant construction and complementation
In-frame deletion strains for S. oneidensis were constructed using the att-based Fusion PCR method as described previously33. In brief, two fragments flanking gene of interest were amplified by PCR with primers containing attB and the gene specific sequence, which were linked by a second round of PCR. The fusion fragments were introduced into plasmid pHGM01 by using Gateway BP clonase II enzyme mix (Invitrogen) according to the manufacturer’s instruction. The resultant plasmids were transformed by electroporation into and maintained in E. coli WM3064, and transferred to S. oneidensis by conjugation with the ratio of the donor to the recipient being 2:1. Integration of the mutagenesis constructs into the chromosome was selected by resistance to gentamycin and confirmed by PCR. Verified transconjugants were grown in LB broth in the absence of NaCl and plated on LB supplemented with 10% sucrose. Gentamycin-sensitive and sucrose-resistant colonies were screened by PCR for deletion of the target gene. Mutants were verified by sequencing the mutated regions.
Promoterless plasmid pHG101 was used in genetic complementation of mutants as described before34. For inducible gene expression, the gene of interest was generated by PCR and introduced into the inducible plasmid pHGE-Ptac under the control of promoter Ptac35. After sequencing verification, the resulting vectors were transferred into the relevant strains via E. coli WM3064-mediated conjugation.
Transposon mutagenesis
Random mutagenesis vector pHGT01, which carries a robust promoter in the transposable region, was used to construct a library in this study20. Briefly, the vector from E. coli WM3064 was transferred into S. oneidensis ΔoxyR via conjugation and growth suppressor strains were screened on plates containing gentamycin and catalase. Plates having 200–300 colonies were analyzed by a specific program to calculate the average size and size ratios of each colony to the average. The colonies, whose size was similar to that of the wild-type, were saved and subsequently subjected to the determination of the transposon insertion sites using the arbitrary PCR36.
Expression analysis
Expression of genes of interest was determined by β-Galactosidase activity assay using integrative lacZ reporter pHGEI0132. In brief, the sequence in sufficient length (~400 bp) upstream of gene of interest was amplified and placed in front of the full-length E. coli lacZ gene. The resulting vector was transformed into and maintained in E. coli WM3064, and transferred to S. oneidensis via conjugation after verification by sequencing. Cells at the mid-log phase (~0.3 of optical density at 600 nM (OD600)) were harvested by centrifugation, washed with phosphate-buffered saline (PBS, pH 7.0) and lyzed with the lysis buffer (0.25 M Tris/HCl, 0.5% Triton X-100, pH 7.5). Soluble proteins were collected after centrifugation and applied to the o-nitrophenyl-β-D-galactopyranoside (ONPG)-based assay as described previously37. The protein concentration of the cell lysates was determined using a Bradford assay with BSA as a standard (Bio-Rad). β-galactosidase activity were determined by monitoring color development at 420 nm using a Synergy 2 Pro200 Multi-Detection Microplate Reader (Tecan), presented as Miller units.
Droplet assays
Droplet assays were employed to evaluate viability and growth inhibition on plates essentially the same as previously described12,22. In brief, cells at the mid-log phase were collected by centrifugation and adjusted to 109 cell/ml, which was set as the undiluted (dilution factor 0). 10-fold serial dilutions were prepared with fresh LB medium. Five micro liters of each dilution was dropped onto LB plates, on which designated agents (catalase and nitrite) may be added. The plates were incubated for 24 h or longer in dark before being read. All experiments were repeated at least three times.
Cytochrome oxidase activity assay
The Nadi test was used for visual analysis of cytochrome cbb3 activity38. Five micro liters of each culture at the mid-log phase under test was dropped onto LB plates w/o catalase, and the plates were incubated for 24 h. A solution of 0.5% α-naphthol in 95% enthanol and 0.5% N,N-dimethyl-ρ-phenyleneidiamine monohydrochloride (DMPD) was applied to cover the droplets developed. Formation of indophenols blue was timed as an indicator of cytochrome cbb3 activity.
Solubilized membranes were prepared for quantitative analysis of the cytochrome oxidase activity as described previously20. In brief, cell pellets were resuspended in 20 mM Tris-HCl (pH 7.6) supplemented with DNase I and protease inhibitors and disrupted by French pressure. After debris and unbroken cells removing, the membranes were pelleted by ultracentrifugation for 1 h at 230,000 × g at 4 °C and subsequently resuspended in 20 mM Tris-HCl pH 7.6 with 5% glycerol to a protein concentration of 10 mg/ml. Solubilization was performed with n-dodecyl β-D-maltoside (DDM) to a final concentration of 1% (w/v) on a rotary tube mixer for 2 h at 4 °C. The DDM-solubilized membranes were obtained by collecting the supernatant after ultracentrifuging for 1 h at 230,000 × g at 4 °C. The cytochrome oxidase activity was assayed as a measure of oxygen consumption rates using an OxyGraph oxygen electrode (Hansatech) using either ubiquinol-1 or N,N,N′,N′-tetramethyl-p-phenylenediamine (TMPD) as electron donor according to the methods described previously20.
Determination of oxygen consumption and NAD+/NADH ratio
For determination of oxygen consumption, a 2-mm O2 sensor and a TBR4100 free radical analyzer (World Precision Instruments) were used. Cells were washed three times in 33 mM potassium phosphate buffer (pH 7.0) and adjusted to an OD600 of 1. Oxygen consumption was induced by addition of either 100 mM disodium succinate as the electron donor. The basal oxygen consumption in the absence of the electron donor was subtracted; oxygen consumption is indicated as nmol oxygen ml−1 min−1.
The ratio of NAD+ to NADH was determined with a NAD/NADH quantitation kit (Sigma-Aldrich). S. oneidensis cultures of the mid-log phase were collected by centrifugation, washed with cold PBS, resuspended in the extraction buffer, and disrupted by sonication (three cycles of 2 min, on/off at 1-s intervals, at 50 W) on ice. After centrifugation at 20,000 × g for 5 min at 4 °C, the supernatants were deproteinated using 3,000-molecular-weight-cutoff (MWCO) spin filters (Millipore Corporation, Bedford, MA), and then subjected to the NAD+/NADH ratio determination according to the manufacturer’s instructions.
Quinone extraction and quantification
Quinone extraction was performed on cultures at the mid-log phase with the methanol and petroleum ether method32,39. The extracted quinone-quinol mixture was resuspended with a glass rod in 80 μl ethanol and fractionated by HPLC (Thermo Accela UHPLC) with a Phenomenex reverse phase C18 (5 μm) column (dimensions 150 × 4.6 mm) with ethanol/methanol (50/50 v/v) as the mobile phase at 0.3 or 0.5 ml min−1. Detection of the quinones was performed at 290 nm for UQs and at 248 nm for MKs. The amount of each quinone species was calculated from the related peak area, using ubiquinone-10 (UQ10) amd menaquinone-4 (MK4) as standards. Peaks were identified by mass spectral analysis as described previously40. Fractions collected from the HPLC were evaporated under nitrogen and redissolved in 90% (wt/vol) acetonitrile, 1% (vol/vol) formic acid, analyzed by using a high solution, high-accuracy mass spectrometer (MS) (Exactive, ThermoFisher).
Site-directed mutagenesis
Plasmid pTRG-OxyR was used as the template for site-directed mutagenesis with a QuikChange II XL site-directed mutagenesis kit (Stratagene) as described previously40. The resulting pTRG-OxyRL197P was verified by sequencing.
Bacterial one-hybrid (B1H) assay
B1H system (Stratagene) was used to investigate DNA-protein interaction in vivo in E. coli cells12,41. Briefly, plasmid constructs were created by cloning the bait DNA (promoter sequence of interest) and target DNA (the oxyR gene) in the pBXcmT and pTRG vectors and verified by sequencing. The resultant plasmids were used to co-transform BacterioMatch II Validation Reporter Competent Cells on M9 salt agar plates containing 25 mg/ml chloramphenicol and 12.5 mg/ml tetracycline with or without 3-amino-1,2,4-triazole (3-AT). Pairs of pBXcmT-PkatB/pTRG-OxyR and pBXcmT-PacpC/pTRG-OxyRL197P were used as positive control and a ~300 bp DNA fragment of the 16 s rRNA gene promoter (pBXcmT-P16s-rRNA) was used as negative control. The plates were incubated for 24 h and then moved to room temperature for an additional 16 h (the colonies indicating positive interaction usually appeared between 18 and 24 h). The positive interactions were confirmed by streaking colonies on plates containing both 3-AT and streptomycin (12.5 mg/ml).
Statistical analyses
Values are presented as means ± SD (standard deviation). In some cases, error bars, which represent SD and less than 10% of the average of experimental values, were omitted for clarity. Student’s t-test was performed with statistical significance set at the 0.05 confidence level.
Results
Defects of ΔoxyR in growth and viability are independent of each other
The plating defect of the S. oneidensis oxyR mutant is due to H2O2 generated abiotically on LB plates12,13. On LB agar plates, only undiluted ΔoxyR culture can grow (Fig. 1A, dilution factor 0, ~109 cell/ml). In LB broth, while the viability defect of ΔoxyR is not evident because of catalase KatB released from lyzed cells10, significant difference in growth rates between the wild-type and ΔoxyR strains was observed (Fig. 1B). The growth defect in LB broth can be fully corrected by a copy of the oxyR gene expressed in trans, but was barely affected by catalase added to levels (2000 U/ml) that were sufficiently high to rescue the plating defect. We then examined effect of catalase on growth of the ΔoxyR strain on plates. Consistently, colony sizes of the wild-type and the ΔoxyR strains differed significantly in the presence of catalase (Fig. 1C). After 24 h, colonies of the ΔoxyR strain were approximately 24% smaller than those of the wild-type, ruling out the possibility that H2O2 generated abiotically is also the cause for the growing defect of the ΔoxyR strain. We then performed the same experiment with superoxide dismutase (SOD) to examine whether superoxide has a role in these two phenotypes. However, the addition of SOD had no effect on alleviating either the plating or growing defect of the ΔoxyR strain (data not shown). These data, all together, suggest that mechanisms for the plating and growing defects of the ΔoxyR strain are distinct.

(A) Plating phenotype of indicated strains. Cultures at the mid-log phase were collected and adjusted to 109 cell/ml (dilution factor 0), which were then serially diluted and 5 μl of each dilution was dropped onto indicated agar plates. Results shown were after growth of the wild-type in the lowest cell density (104 cell/ml, dilution factor 5) became evident. (B) Growth of indicated strains in LB without or with catalase (CAT), including the wild-type (WT), ∆oxyR, genetically complemented ∆oxyR (∆oxyR/oxyR). OD600 values of the five time points during the exponential phase were plotted, with exponential trendlines (representing growth rate) being displayed (the same thereafter). (C) Growth of indicated strains on LB plates containing CAT. Cultures at the mid-log phase were serially diluted and spread onto LB plates containing CAT. After 24 h, plates having ~100 colonies were photographed and the averaged size of 21 colonies on (7 per plate) three plates for each strain was calculated out. Presented was the ratio of the averaged sizes of mutants to that of the wild-type. **p < 0.01. Experiments were performed at least three times, with representative results and the average ± error bars representing standard deviation being presented.
Loss of cytochrome bd suppresses the growth defect of the oxyR mutant
To search for genes whose products are related to the growing defect of the ΔoxyR strain, we employed transposon-based mutational screening. As the ΔoxyR strain grows significantly slower than the wild-type, we reasoned that we may be able to obtain suppressor strains with elevated growth rate. Vector pHGT01, which can be used for construction of transposon insertion libraries as well as for identification of cryptic operons because of an embedded promoter in the transposable sequence, was used in this study20. The resulting library was diluted properly (100–300 colonies per plate) and spread out on LB plates containing catalase and required antibiotics. A total of ~15,000 colonies on plates were screened and 323 mutants grew to significantly increased size (at least 15% larger than the average size of the colonies on plates) (Fig. 2A). Most of these (276 of 323), however, were not stable, growing into colonies varying in size during confirmation (by re-plated on the same plate), and were thus discarded. Among the remaining, 18 displaying largest colony size (indistinguishable from the wild-type) were subject to transposon insertion mapping. In 16 out of them, the transposon was successfully identified. Interestingly, 9 were found to have transposon insertions disrupting genes in the cydABX operon, which encodes cytochrome bd19,22 (Fig. 2A). The insertions were not only in the essential cydA or cydB gene but also in the operon promoter, implicating that the functional loss of the enzyme may dictate the suppression. As disruption of the cydABX operon in the screening was repeatedly identified, it is highly possible that cytochrome bd is associated with the growth defect of the oxyR mutant.

(A) Suppressors for growth defect of ∆oxyR. Shown were representatives of stable suppressor strains derived from ∆oxyR on CAT-containing LB plates. Schematics indicating the approximate locations of the transposon insertions were shown below. Arrows represent transposon insertion points. X represents cydX. (B) Nitrite sensitivity assay. Mid-log phase cultures were subject to 10-fold serial dilution, and 5 μl of each dilution was spotted onto plates containing 2 mM nitrite and CAT. Double mutant ∆oxyR∆cyd was complemented by either of the missing genes (∆oxyR∆cyd/oxyR and ∆oxyR∆cyd/cyd) on indicated plates to confirm effects of the additional loss of Cyd. (C) Growth of indicated strains in LB without or with catalase (CAT), including the wild-type (WT), ∆oxyR, ∆oxyR∆cyd, and genetically complemented ∆oxyR∆cyd (∆oxyR∆cyd/cyd). Experiments were performed at least three times and either representative results or error bars representing standard deviation.
To confirm that the suppression is due to the loss of cytochrome bd, we deleted the entire cyd operon from the ΔoxyR strain. In S. oneidensis, cytochrome bd confers cells resistance to nitrite22, thus the additional mutation was validated by the finding that the ΔoxyRΔcyd strain was sensitive to nitrite the same as the Δcyd strain (Fig. 2B). As expected, the removal of cytochrome bd enabled the ΔoxyR strain to grow indistinguishably from the wild-type (Fig. 2C). In the case of the plating defect, however, the ΔoxyRΔcyd strain was similar to the ΔoxyR strain (Fig. 2B). Importantly, the growth and the plating defects of the ΔoxyRΔcyd strain were relieved, when the cyd operon and the oxyR gene were expressed in trans, respectively (Fig. 2B,C). These data firmly established that the loss of cytochrome bd suppresses the growth defect of the oxyR mutant.
The cyd operon is up-regulated in the oxyR mutant
Notably, when complemented by the cyd operon within pHG101 the ΔoxyRΔcyd strain grew even slower than the ΔoxyR strain (Fig. 2C). This is probably due to overproduction of cytochrome bd as the plasmid for complementation is present ~5 copies per cell22. Cytochrome bd is characterized by its high affinity for O2 and usually is induced under microaerobic conditions. Hence, cytochrome bd levels are rather low in S. oneidensis under normal aerobic conditions as adopted in this study18. Given these features, we reasoned that cellular levels of cytochrome bd are likely to be up-regulated in the oxyR mutant.
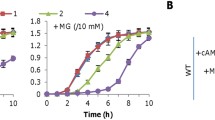
The cyd promoter activity was assayed using an integrative lacZ reporter as before32. In the mid-log phase wild-type cells, the promoter activity was rather low, at levels of approximately 40 Miller units (Fig. 3A). In comparison, it increased more than three times in the oxyR mutant, implying that OxyR represses expression of the cyd operon. We then manipulated production levels of cytochrome bd by IPTG-controlled promoter Ptac within pHGE-Ptac to assess effect of the enzyme in varying amounts on growth of the Δcyd and ∆oxyRΔcyd strains. The expression system is highly reliable, producing protein of interest proportional to IPTG up to 1 mM13,42. Despite this, in order to precisely interpret data, levels of cytochrome bd proteins produced in these strains by IPTG induction were estimated by nitrite sensitivity assay. In agreement with previous calibration, the higher the IPTG level, the stronger resistance the cells to nitrite (Fig. S1).

(A) Promoter activity of Pcyd revealed by an integrated lacZ reporter in indicated strains grown in CAT-containing LB. Cells of mid-log phase cultures (~0.3 of OD600) were pelletted, processed, and subjected to β-galactosidase activity assay as described in Experimental procedures. (B) Effects of cytochrome bd in varying amounts on growth of ∆oxyR∆cyd in CAT-containing LB. Cytochrome bd was produced in cells from a copy of the cyd operon under control of IPTG-inducible promoter Ptac. The numbers shown represent concentrations of IPTG. Experiments were performed at least three times and error bars representing standard deviation were presented.
With IPTG at 0.01 mM the ∆oxyRΔcyd strain did not display any defect in growth (Fig. 3B). When IPTG was added to 0.05 mM, which confers the promoter ~120 Miller units according to previous calibration13,42, expression of cyd slowed growth of ∆oxyRΔcyd to extent comparable to that the ∆oxyR strain (Fig. 3B). Growth defect became more substantial with IPTG at higher concentrations, indicating that cytochrome bd in excess does have a negative influence on growth.
Loss of OxyR results in electron shortage to cytochrome cbb3
In S. oneidensis, although cytochrome bd is able to respire oxygen to support growth in the absence of cytochrome cbb3, its loss does not significantly affect aerobic growth18. Clearly, given that the oxyR mutation causes enhanced production of cytochrome bd, which promotes oxygen respiration, activity of cytochrome bd per se may not be a factor accountable for the growth defect caused by the OxyR loss. We therefore turned to cytochrome cbb3 because it is predominant factor for aerobic growth. We used the Nadi assay to examine activity of cytochrome cbb3 in relevant strains because it is the only enzyme reacting with Nadi reagents in S. oneidensis18. In the presence of catalase, the ΔoxyR strain generated a clearly visible blue ring in 2 min, stronger in intensity than that produced by the wild-type in 5 min (Fig. 4A). The negative control, a Δcco strain, which lacks entire cytochrome cbb3, did not generate a blue ring after an incubation of 5 min. Thus, there appears a significant difference in cytochrome cbb3 activity between the ΔoxyR and wild-type strains. While the loss of cytochrome bd (Δcyd) had no impact on the activity of cytochrome cbb3 as previously reported18, it restored the activity of cytochrome cbb3 of ΔoxyR to the wild-type level (Fig. 4A). It is worth noting that the additional loss of cytochrome cbb3 had an extremely weak influence on the growth defect of the oxyR mutant, implicating that contribution of cytochrome cbb3 to oxygen respiration in the ΔoxyR strain is negligible (Fig. S2).

(A) Activities of cytochrome cbb3 in indicated strains grown on CAT-containing LB plates by the Nadi assay. The method is based on the rapid formation of indophenol blue from colorless a-naphtol catalyzed by cytochrome c oxidase, using N’,N’-dimethyl-p-phenylenediamine monohydrochloride as an exogenous electron donor. WT and previously verified ∆cco served as positive and negative controls. Shown on the right panel were effects of cytochrome bc1 at varying amounts on activities of cytochrome cbb3. Expression of the pet operon was driven by IPTG-inducible Ptac with IPTG at indicated concentrations (mM). (B) Activities of cytochrome cbb3 in membranes of indicated strains by the cytochrome c oxidase activity analysis. The pet mutant and its complemented strain were included for comparison. Asterisks indicate statistically significant difference (**P < 0.01). (C) Effects of cytochrome bc1 at varying amounts on growth of the ∆oxyR strain. Expression of the pet operon in the ∆oxyR strain was conducted as in (A) The numbers shown represent concentrations of IPTG. (D) Oxygen consumption rates in resting cells of the relevant strains. Oxygen consumption rates were determined as described in Methods. In (A) shown were representative results from multiple experiments at indicated reaction times. In others, the experiment was performed at least three times and error bars indicate standard deviation.
The enhanced cytochrome cbb3 activity in the ΔoxyR strain detected by the Nadi reagents may be due to increased production of the enzyme. However, we found that expression levels of the cco operon were comparable in the wild-type, ΔoxyR, and ΔoxyRΔcyd by using transcriptional fusion (Fig. S3A). The quantity of the enzyme was also assessed by measuring cytochrome c oxidase activity in solubilized membranes, which is independent of the cellular electron transport chain. As shown in Fig. 4B, differences in the cytochrome cbb3 activities among ΔoxyR, Δcyd, and ΔoxyRΔcyd strains were insignificant. Thus, we conclude that production of cytochrome cbb3 is not affected by the oxyR mutation.
Alternatively, it is possible that there is electron shortage to cytochrome cbb3. We have previously demonstrated that electrons from both the electron transport chain and Nadi reagents compete for cytochrome cbb3; as a result, cytochrome cbb3 is highly active with Nadi reagents when the electron transport chain is impaired20. The scenario can be best illustrated by a strain (∆pet) lacking cytochrome bc1. In S. oneidensis, cytochrome bc1 encoded by the petABC operon is responsible for transporting electrons from the quinone pool to cytochrome cbb3, and its loss results in a growth defect comparable to that of the Δcco32. Consistent with our previous data20, the loss of cytochrome bc1 drastically increased the activity of cytochrome cbb3 based on the Nadi assay, with the whole colony turning blue in 2 min (Fig. 4A). However, this was not observed in disrupted cells, in which the cbb3 activity of the pet mutant was actually lower than that of the wild-type (Fig. 4B). This is not surprisingly because the loss of cytochrome bc1 down-regulates expression of the cco genes18.
One possibility for reduced electron transfer to cytochrome cbb3 is that cytochrome bc1 is produced less in the ΔoxyR strain. But the promoter activity of the pet operon did not significantly change in ΔoxyR comparing to that of the wild-type (Fig. S3A). We then made attempts to manipulate production of cytochrome bc1 such that the electron flow to cytochrome cbb3 can be altered. The pet operon was placed under the control of the IPTG– induced Ptac promoter within pHGE-Ptac and the resulting vector was introduced into the pet mutant. As expected, the cytochrome cbb3 activity revealed by the Nadi assay decreased with IPTG levels inversely (Fig. 4A), implicating that electrons delivered to cytochrome cbb3 increase in quantity when cytochrome bc1 is overproduced, albeit not necessarily in a proportion manner. Despite this, overproduction of cytochrome bc1 with IPTG up to 0.5 mM had no effect on correcting the growth defect of the ΔoxyR strain (Fig. 4C).
All of these observations suggest that in the ΔoxyR strain the majority of electrons are transferred to cytochrome bd whose production is enhanced, resulting in low efficacy of oxygen respiration. If so, we would expect that oxygen consumption in the oxyR mutant may not be significantly altered. Indeed, the oxyR mutant had an oxygen consumption rate comparable to the wild-type, Δcyd, and ΔoxyRΔcyd which was about 90% higher than the strain lacking cytochrome cbb3 (Fig. 4D). The additional removal of OxyR in the cco mutant did not improve the oxygen consumption rate. This is conceivable given that cytochrome bd is overproduced in the absence of cytochrome cbb318. Hence, the suppression of the ΔoxyR growth defect by the loss of cytochrome bd is likely due to that more electrons from the quinone pool are delivered to cytochrome cbb3 as a result of the shutdown of the alternative route.
Lack of CymA partly rescues the growth defect of the oxyR mutant
Given that the bc1 complex in overabundance did not influence the electron flow toward cytochrome cbb3, we proposed the quinone pool as the next candidate responsible for the growth defect of ΔoxyR. If so, it is possible that other quinone oxidases, at least the major one, in their absence may also suppress the growth defect resulting from the oxyR mutation. In S. oneidensis, the quinol pool passes electrons to many quinone oxidases, including CymA, TorC, SirCD, PsrBC, cytochrome bc1, as well as cytochrome bd30,43,44,45. While CymA is linked to respiration of a variety of non-oxygen EAs, TorC, SirCD, and PsrBC are specific for TMAO, sulfite, and thiosulfate respiration, respectively. Moreover, CymA but not TorC, SirCD, or PsrBC is produced considerably in the presence of oxygen30,46,47, implying that CymA may be capable of consuming electrons from the quinone pool under aerobic conditions.
To test this, we deleted cymA from the ΔoxyR strain. The Nadi assay revealed that ΔoxyRΔcymA generated a blue ring by a significantly slower rate than the ΔoxyR strain, but still faster than the wild-type did (Fig. 5A), supporting an alleviation of the electron shortage for the cytochrome cbb3. Consistently, the growth defect of ΔoxyR was partially corrected by the additional removal of cymA (Fig. 5B). These data suggest that CymA and cytochrome bd likely function in a similar manner in regard of the growth defect of ΔoxyR, and strongly support that electron deficiency for cytochrome cbb3 is the main reason for the growth defect of ΔoxyR. Although EA-specific quinone oxidases (TorC, SirCD, and PsrBC) are barely produced under test conditions, we removed each from the ΔoxyR strain and assessed impacts of the additional removal on the growth defect. Expectedly, all resulting double mutants were indistinguishable from ΔoxyR with respect to growth (data not shown), ruling out the possibility that these proteins play an indispensable role in consuming electrons from the quinone pool under test conditions. Importantly, production of CymA in the ΔoxyR strain was similar to that in the wild-type (Fig. S3B), indicating that the electron shortage to cytochrome cbb3 was not due to excessive CymA. All together, these results indicate that quinone oxidase CymA has ability to draw electrons from the quinone pool under test conditions, contributing to an electron shortage for cytochrome cbb3.

(A) Activities of cytochrome cbb3 in indicated strains grown on CAT-containing LB plates by the Nadi assay. Shown were representative results from multiple experiments at indicated reaction times. (B) Growth of indicated strains in CAT-containing LB. Experiments were performed at least three times with error bars representing standard deviation.
The ratio of MKs to UQs increases in the oxyR mutant
Results presented thus far suggest that respiratory quinones are likely a key factor for the growth defect resulting from the oxyR mutation. Given that loss of either cytochrome bd or CymA improves ΔoxyR growth, it is possible that electron deficiency for cytochrome cbb3 in ΔoxyR is due to the changes in the composition of quinones. To test this possibility, we determined levels of quinones from the wild-type, ΔoxyR, Δcyd, ΔcymA, ΔoxyRΔcyd, and ΔoxyRΔcymA strains grown to the mid-log phase under aerobic conditions with high-performance liquid chromatography (HPLC) and mass spectrometer (MS) (Fig. 6). The ratio of UQs to MKs in the wild-type was approximately 52:48 under test conditions, which is in excellent agreement with previous data27,32. Similar results were obtained from Δcyd and ΔcymA, indicating that the loss of either gene alone would not significantly affect production of UQs and MKs. In contrast, the ratio was about 43:57 in the ΔoxyR strain, suggesting that the oxyR mutation results in substantially changes in production of these quinones. Strikingly, the additional removal of cytochrome bd in the oxyR mutant had a significant influence (p < 0.05) on the ratio, restoring it (48:52) to levels close to that of the wild-type. In contrast, the depletion of CymA had no such effect. These data suggest that cytochrome bd functions more than a competing quinone oxidase for electrons against cytochrome bc1 (for cytochrome cbb3).

UQ/MK profiles in indicated strains were compared. Cells of mid-log phase cultures (~0.3 of OD600) grown in CAT-containing LB were pelletted, processed, and subjected to Quinone extraction as described in Experimental procedures. Quinone identification and quantification by LC-MS were described in the Experimental procedure and text. Experiments were performed at least three times and standard variations were within 5% range.
Disruption of MK biosynthesis partially rescues the growth defect of the oxyR mutant
To confirm that the ratio of UQs to MKs is linked to the oxyR mutant growth phenotype, we assessed effects of the menA mutation on the growth defect of the oxyR mutant32. The menA gene encodes 1,4-dihydroxynahthoate (DHNA) octaprenyltransferase, an essential component of MK-7 biosynthesis pathway that converts DHNA to dimethylmenaquinone48. The ΔoxyRΔmenA strain grew apparently faster than ΔoxyR did, at a rate similar to that of ΔoxyRΔcymA (Fig. 7A). Consistently, cytochrome cbb3 activities based on the Nadi reaction in ΔoxyRΔmenA and ΔoxyRΔcymA were also comparable (Fig. 5A). The same analysis could not be applied to UQ biosynthesis, because we failed to knock out any gene essential for UQ biosynthesis after many tries32. Instead, we performed chemical complementation. In the presence of UQ (Q10) up to 8 μM, growth of the wild-type was not affected (Fig. 7B). In contrast, an improvement in growth of ΔoxyR was observed with UQ at 4 and 8 μM, suggesting that exogenous UQ can partially correct the growth defect resulting from the oxyR mutation.

(A) Growth of indicated strains in CAT-containing LB. (B) Growth of indicated strains in CAT-containing LB with UQ-10 addition. UQ-10 concentrations used in the experiment were shown. Experiments were performed at least three times with error bars representing standard deviation.
A remaining possibility that could address the growth defect of the oxyR mutant is that electrons entering the quinone pool are reduced in quantity. To test this, we monitored the NAD+/NADH ratio as these redox chemicals are predominant electron carriers, especially for aerobic respiration. However, the difference between the wild-type and ΔoxyR strains was insignificant (Fig. S4A). In addition, we compared in these two strains the promoter activities of the nuo operon, which encodes NADH dehydrogenase that couples the electron transfer from NADH to quinones with a proton translocation49. Similarly, the results were comparable (Fig. S4B). By ruling out the difference in electrons entering the quinone pool, the data conclude that the ubiquinone/menaquinone levels, which are altered in the oxyR mutant, are associated with the growth defect.
OxyR represses cytochrome bd and MK production in an indirect manner
To explain why the ratio of UQs to MKs decreases in the oxyR mutant, we analyzed the activity of promoters for some ubi (UQ biosynthesis) and men (MK biosynthesis) genes. In line with the unchanged levels of UQs in the wild-type and ΔoxyR strains, activities of promoters for all ubi operons were not significantly affected by the oxyR mutation (data not shown). The MK biosynthetic pathway entails 7 reactions that convert chorismate to MK-7 by 7 genes in 4 predicted operons, MenF, MenDHCE, MenB, and MenA (Fig. 8). Although most men promoters were unresponsive to the oxyR mutation the menA promoter displayed enhanced activity, approximately 2-fold relative to that in the wild-type (Fig. 8). Because MenA is specific for MK-7 synthesis while all other Men proteins are responsible for production of 1,4-dihydroxy-2-naphthoate, an intermediate for all MKs, the result supports that MK-7 is probably only MK species in increased production in the oxyR mutant as revealed above (Fig. 6).

Activity of promoters for MK-7 biosynthesis operons were assayed by an integrated lacZ reporter in indicated strains grown in CAT-containing LB. The MK-7 biosynthesis pathway was outlined. MenDHCE, underlined, are predicted to be co-transcribed (www.biocyc.org). Cells of mid-log phase cultures (~0.3 of OD600) were prepared the same as in Fig. 3A. Experiments were performed at least three times with error bars representing standard deviation.
OxyR is regarded as a global regulator but its regulon remains poorly defined in S. oneidensis12. To determine whether OxyR regulates cyd and menA transcription in a direct manner, we employed bacterial one-hybrid (B1H) to assess the interaction between OxyR and the menA promoter sequence. The B1H system used here is particularly effective and efficient as it is derived from the BacterioMatch II two-hybrid system41. Vectors containing ‘bait’ (DNA) and ‘target’ (OxyR) were co-transformed into the reporter strain, of which those having positive DNA-protein interactions are able to grow on selective plates. The reporter strains carrying positive control pair PkatB and OxyR (OxyR in reduced form repressing the katB gene) formed a large number of colonies on selective plates (Table 2). A similar result was obtained with the reporter strains carrying another positive control pair PahpC and OxyRL197P, an OxyR mutant that is locked in the activated state for transcription of the ahpC gene50. In contrast, the reporter strain having negative control pair (P16s-rRNA/OxyR and P16s-rRNA/OxyRL197P) developed a few colonies, which, importantly, were unable to grow on confirmation plates. When Pcyd/OxyR, Pcyd/OxyRL197P, PmenA/OxyR, and PmenA/OxyRL197P, were under examination, the reporter strains failed to form any colonies that can pass through confirmation. These data suggest that the regulatory region of the menA gene unlikely interacts with OxyR in S. oneidensis.
Discussion
In many bacteria, OxyR is now regarded as a pleiotropic regulator and its loss causes multiple defects in various biological processes1,51. While some are species-dependent, two are profound and common: viability deficiency and growth defect. This is not surprising as most OxyR proteins studied to date function as an activator, at least for H2O2–scavenging genes. Because it seems logic to attribute these two defects of oxyR mutants to reduced ability to scavenge endogenous H2O2, direct experimental validation lags behind, if not totally overlooked. The scenario is different in S. oneidensis, whose OxyR acts as a repressor for major catalase gene katB such that KatB levels in the oxyR mutant are high, leading to rapid H2O2 degradation12. Thus, we were surprised when we found that the S. oneidensis oxyR mutant also carries these defects. After illustrating the mechanism accountable for viability deficiency13, we made efforts to identify factors underlying the growth defect of the oxyR mutant.
Viability deficiency is largely due to cellular damages caused by ambient H2O2, which is spontaneously generated in rich media13. As exogenous catalase is able to fully suppress this defect but fails in correcting the growth defect, it is apparent that different factors are responsible for the latter. Upon revelation of reduced electron supply from the quinone pool to cytochrome cbb3 in the oxyR mutant as a main cause to the growth defect, we suggest a model to explain what we have observed in this study (Fig. 9). In S. oneidensis, cytochrome cbb3, which obtains electrons from ubiquinones via cytochrome bc1, is the primary system for oxygen respiration, and thus dictates aerobic growth18. Although abundance of both cytochrome cbb3 and cytochrome bc1 is not affected by the oxyR mutation, fewer electrons are passed to these protein complexes, resulting in growth defect of the oxyR mutant. Quantities of electrons in the quinone pool may not be significantly altered, based on the observation that the mutation has a negligible impact on the NAD+/NADH ratio. Instead, substantial increase in production of cytochrome bd and MK-7, especially the former, leads to drastic reduction in the number of electrons that are passed to cytochrome cbb3. By removing other quinone oxidases, cytochrome bd and CymA, electron flow to cytochrome bc1 augments, leading to suppression of the growth defect.

The electron transport chain in the cytoplasmic membrane links the quinone pool with cytochrome cbb3, cytochrome bd, and various terminal reductases. All relevant components were included. The quinone pool is composed of UQs and MKs, which act as electron donors specifically for terminal oxidases (cytochrome cbb3 and bd) and CymA, respectively. (A) In the wild-type, UQs are slightly more abundant than MKs. The majority of electrons (by thick red lines) in the quinone pool are delivered to energy-efficient cytochrome cbb3 for oxygen respiration. (B) In the oxyR mutant, cytochrome bd is overproduced, impairing energy conservation (proton gradient) because of its low efficiency (dash line represents non-proton-pump mechanism for proton translocation) and low activity of cytochrome cbb3 due to an electron shortage. In addition, the UQs/MKs ratio is reduced, leading to less electrons available for oxygen respiration. The terminal phenotype for the oxyR mutant is reduced growth rate.
In the electron transport chain for respiration, the quinone pool plays a central role as a recyclable electron reservoir which accepts electrons from donors such as NADH and passes them to quinone oxidases, and eventually to electron accepting molecules25. This is particularly important for S. oneidensis, in which respiration is the only means of energy generation to support growth52. Given that Shewanella thrive in redox-stratified environments, evolution has arranged to produce UQs and MKs in considerable quantities under any circumstance and at least three quinone oxidases, cytochrome bd, cytochrome bc1, and CymA during aerobiosis, such that simultaneous respiration of multiple EAs could proceed. For example, nitrate is reducible by cells at any growth phase under aerobic conditions and many EAs can be respired after aerobic-growing cells enter the stationary phase53,54. In the oxyR mutant, the composition of UQs and MKs is altered significantly, largely due to increased levels of MKs. Given that UQs are preferable, if not exclusive, electron donors for oxygen respiration, reduction in UQ contents, or at least in proportion, diminishes electron flow to cytochrome bc1 and then to cytochrome cbb3, resulting in the growth defect. In line with this, the defect is relieved by either depleting MKs or adding exogenous UQ.
Under aerobic conditions, oxidative phosphorylation, the predominant way for the conservation of energy, is linked to an energized state of the membrane, which is established by electron transport reactions between electron carriers25. The process is initiated with NADH, a molecule generated mainly through the tricarboxylic acid (TCA) cycle. NADH dehydrogenase transfer electrons from NADH (oxidation) to quinones such as UQs or MKs. This electron input route of S. oneidensis into the quinone pool largely resembles that established in the model organisms. More importantly, the electron input into the quinone pool is hardly affected by the oxyR mutation based on the finding that the intracellular ratio of NAD+ to NADH is unchanged in the oxyR mutant. In contrast, the electron output from the quinone pool is carried out by remarkably diverse electron transport pathways20,30,32. As a consequence, a competition exists among quinone oxidases that couple the quinone pool to different electron accepting molecules. Among three quinone oxidases produced during aerobiosis, CymA not only requires MK-7 as a co-factor but also has a clear catalytic bias towards the reduction of MK-728,30. Moreover, given the reduction potentials of CymA and UQs, oxidation of UQs by CymA is thermodynamically unfeasible30. Hence, it is not surprising that the impact of the CymA loss on the growth defect of the oxyR mutant is rather limited. In contrast, there is direction competition between cytochromes bd and bc1 for electrons carried by UQs. When the ratio of cytochromes bd to UQs increases, less electrons are transferred to cytochrome bc1. Our data also suggest that cytochrome bd is advantageous of cytochrome bc1 in reducing UQs. Overproduction of cytochrome bd worsens its inhibitory effect on the growth of the oxyR mutant, but cytochrome bc1 in excess could not alleviate the defect. This observation is in line with the differences in oxygen affinity between cytochrome bd and cbb3; cytochrome bd has a higher affinity to oxygen than cytochrome cbb3. As such, when cytochrome bd is produced, it consumes electrons in the quinone pool to respire oxygen over cytochrome cbb3. To compete under aerobic conditions, S. oneidensis tends to repress production of cytochrome bd in the presence of cytochrome cbb318. Derepression can be achieved when cytochrome cbb3 is depleted or cells grow in certain conditions, such as in the presence of nitrite or cAMP at elevated levels22,23.
UQs appear to be essential to aerobic respiration in S. oneidensis. This is not surprising because the bacterium that is unable to ferment52. Although levels of UQs in the oxyR mutant do not change significantly, the ratio of UQs to MKs is substantially reduced due to enhanced production of the latter. Commonly, only a few steps, especially those rate-limiting ones, in a biosynthesis pathway are conditionally inducible, such as hemA, hemF, and hemH in heme synthesis23,55. In line with this, the presented data demonstrate that menA, out of 7 men genes for the MK biosynthetic pathway56, is only one subjected to repression by OxyR. Coincidently, the cyd operon is also under repression of OxyR. The repression, however, appears indirect because OxyR fails to interact with the menA promoter region. Like the cyd operon18,22, the menA gene is probably to be directly regulated by cAMP-Crp as a conserved Crp-binding motif (5’-ATCTGTGCGCCAATTCAAACTC) resides in the upstream region57. In E. coli, a key physiological role of the cAMP-Crp complex is to ensure resources to be spent on distinct metabolic sectors as needed in different nutrient environments58. But this may not be the case in S. oneidensis given that the cAMP-Crp complex critically deviates from the E. coli paradigm with respect to regulons57. Nevertheless, given the consistent responses of the menA gene and the cyd operon to the oxyR mutation, it is likely that OxyR plays a role in mediating respiration. Efforts to test this notion are underway.
Why does the oxyR mutation up-regulate production of cytochrome bd and MKs ? Given that the ultimate consequence is the reduced growth rate, we propose that the oxyR mutant probably exploits both to diminish production of endogenous ROS. There is a large body of evidence to show that cytochrome bd enhances bacterial resistance to oxidative stress24. Although not completely understood, two mechanisms underpinning it are proposed: cytochrome bd may act as an O2-scavenger and electron sink as well as ROS metabolizer59,60,61. Due to its high affinity for O2 and low energetic efficiency, cytochrome bd is able to reduce the intracellular O2 levels without generating sufficient energy to support fast growth, and thus repress ROS formation, H2O2 in particular. Moreover, ROS formation is also repressed by lowered electron availability for ROS-generation enzymes because electrons in the quinone pool are simultaneously dissipated. As a result, although the oxyR mutant grows at a reduced rate, its ability to consume oxygen is not significantly compromised. On the other hand, cytochrome bd can directly remove H2O2 and other ROS62. But this may not be critical in S. oneidensis because the oxyR mutant has elevated ability to remove H2O2, which overshadows the ROS-scavenger activity of cytochrome bd12,13. In comparison with cytochrome bd, which is no doubts the major system accountable for the growth defect of the oxyR mutant, MKs seem auxiliary. The Increased levels of MKs resulting from the oxyR mutation may prompt dissipation of electrons onto non-oxygen EAs, leaving less to react with oxygen, thus limiting growth rate and ROS formation.
Additional Information
How to cite this article: Wan, F. et al. Loss of OxyR reduces efficacy of oxygen respiration in Shewanella oneidensis. Sci. Rep. 7, 42609; doi: 10.1038/srep42609 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Imlay, J. A. The molecular mechanisms and physiological consequences of oxidative stress: lessons from a model bacterium. Nat Rev Micro 11, 443–454, doi: 10.1038/nrmicro3032 (2013).
Korshunov, S. & Imlay, J. A. Two sources of endogenous hydrogen peroxide in Escherichia coli . Mol Microbiol 75, 1389–1401, doi: 10.1111/j.1365-2958.2010.07059.x (2010).
Ravindra Kumar, S. & Imlay, J. A. How Escherichia coli tolerates profuse hydrogen peroxide formation by a catabolic pathway. J Bacteriol 195, 4569–4579, doi: 10.1128/jb.00737-13 (2013).
Imlay, J. A. Cellular defenses against superoxide and hydrogen peroxide. Annu Rev Biochem 77, 755–776, doi: 10.1146/annurev.biochem.77.061606.161055 (2008).
Mishra, S. & Imlay, J. Why do bacteria use so many enzymes to scavenge hydrogen peroxide? Arch Biochem Biophys 525, 145–160, doi: 10.1016/j.abb.2012.04.014 (2012).
Seib, K. L. et al. Characterization of the OxyR regulon of Neisseria gonorrhoeae . Mol Microbiol 63, 54–68, doi: 10.1111/j.1365-2958.2006.05478.x (2007).
Fredrickson, J. K. et al. Towards environmental systems biology of Shewanella . Nat Rev Micro 6, 592–603 (2008).
Ghosal, D. et al. How radiation kills cells: Survival of Deinococcus radiodurans and Shewanella oneidensis under oxidative stress. FEMS Microbiol Rev 29, 361–375, doi: 10.1016/j.fmrre.2004.12.007 (2005).
Li, N., Luo, Q., Jiang, Y., Wu, G. & Gao, H. Managing oxidative stresses in Shewanella oneidensis: intertwined roles of the OxyR and OhrR regulons. Environ Microbiol 16, 1821–1834, doi: 10.1111/1462-2920.12418 (2014).
Fu, H., Yuan, J. & Gao, H. Microbial oxidative stress response: Novel insights from environmental facultative anaerobic bacteria. Arch Biochem Biophys 584, 28–35, doi: 10.1016/j.abb.2015.08.012 (2015).
Wu, G., Wan, F., Fu, H., Li, N. & Gao, H. A matter of timing: Contrasting effects of hydrogen sulfide on oxidative stress response in Shewanella oneidensis . J Bacteriol 197, 3563–3572, doi: 10.1128/jb.00603-15 (2015).
Jiang, Y. et al. Protection from oxidative stress relies mainly on derepression of OxyR-Dependent KatB and Dps in Shewanella oneidensis . J Bacteriol 196, 445–458, doi: 10.1128/JB.01077-13 (2014).
Shi, M., Wan, F., Mao, Y. & Gao, H. Unraveling the mechanism for the viability deficiency of Shewanella oneidensis oxyR null mutant. J Bacteriol 197, 2179–2189, doi: 10.1128/jb.00154-15 (2015).
Christman, M. F., Storz, G. & Ames, B. N. OxyR, a positive regulator of hydrogen peroxide-inducible genes in Escherichia coli and Salmonella typhimurium, is homologous to a family of bacterial regulatory proteins. Proc Natl Acad Sci USA 86, 3484–3488 (1989).
Maciver, I. & Hansen, E. J. Lack of expression of the global regulator OxyR in Haemophilus influenzae has a profound effect on growth phenotype. Infect Immuni 64, 4618–4629 (1996).
Brzezinski, P. & Gennis, R. B. Cytochrome c oxidase: exciting progress and remaining mysteries. J Bioenerg Biomembr 40, 521–531, doi: 10.1007/s10863-008-9181-7 (2008).
Puustinen, A., Finel, M., Haltia, T., Gennis, R. B. & Wikstrom, M. Properties of the two terminal oxidases of Escherichia coli . Biochemistry 30, 3936–3942 (1991).
Zhou, G. et al. Combined effect of loss of the caa 3 oxidase and Crp regulation drives Shewanella to thrive in redox-stratified environments. ISME J 7, 1752–1763, doi: 10.1038/ismej.2013.62 (2013).
Chen, H., Luo, Q., Yin, J., Gao, T. & Gao, H. Evidence for the requirement of CydX in function but not assembly of the cytochrome bd oxidase in Shewanella oneidensis . Biochim Biophys Acta 1850, 318–328 (2015).
Yin, J. et al. Regulation of nitrite resistance of the cytochrome cbb 3 oxidase by cytochrome c ScyA in Shewanella oneidensis . MicrobiologyOpen 4, 84–99, doi: 10.1002/mbo3.224 (2015).
Le Laz, S. et al. A biochemical approach to study the role of the terminal oxidases in aerobic respiration in Shewanella oneidensis MR-1. PLoS One 9, e86343 (2014).
Fu, H. et al. Crp-dependent cytochrome bd oxidase confers nitrite resistance to Shewanella oneidensis . Environ Microbiol 15, 2198–2212, doi: 10.1111/1462-2920.12091 (2013).
Zhang, H. et al. Impacts of nitrate and nitrite on physiology of Shewanella oneidensis . PLoS One 8, e62629 (2013).
Giuffrè, A., Borisov, V. B., Arese, M., Sarti, P. & Forte, E. Cytochrome bd oxidase and bacterial tolerance to oxidative and nitrosative stress. Biochim Biophys Acta 1837, 1178–1187, doi: 10.1016/j.bbabio.2014.01.016 (2014).
Aussel, L. et al. Biosynthesis and physiology of coenzyme Q in bacteria. Biochim Biophys Acta 1837, 1004–1011, doi: 10.1016/j.bbabio.2014.01.015 (2014).
Unden, G. & Bongaerts, J. Alternative respiratory pathways of Escherichia coli: energetics and transcriptional regulation in response to electron acceptors. Biochim Biophys Acta 1320, 217–234, doi: 10.1016/s0005-2728(97)00034-0 (1997).
Venkateswaran, K. et al. Polyphasic taxonomy of the genus Shewanella and description of Shewanella oneidensis sp. nov. Inter J Syst Bacteriol 49, 705–724, doi: 10.1099/00207713-49-2-705 (1999).
McMillan, D. G. G., Marritt, S. J., Butt, J. N. & Jeuken, L. J. C. Menaquinone-7 is specific cofactor in tetraheme quinol dehydrogenase CymA. J Biol Chem 287, 14215–14225, doi: 10.1074/jbc.M112.348813 (2012).
Borisov, V. B., Gennis, R. B., Hemp, J. & Verkhovsky, M. I. The cytochrome bd respiratory oxygen reductases. Biochim Biophys Acta 1807, 1398–1413, doi: 10.1016/j.bbabio.2011.06.016 (2011).
Marritt, S. J. et al. A functional description of CymA, an electron-transfer hub supporting anaerobic respiratory flexibility in Shewanella . Biocheml J 444, 465–474, doi: 10.1042/bj20120197 (2012).
Safarian, S. et al. Structure of a bd oxidase indicates similar mechanisms for membrane-integrated oxygen reductases. Science 352, 583–586, doi: 10.1126/science.aaf2477 (2016).
Fu, H., Jin, M., Ju, L., Mao, Y. & Gao, H. Evidence for function overlapping of CymA and the cytochrome bc 1 complex in the Shewanella oneidensis nitrate and nitrite respiration. Environ Microbiol 16, 3181–3195, doi: 10.1111/1462-2920.12457 (2014).
Jin, M. et al. Unique organizational and functional features of the cytochrome c maturation system in Shewanella oneidensis . PLoS One 8, e75610, doi: 10.1371/journal.pone.0075610 (2013).
Wu, L., Wang, J., Tang, P., Chen, H. & Gao, H. Genetic and molecular characterization of flagellar assembly in Shewanella oneidensis . PLoS One 6, doi: 10.1371/journal.pone.0021479 (2011).
Luo, Q., Dong, Y., Chen, H. & Gao, H. Mislocalization of Rieske protein peta predominantly accounts for the aerobic growth defect of tat mutants in Shewanella oneidensis . PLoS One 8, e62064, doi: 10.1371/journal.pone.0062064 (2013).
Das, S., Noe, J. C., Paik, S. & Kitten, T. An improved arbitrary primed PCR method for rapid characterization of transposon insertion sites. J Microbiol Meth 63, 89–94, doi: 10.1016/j.mimet.2005.02.011 (2005).
Shi, M. et al. Exoprotein production correlates with morphotype changes of nonmotile Shewanella oneidensis mutants. J Bacteriol 195, 1463–1474, doi: 10.1128/jb.02187-12 (2013).
Marrs, B. & Gest, H. Genetic mutations affecting the respiratory electron-transport system of the photosynthetic bacterium Rhodopseudomonas capsulata . J Bacteriol 114, 1045–1051 (1973).
Myers, J. M. & Myers, C. R. Role of the tetraheme cytochrome CymA in anaerobic electron transport in cells of Shewanella putrefaciens MR-1 with normal levels of menaquinone. J Bacteriol 182, 67–75 (2000).
Sun, L. et al. Two residues predominantly dictate functional difference in motility between Shewanella oneidensis flagellins FlaA and FlaB. J Biol Chem 289, 14547–14559 (2014).
Guo, M. et al. Dissecting transcription regulatory pathways through a new bacterial one-hybrid reporter system. Genome Res 19, 1301–1308, doi: 10.1101/gr.086595.108 (2009).
Li, M., Meng, Q., Fu, H., Luo, Q. & Gao, H. Suppression of fabB mutation by fabF1 is mediated by transcription read-through in Shewanella oneidensis . J Bacteriol 198, 3060–3069, doi: 10.1128/jb.00463-16 (2016).
Schwalb, C., Chapman, S. K. & Reid, G. A. The tetraheme cytochrome CymA is required for anaerobic respiration with dimethyl sulfoxide and nitrite in Shewanella oneidensis . Biochemistry 42, 9491–9497, doi: 10.1021/bi034456f (2003).
Burns, J. L. & DiChristina, T. J. Anaerobic respiration of elemental sulfur and thiosulfate by Shewanella oneidensis MR-1 requires psrA, a homolog of the phsA gene of Salmonella enterica Serovar Typhimurium LT2. Appl Environ Microbiol 75, 5209–5217, doi: 10.1128/aem.00888-09 (2009).
Shirodkar, S., Reed, S., Romine, M. & Saffarini, D. The octahaem SirA catalyses dissimilatory sulfite reduction in Shewanella oneidensis MR-1. Environ Microbiol 13, 108–115, doi: 10.1111/j.1462-2920.2010.02313.x (2011).
Beliaev, A. S. et al. Global transcriptome analysis of Shewanella oneidensis MR-1 exposed to different terminal electron acceptors. J Bacteriol 187, 7138–7145, doi: 10.1128/jb.187.20.7138-7145.2005 (2005).
Gao, H. C. et al. Reduction of nitrate in Shewanella oneidensis depends on atypical NAP and NRF systems with NapB as a preferred electron transport protein from CymA to NapA. ISME J 3, 966–976, doi: 10.1038/ismej.2009.40 (2009).
Suvarna, K., Stevenson, D., Meganathan, R. & Hudspeth, M. E. S. Menaquinone (Vitamin K(2)) biosynthesis: Localization and characterization of the menA gene from Escherichia coli. J Bacteriol 180, 2782–2787 (1998).
Friedrich, T., Dekovic, D. K. & Burschel, S. Assembly of the Escherichia coli NADH:ubiquinone oxidoreductase (respiratory complex I). Biochim Biophys Acta 1857, 214–223, doi: 10.1016/j.bbabio.2015.12.004 (2016).
Binnenkade, L., Teichmann, L. & Thormann, K. M. Iron Triggers λ So prophage induction and release of extracellular DNA in Shewanella oneidensis MR-1 Biofilms. Appl Environ Microbiol 80, 5304–5316, doi: 10.1128/aem.01480-14 (2014).
Dubbs, J. M. & Mongkolsuk, S. Peroxide-sensing transcriptional regulators in bacteria. J Bacteriol 194, 5495–5503, doi: 10.1128/jb.00304-12 (2012).
Serres, M. H. & Riley, M. Genomic analysis of carbon source metabolism of Shewanella oneidensis MR-1: Predictions versus experiments. J Bacteriol 188, 4601–4609 (2006).
Dong, Y. et al. A Crp-dependent two-component system regulates nitrate and nitrite respiration in Shewanella oneidensis . PLoS One 7, doi: 10.1371/journal.pone.0051643 (2012).
Yuan, J., Chen, Y., Zhou, G., Chen, H. & Gao, H. Investigation of roles of divalent cations in Shewanella oneidensis pellicle formation reveals unique impacts of insoluble iron. Biochim Biophys Acta 1830, 5248–5257, doi: 10.1016/j.bbagen2013.07.023 (2013).
Mancini, S. & Imlay, J. A. The induction of two biosynthetic enzymes helps Escherichia coli sustain heme synthesis and activate catalase during hydrogen peroxide stress. Mol Microbiol 96, 744–763, doi: 10.1111/mmi.12967 (2015).
Yin, J., Meng, Q., Fu, H. & Gao, H. Reduced expression of cytochrome oxidases largely explains cAMP inhibition of aerobic growth in Shewanella oneidensis . Sci Rep 6, 24449, doi: 10.1038/srep24449 (2016).
Gao, H. et al. Physiological roles of ArcA, Crp, and EtrA and their interactive control on aerobic and anaerobic respiration in Shewanella oneidensis . PLoS One 5, e15295, doi: 10.1371/journal.pone.0015295 (2010).
You, C. et al. Coordination of bacterial proteome with metabolism by cyclic AMP signalling. Nature 500, 301–306, doi: 10.1038/nature12446 (2013).
Goldman, B. S., Gabbert, K. K. & Kranz, R. G. The temperature-sensitive growth and survival phenotypes of Escherichia coli cydDC and cydAB strains are due to deficiencies in cytochrome bd and are corrected by exogenous catalase and reducing agents. J Bacteriol 178, 6348–6351 (1996).
Lindqvist, A., Membrillo-Hernández, J., Poole, R. K. & Cook, G. M. Roles of respiratory oxidases in protecting Escherichia coli K12 from oxidative stress. Antonie van Leeuwenhoek 78, 23–31, doi: 10.1023/a:1002779201379 (2000).
Forte, E. et al. Cytochrome bd oxidase and hydrogen peroxide resistance in Mycobacterium tuberculosis . mBio 4, 01006–13, doi: 10.1128/mBio.01006-13 (2013).
Borisov, V. B. et al. Cytochrome bd oxidase from Escherichia coli displays high catalase activity: An additional defense against oxidative stress. FEBS Lett 587, 2214–2218, doi: 10.1016/j.febslet.2013.05.047 (2013).
Dairi, T. In Methods in Enzymology, pp. 107–122 David, A. H. (ed), Chapter Six - Menaquinone biosyntheses in microorganisms, Academic Press (2012).
Acknowledgements
This research was supported by National Natural Science Foundation of China (41476105), Natural Science Foundation of Zhejiang Province (LZ17C010001), and the Fundamental Research Funds for the central Universities (2015FZA6001, 2016FZA6003).
Author information
Authors and Affiliations
Contributions
H.G. conceived the idea and designed the project. F.W. and M.S. carried out the experiments. F.W. and H.G. analyzed data. F.W. and H.G. wrote the paper. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Wan, F., Shi, M. & Gao, H. Loss of OxyR reduces efficacy of oxygen respiration in Shewanella oneidensis. Sci Rep 7, 42609 (2017). https://doi.org/10.1038/srep42609
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep42609
- Springer Nature Limited
This article is cited by
-
Mechanisms of oxidative response during biodegradation of malathion by S. oneidensis MR-1
Environmental Science and Pollution Research (2024)
-
Regulation of fadR on the ROS defense mechanism in Shewanalla oneidensis
Biotechnology Letters (2024)