
Abstract
Dagger and needle nematodes included in the family Longidoridae (viz. Longidorus, Paralongidorus, and Xiphinema) are highly polyphagous plant-parasitic nematodes in wild and cultivated plants and some of them are plant-virus vectors (nepovirus). The mitochondrial (mt) genomes of the dagger and needle nematodes, Xiphinema rivesi, Xiphinema pachtaicum, Longidorus vineacola and Paralongidorus litoralis were sequenced in this study. The four circular mt genomes have an estimated size of 12.6, 12.5, 13.5 and 12.7 kb, respectively. Up to date, the mt genome of X. pachtaicum is the smallest genome found in Nematoda. The four mt genomes contain 12 protein-coding genes (viz. cox1-3, nad1-6, nad4L, atp6 and cob) and two ribosomal RNA genes (rrnL and rrnS), but the atp8 gene was not detected. These mt genomes showed a gene arrangement very different within the Longidoridae species sequenced, with the exception of very closely related species (X. americanum and X. rivesi). The sizes of non-coding regions in the Longidoridae nematodes were very small and were present in a few places in the mt genome. Phylogenetic analysis of all coding genes showed a closer relationship between Longidorus and Paralongidorus and different phylogenetic possibilities for the three Xiphinema species.
Similar content being viewed by others


Introduction
The phylum Nematoda is one of the largest and most diverse groups of animal organisms, with a global distribution. Most species are found in oceanic, freshwater and soil ecosystems, and only a small number are pathogens of animals or plants. They cause reductions in agricultural productivity and disease in humans, and animals1. Plant-parasitic nematodes (PPNs) are distributed between Classes Chromadorea and Enoplea in only three orders viz. Rhabditida, Dorylaimida and Triplonchida2. The order Dorylaimida within Enoplea includes several genera of dagger and needle nematodes belonging to the family Longidoridae (viz. Australodorus, Longidoroides, Longidorus, Paralongidorus, Paraxiphidorus, Xiphidorus and Xiphinema)2. Longidoridae nematodes are highly polyphagous on wild and cultivated plants, and some are plant-virus vectors (nepovirus)3. Also some of the Longidoridae species are listed as A1 and A2 quarantine pests by the European and Mediterranean Plant Protection Organisation (EPPO, www.eppo.int/QUARANTINE/).
Mitochondrial (mt) genomes and sequences of individual mt genes are used to infer phylogenetic relationships among species at different taxonomic levels4,5,6,7. Animal mtDNAs are relatively constant in gene content and order, maternally inherited, and have a reduced recombination rate and high evolutionary rate8. There are fourteen mt genomes of PPNs sequenced to date, and only one of them was included in the class Enoplea (Xiphinema americanum) and fourteen in the class Chromadorea (Aphelenchoides besseyi, Bursaphelenchus mucronatus, B. xylophilus, Globodera ellingtonae, G. pallida, G. rostochiensis, Heterodera glycines, Pratylenchus vulnus, Meloidogyne chitwoodi, M. floridensis, M. graminicola, M. hapla, M. incognita, Radopholus similis) and some of them display some unusual features9,10,11,12,13,14,15,16,17,18,19,20,21. Xiphinema americanum mt genome encodes genes in both strands and has short coding regions18. On the other hand, within PPN Chromadorean species, Globodera ellingtonae, G. pallida and G. rostochiensis have multipartite mt genomes17,19,20, Meloidogyne spp. and Pratylenchus vulnus have a large non-coding region with tandem repeats and the control region9,10,11,12,13,14,21 and Radopholus similis has a unique genetic code, and uses the UAA codon for the aminoacid Tyr (Y) instead of a termination site16. Thus, there is a lack of information concerning the mt genomes for other genera within Longidoridae which is needed to derive insights into their taxonomy, phylogeny and possible molecular markers.
The main objectives of this research were: (i) to determine the mitochondrial genomes in three different genera within important PPNs from Longidoridae (Xiphinema, Longidorus and Paralongidorus); and (ii) to associate their gene structures and protein-coding gene sequences with their phylogeny.
Results and Discussion
General features of the mitochondrial genomes of Longidorus vineacola, Paralongidorus litoralis, Xiphinema pachtaicum and Xiphinema rivesi
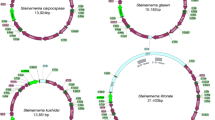
A summary of the mt genomes in Longidoridae is shown in Table 1. The complete mt genomes of P. litoralis, L. vineacola, X. pachtaicum and X. rivesi were 12,763 bp (KU746819), 13,519 bp (KU746818), 12,489 bp (KU746821) and 12,624 bp (KU746820) in size, respectively (Fig. 1 and Table 1). The four new mt genomes showed a similar size and gene number complement to X. americanum (NC_005928) (12,626 bp). They are smaller in size than other Enopleans species such as Romanomermis culicivorax (26,194 bp) or Hexamermis agrotis (24,606 bp). The mt genome of X. pachtaicum represents the smallest mt Nematoda genome known so far (search done in GenBank, November 9, 2016).

Positive or negative after the gene name abbreviation depends on the sense in the genome. Protein and rRNA genes have standard nomenclature. tRNA genes were designated by single-letter abbreviations. Two tRNAs for Leucine (L) and Serine (S) are present. Non-coding regions, with the exception of possible replication control region (CR), were not included.
The nucleotide composition of the mtDNA genomes studied showed an A + T content similar among dagger nematode species (66.50%, 68.86% and 68.50% for X. americanum, X. rivesi and X. pachtaicum, respectively), but slightly lower in needle nematode species (L. vineacola and P. litoralis, 63.64% and 63.89%, respectively) (Table 1). These levels were lower than that for other members of the class Enoplea such as Romanomermis culicivorax (79.34%) or Hexamermis agrotis (78.42%). The GT-rich sequences in Xiphinema species were 54.00% and 56.63% (G + T) in one of the strands for X. rivesi (GT rich strand not containing the cox1 gene) and X. pachtaicum (strand containing the cox1 gene), respectively; while, for Longidorus and Paralongidorus these differences were minimal (50.51% and 50.75%, respectively). These differences influence the coding genes, rRNA and tRNA distribution in the genome. In X. rivesi, the GT rich strand had sense sequences of 8 protein coding genes (PCGs), 12 tRNAs and the 2 rRNA genes whereas for X. pachtaicum 10 PCGs, 12 tRNAs and the 2 rRNA genes were detected. These differences were minimal for Longidorus and Paralongidorus between AC-rich vs GT-rich strands with 6 vs 6 PCGs, 10 vs 11 tRNAs and 1 rRNA gene in each of the strands in the case of P. litoralis (GT-rich strand containing the cox1 gene) and 5 vs 7 PCGs proteins, 14 vs 8 tRNAs, and the 2 vs 0 rRNA genes in the case of L. vineacola (GT-rich strand containing the cox1 gene).
The mtDNA genomes of L. vineacola, P. litoralis, X. pachtaicum and X. rivesi contained 12 PCGs, viz. cox1-3, nad1-6, nad4L, atp6 and cob; and two ribosomal RNA genes (rrnL and rrnS) but the atp8 gene was not detected (Fig. 1; Table 2). The gene arrangement within Longidoridae was very different within dagger and needle nematode species (Fig. 1), with the exception of X. americanum and X. rivesi, in which it was identical. This is in concordance with the very high degree of variation in mtDNA genome gene arrangements across the Metazoa22. A comparison of closely related species with different gene orders suggests that there are several types of “elementary” rearrangement events5: inversions, transpositions, inverse transpositions (i.e. a transposition in which the re-inserted fragment is inverted), and tandem duplications followed by the random loss of one of the copied genes.
Protein encoding genes and codon usage
Protein encoding genes were transcribed from both strands in the four mt genomes sequenced. Genes nad4L and nad3 were always together and separated by a non-coding region in all the studied species (Fig. 1). The gene order of two genes in nad5-nad6, atp6-nad4 and nad1-cox1 were conserved between X. americanum/X. rivesi vs X. pachtaicum. On the other hand, the gene order between Longidorus and Paralongidorus species was kept in two regions: cob-nad4L-nad3-cox1 and cox2-cox3-nad2. Only the gene associations of nad5-nad6 (inverted gene sense in L. vineacola) and atp6-nad4 were kept between Paralongidorus and Xiphinema species. We could not find coincidences for arrangement of PCGs between the genus Longidorus and Xiphinema. For instance, the association between nad5-nad6 and atp6-nad4 seem derived from an inversion between Longidorus and Paralongidorus.
All PCGs shared an ATA start codon. The PCGs all terminated with a potential TAA stop codon with the exception of cox3 and nad3 for X. rivesi; cox1, cox3, nad4, nad4L, nad6 and cob for X. pachtaicum, and nad1 and nad3 for P. litoralis (Table 2). Some genes in all the nematode species studied partially overlapped and probably terminated with T/TA (Table 2). These features were not conserved in these genes among the studied species. Only the cox2 termination codon was conserved in X. americanum, X. rivesi, X. pachtaicum and L. vineacola. However, they were partially conserved between X. americanum and X. rivesi for cox1 and atp6 genes. Additionally to these termination codons, some genes overlap several tRNA codons and a few bases (1 or 2) with other genes (Table 2). Coding gene overlapping seems to be a common feature in Longidoridae, since the five sequenced species showed this feature in their genomes. Overlaps were detected in the same or in the opposite direction. However, gene overlap in the same sense strand was not detected in P. litoralis. In X. americanum, X. rivesi and X. pachtaicum a 1 bp overlap was detected between nad2 and cox2 in the same sense strand, X. pachtaicum has a 4 bp overlap between nad4 and cox3, and L. vineacola has a 1 bp overlap between cox2 and cox3 in the same sense strand in both cases. In the case of overlapping coding genes in humans (atp8/atp6 and nad4L/nad4) both pairs of genes result in dicistronic messengers23. In Ascidian species, two cases of overlapped genes, expressed as transcripts with no poly(A) tails downstream of the first open reading frame, suggested the presence of mature bicistronic transcripts24.
The majority of genes kept the transcription sense with 2 or more consecutive genes, but there are exceptions where unique genes have a different sense to other genes in the mitochondria, as cox1 in the case of X. americanum and X. rivesi, and nad1 in the case of L. vineacola and P. litoralis. It is suggested that gene arrangement is affected by the transcription mechanism, for example by the need to co-regulate the expression of some genes or the required stoichiometry of the gene products22.
As in other published nematode mtDNAs, the mtPCGs of our sequenced species were notably biased toward using amino acids encoded by T-rich codons13 (Table 3). The three most frequently used codons were TTT, TTA and ATT (Table 3) in Xiphinema species. These T-rich codons accounted for 21.97% (X. americanum), 24.37% (X. rivesi) and 23.68% (X. pachtaicum) of the total codons used. In the case of Longidorus and Paralongidorus the most used triplets were TTA, TTT and ATA. These codons account for 18.61% (P. litoralis) and 17.01% (L. vineacola) of the total codons used. However, these differences in T-rich codons were smaller in comparison to other Chromadorean nematodes such as B. xylophilus or P. vulnus, which account for 40.30% and 31.44% of the total codons used, respectively, while our range of the most used codons was similar to other Enoplean nematodes such as Trichinella or Trichuris species25,26. The higher frequency of amino acids encoded by T-rich codons, and unequal synonymous codon usage with bias against C-rich codons is consistent with the high percentage of A + T content in the nucleotide composition of PCGs as in other mt genomes for nematodes13.
Transfer RNA (tRNA) and ribosomal RNA genes
The mt genome of metazoans typically encodes 22 tRNAs8. However, only L. vineacola showed the typical 22 tRNAs, whereas 21 tRNAs were identified in X. rivesi and in P. litoralis in which we could not identify the mt-tRNAN gene, and 19 tRNAs were identified in X. pachtaicum in which we could not identify the mt-tRNAN, mt-tRNAA and mt-tRNAR genes. The position predicted for mt-tRNAN in mt genomes of X. rivesi and P. litoralis based on sequence similarity in the genes predicted by Jühling et al. was deep inside the l-rRNA annotation and in the same sense strand27. The annotation of our l-rRNA was based on similarity to sequences deposited in GenBank and mainly with the X. americanum l-rRNA annotation done by He et al. using mRNA sequencing18. The same situation was also found in X. pachtaicum for mt-tRNAN where additionally, the other two mt-RNAs, mt-tRNAA and mt-tRNAR, were not found. These tRNAs would therefore need to be imported from the nucleus, implying that a mechanism of this sort exists in nematodes. Import of tRNA from the nucleus to the mitochondrion has been demonstrated in marsupials28 and in a protozoan (Trypanosoma bruceii)29. Another possibility is that the tRNA detection methods used in this study (Mitos-MITFI and Arwen v1.2) could not identify them.
The tRNA structures detected in the four studied mtDNA genomes shared some features with those of other metazoans30 including a 5 bp anticodon stem, a 7 base anticodon loop with a T always preceding an anticodon as well as a purine always following an anticodon (Figs S1–S3). Four secondary structures have been found in Longidoridae (Table 4; Figs S1–S3): (i) typical nematode tRNAs structure with a D-arm but no T-arm; (ii) structure retaining their T-arm and lacking the D-arm; (iii) structures lacking both the T-arm and the D-arm; and (iv) the intact clover-leaf structure is also present. The conventional cloverleaf structure it is also present in the mt-tRNAs of Trichuris ovis, Trichuris discolor26 and Trichinella spiralis31. All of the tRNAs with a clover-leaf structure found in the species included in this study were coincident with those found in T. spiralis and only the tRNAY which showed a clover-leaf structure in X. rivesi, does not appear with this secondary structure in T. spiralis. These diversity of structures in our mt tRNAs support the hypothesis expressed by Jühling et al., that mt-tRNA not only evolve rapidly at the sequence level but also exhibit a variety of deviations from the common clover-leaf structure27.
Non-coding regions
The sizes of the non-coding regions in Longidoridae nematodes in this study were very small and in few places in the mt genome (Table 5). In the mt genome of X. americanum, the longest noncoding region was just 96 bp in length between the nad3 and the nad4L genes with an A + T content of 72%, and inverted or direct repeats were not present32. A sequence motif (5′-GAGACCTGAGCCCAAGATA-3′) was present in this 96-bp noncoding region for X. americanum32 that was similar to the conserved promoter element sequence (5-CA(G)ACC(G)CC(A)AAAGATA-3) around the transcription start site in the D-loop region of the human mt genome33. We could not find a clear similarity in our non-coding sequences to this promoter element sequence. However, the position of a non-coding region and a stable gene arrangement between nad3 and nad4l within all the studied Longidoridae nematodes pointed out the importance of this region in the viability of the nematode mitochondria. This feature, the strong secondary structure (Figs 2 and S4) and the location of the Control Region (CR) for X. americanum were the basis for annotating this sequence as CR. The CRs were of different size and composition in comparison to the A + T rich sequence found in X. americanum. Sizes of 93, 103, 96, 110, and 140 bps with A + T contents of 70%, 74%,72%, 73% and 75% were found in the case of L. vineacola, P. litoralis, X. americanum, X. rivesi and X. pachtaicum, respectively. For X. americanum, X. rivesi and X. pachtaicum, the A quantity was higher than T (X. americanum = A: 55.21% vs T: 16.67%; X. rivesi = A: 55.21% vs T: 17.71%; X. pachtaicum = A: 50.71% vs T: 24.29%), while for L. vineacola and P. litoralis it was more balanced between A and T (L. vineacola = A: 34.41% vs T: 35.48%; P. litoralis = A: 33.01% vs T: 40.78%). We could not find a good explanation for these ratios, but in the case of P. litoralis and L. vineacola, this region was accompanied with strong secondary structures and a similar number of coding genes in both strands. Probably, this composition has strong effects on replication and transcription. Lewis et al. found that mtDNA synthesis in the C. elegans gonad produces branched-circular lariat structures with multimeric DNA tails; they were able to detect multimers up to four mtDNA genome unit lengths34. These authors have raised the possibility that the rolling circle mtDNA replication mechanism may be an ancestral trait among metazoans. However, C. elegans has two well-defined non-coding regions and with our data only one was detected and in a similar position in the studied Longidoridae species. Many species of Arthropoda, Nematoda, Mollusca and Annelida harbor palindromes and inverted repeats in their CRs35. Length of the CR in the nematode mtDNA genome could be variable because of the presence of repeated sequences22. We only found a partially conserved palindrome sequence in the CR of P. litoralis (5′-TAGTAATAACTATTTTCAGTA-3′) applying the procedure explained in Arunkumar and Nagaraju35.

Stem-loop structures predicted for the noncoding region between the nad4L and nad3 genes (replication control region, CR) for Longidorus vineacola and Paralongidorus litoralis.
Another interesting aspect of these mt genomes was in the number of short non-coding regions and in their distribution in different positions in the genome ranged from 1 to 6. We found important differences between the studied species (excluding CR) (Table 5). The number of non-coding regions longer than 30 bp ranged from 1 to 6 non-coding regions. Non-coding regions longer than 50 bp were only found in L. vineacola and P. litoralis (4 and 1, respectively; Table 5). Secondary structures were drawn in Figs S5 and S6. Long non-coding regions showed strong structures, but they were located in different regions in L. vineacola and P. litoralis. Some non-coding regions were at the same position in all the studied species, as it is the case between tRNAN and tRNAQ, with a similar size (from 36 to 42 bp). This is coincident with the non-coding region having a strong arm structure (Figs S5 and S6). It is possible that these regions function as splicing recognition sites during processing of the transcripts. Mitochondrial genomes in Longidoridae have the important trait of genome size economy and in having fewer non-coding regions. These features were retained even with the important differences in gene arrangements between the studied sequences.
Mitochondrial phylogeny of Enoplea nematodes
We conducted BI and ML phylogenetic analyses of an amino acid sequence dataset (13 protein-coding genes) for 25 nematode species. Phylogenetic analysis separated four different clades and subclades including Trichinellidae, Trichuridae, Mermithidae and Longidoridae (Figs 3 and 4). In our analysis, Trichinellidae and Trichuridae formed a well-supported group which is sister to Mermithidae, and all these three groups formed a clade weakly supported in both analysis (BI and ML). This phylogenetic position is mainly consistent with recent reports based on mt genome analysis11,36, but those were only based on one sequence for Longidoridae (viz. X. americanum). Meanwhile, Longidoridae clade, with 5 representatives in our analysis, was a sister clade with the other groups in the Bayesian inference (BI) and Maximum likelihood (ML) analyses in the amino acid dataset (Figs 3 and 4). In Longidoridae, two well-supported subclades were formed, one of them with Longidorus, Paralongidorus and X. pachtaicum with Bayesian probability values (BPP) of 0.97 in the BI analysis, while the position of X. pachtaicum was not associated to any specific clade in the ML analysis, and another with the other Xiphinema species. Xiphinema americanum and X. rivesi were closely related phylogenetically and this is shown with the strong support of their relationship in both analyses (100% bootstrap support (BS) and 1.00 BPP), while the position of X. pachtaicum was well resolved in this clade by BI (1.00 BPP), but not by ML (65% BS). Nucleotide phylogenetic analysis showed a similar pattern of well-supported phylogenetic clades as in the aminoacid dataset forming two sister clades, one with Trichinellidae and Trichuridae and another with Mermithidae and Longidoridae in BI and ML analysis (Figs S7 and S8). However, this phylogenetic position with Longidoridae as a sister clade to Mermithidae was well-supported in BI and weakly supported in ML analysis. Longidoridae clade was formed by two subclades, one for Paralongidorus and Longidorus and the other for Xiphinema species. This clade was strongly supported in the BI analysis and weakly supported in the ML analysis. Xiphinema americanum and X. rivesi were closely related phylogenetically as in the case of amino acid dataset (100% BS and 1.00 BPP). This different position of the Longidoridae clade was shown by other researchers with only one sequence from Longidoridae (X. americanum)11,36. The results of Kim et al.36 were similar to our phylogenetic analysis using a BI approach and using amino acid and nucleotides datasets, while the phylogenetic approach used by Humphreys-Pereira and Elling11 was similar in the amino acid dataset and different in the nucleotide dataset excluding the third codon position. In order to assess whether the phylogenetic relationships recovered by the mt genome sequences were influenced by the use of an entirely maternal marker, we assessed the phylogeny of the Enoplea using an available nuclear marker, partial 18S rRNA. Enoplea partial 18S (Fig. S9) showed two main well-supported clades (100% BPP) for Enoplia and Dorylaimia. Longidoridae species were closely related phylogenetically and forming a well-supported clade (100% BPP) with Nordiidae, Qudsianematidae, Dorylaimidae and Aporcelaimidae. However, no member of these families has a complete mt genome currently available. The Longidoridae clade was formed by two subclades, the superior subclade is well-supported (BPP = 0.99) comprising four different genera, Longidorus, Paralongidorus, Xiphinema americanum group and Xiphidorus. Longidorus species were phylogenetically related with Paralongidorus species forming a well-supported clade (BPP = 1.00) and the Xiphinema americanum group which formed a sister-clade with Xiphidorus, however the BPP values for this sister-clade is moderately supported (BPP = 0.95). The second subclade is well-supported (BI = 1.00) and was formed by Xiphinema non-americanum group species. The relationships between these three groups were not well-defined in these analyses or in other phylogenetic analysis using other phylogenetic markers with more Longidoridae species37. Unfortunately, we could not obtain a complete mt genome sequence for a Xiphinema non-americanum group species, so this point could not be resolved. Taking into account the sequence evolution in both clades (Longidorus-Paralongidorus) and (Xiphinema-Xiphidorus) neither of these clades seems more ancient than the others. But, clearly there are parallels in gene arrangements, phylogenetic relationships and non-coding regions between Longidorus and Paralongidorus. Additionally, L. vineacola has the longest non-coding regions followed by P. litoralis. For this reason, we hypothesized a less evolved mt genome or different selection pressure in the evolution of these species. Another observation found in recent phylogenetic analysis, is the extreme diversity of some regions (cox1 gene) with Longidorus orientalis showing incongruence of phylogenies inferred from ITS1 rRNA and cox1 genes38 while other species show low differences within the populations sampled (i.e. X. pachtaicum and X. index)39. The high variation observed in the cox1 priming sites in L. orientalis can adversely affect the certainty of the nematode identification by barcoding38 and thus integrative taxonomical approaches are needed for an accurate identification of these and other plant-parasitic nematodes40,41,42. Our complete mt sequences for Xiphinema, Longidorus and Paralongidorus genera could help in resolving these problems by comparing sequences for primer design.

Bayesian probability values (BPP) shown above the node (≥70).

Analysis of amino acid sequences for 13 protein-coding genes inferred using RAxML (see methods for analysis details). Bootstrap values shown above the node (≥70).
While mapping the gene arrangement with the species in this study, we found an important variability in Enoplea (Figs 3 and 4). Some genera such as Trichinella and Trichuris were homogenous with their PCG arrangements, however, the genus Romanomermis showed an important variability in its arrangement in a similar way to Longidoridae species. We could not find similarities in the PCG arrangement between the species studied, with the exception of very closely related species (X. americanum and X. rivesi).
Material and Methods
Samples and nematode extraction
Soil samples from which nematodes were extracted were collected in 2015 in Spain from several crops and wild habitats (Table 6). Soil samples were collected with a shovel discarding the upper 5-cm topsoil profile, from a depth of 5- to 40-cm, in the close vicinity of active roots. Nematodes from the soil were extracted from a 500-cm3 sub-sample using the magnesium sulphate centrifugal-flotation method43. Xiphinema pachtaicum, X. rivesi, L. vineacola, and P. litoralis were identified using integrative taxonomy as described in previous studies37,42,44,45. Only live and individual nematodes were used for DNA extraction. No pure populations were multiplied in pots in greenhouse and nematodes were extracted from original sampling points.
Mitochondrial DNA extraction and amplification
For the molecular analyses, in order to avoid complications from mixed populations in the same sample, at least two live nematodes from each sample were temporarily mounted in a drop of 1 M NaCl containing glass beads (to avoid crushing the nematode) and diagnostic morphological characters were observed and measurements were taken to confirm the species identity. The slides were then dismantled and DNA extracted. Nematode DNA was extracted from single individuals and PCR assays were conducted as described by Subbotin et al.46. A portion of the cox1 gene was amplified as described by Lazarova et al. using primers COIF (5′-GATTTTTTGGKCATCCWGARG-3′) and COIR (5′-CWACATAATAAGTATCATG-3′)47 and PCR cycling conditions as described by He et al.32. PCR products were purified using ExoSAP-IT (Affmetrix, USB products) and used for direct sequencing in both directions. The resulting products were run on a DNA multicapillary sequencer (Model 3130XL genetic analyzer; Applied Biosystems, Foster City, CA, USA), using the BigDye Terminator Sequencing Kit v.3.1 (Applied Biosystems, Foster City, CA, USA), at the Stab Vida sequencing facilities (Caparica, Portugal).
For all mt DNA amplification, the DNA extraction protocol was similar to that described in Subbotin et al.38 with the exception that several live nematodes previously identified under microscope were used for each extraction and the proteinase K digestion was performed at 50 °C. Primers were designed using the cox1 sequences for each species. The primer design was performed using Primer348 (Table 6) with the correct sense for long-range PCR which was carried out using Advantage® 2 PCR Kit (Clontech, Takara Biotechnology, Japan). Each reaction contained 0.3 μM primer, 1X BD Advantage 2 PCR buffer and 2 μl of DNA nematode extraction in a final PCR volume of 25 μl. Long-range PCR conditions were as follows: initial denaturation at 94 °C for 3 min followed by 40 cycles of 94 °C for 15 s, annealing between 55 and 57 °C depending on the primer (Table 6) for 30 s and extension at 68 °C for 15 min during the first 10 cycles and after 15 min + 15 s/cycle. The product was visualized using an agarose gel; gel purified using Cut & Spin (Grisp, Portugal) and quantified using a Nanodrop spectrophotometer.
Ion-torrent Sequencing and read processing
Ion-torrent sequencing platform was performed at Stab Vida sequencing facilities (Caparica, Portugal). The 200–300 bp insert size library was constructed using enzymatic fragmentation of amplified DNA, Ion-torrent specific adapter ligation, size selection and amplification. The concentration and size distribution of library DNA fragments was determined using Qubit® fluorometer 2.0 (Invitrogen) and Bioanalyzer 2100 (Agilent Technologies). The numbers of reads obtained were 351,698; 665,110; 339,777; and 596,021 for X. rivesi, X. pachtaicum, L. vineacola and P. litoralis, respectively. Raw data obtained were analyzed and trimmed using CLC Genomics Workbench 7.5.1 (Qiagen) following standard procedures described by the manufacturer in Stab Vida facilities.
Mitochondrial genome assembly and annotation
Filtered data was de novo assembled using CLC Genomics Workbench 7.5.1 (Qiagen). Prediction of protein-coding genes and rRNA genes was done by using a combination of BLAST searches in Artemis v. 16.0.049, and MITOS online software50. Putative tRNA (transfer RNA) genes were identified in the MITOS online software50, which uses the strategy presented in Jühling et al.27. Additionally, these tRNA predictions were checked using the program Arwen v1.251. The assembled genomes were annotated using Artemis v. 16.0.049. Annotated sequences were submitted to GenBank with the accession numbers KU746820, KU746821, KU746818 and KU746819 for X. rivesi, X. pachtaicum, L. vineacola and P. litoralis, respectively. Codon usage was studied on-line using the server http://www.bioinformatics.org/sms2/codon_usage.html.
Phylogenetic analyses
Phylogenetic analyses were performed on amino acid (AA) and nucleotide data sets and the nuclear partial 18S rRNA. The newly obtained and published sequences for mt coding genes in complete Enoplea mt genomes were used for phylogenetic reconstruction. Lithobius forficatus (NC002629) and Limulus polyphemus (NC003057) were used as outgroups, according to previous studies11,36. For multiple alignments of AA sequences, the nucleotide sequences of each of the protein coding genes (PCG) were initially translated into AA with MEGA652 using the invertebrate mt genetic code setting. The amino acid sequences for each PCG were aligned individually using ClustalW53, implemented in MEGA6 under default settings. Conserved regions in the alignments of the 13 PCGs were selected using the Gblocks v 0.91b server set at the “less stringent” option (allow smaller final blocks, allow gap positions within the final blocks and allow less strict flanking positions)54. Each individual gene alignment was tested for the best-fit substitution model using ProtTest 2.455 based on the Akaike information criterion (AIC). All AA alignments of the 13 PCGs were concatenated into a single alignment using Mesquite v3.0456. The final alignment included 2468 out of 3926 AA, representing 63% of the original sequence alignment. Similarly, aligned nucleotide sequences from the PCG using the aligned amino acid sequences were trimmed, concatenated in a similar procedure as for AA dataset. The best fitted model of DNA evolution for each individual gene alignment was obtained using jModelTest v. 2.1.757 with AIC. Model used in both datasets are shown in Table S1. Selected models from the available programs in MrBayes 3.1.2 and RAxML 8.2.2 which best fit our dataset from the ranked models in Protest 3.2 and jModelTest 2.1.7. were used in the phylogenetic analysis
The partial 18S rRNA data set consisted of 88 Enoplea sequences from GenBank and was 1666 bp in length, and comprised representatives of the main families. Lithobius forficatus (EU024571) and Limulus polyphemus (HQ588741). Sequences for partial 18S were aligned using MAFFT v. 7.20558. Conserved regions in the alignments were trimmed using the Gblocks v 0.91b server set at the “less stringent” option54. The best fitted model of DNA evolution was obtained using jModelTest v. 2.1.757 with AIC.
Phylogenetic analyses of the AA data sets were performed based on maximum likelihood using the rapid bootstrap algorithm in RAxML v. 8.2.259 with 200 bootstrap replicates. BI was performed using MrBayes 3.1.260. In both cases, the models were included in the analysis. BI was performed including the model in a partition setting with the model for each PCG and for nucleotides and was run under a general time reversible of invariable sites and a gamma-shaped distribution (GTR + I + G) model with four chains for 1 × 106 generations. After discarding burn-in samples and evaluating convergence, the remaining samples were retained for further analyses. The topologies were used to generate a 50% majority rule consensus tree. Trees were visualized using TreeView61 and FigTree v1.4.2 (http://tree.bio.ed.ac.uk/software/figtree/).
Additional Information
How to cite this article: Palomares-Rius, J. E. et al. Mitochondrial genome diversity in dagger and needle nematodes (Nematoda: Longidoridae). Sci. Rep. 7, 41813; doi: 10.1038/srep41813 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Baldwin, J. G., Nadler, S. A. & Adams, B. J. Evolution of plant parasitism among nematodes. Annu Rev Phytopathol 42, 83–105 (2004).
Manzanilla-López, R. H. & Hunt, D. In Practical Plant Nematology (ed Manzanilla-López, R. H. & Marbán-Mendoza, N. ) 65–87 (Mundi-Prensa-España, 2012).
Decraemer, W. & Chaves, E. In Practical Plant Nematology (ed Manzanilla-López, R. H., Marbán-Mendoza, N. ) 25–62 (Mundi-Prensa-España, 2012).
Rota-Stabelli, O. et al. Ecdysozoan Mitogenomics: Evidence for a Common Origin of the Legged Invertebrates, the Panarthropoda. Genome Biology and Evolution 2, 425–440, doi: 10.1093/gbe/evq030 (2010).
Bernt, M., Braband, A., Schierwater, B. & Stadler, P. F. Genetic aspects of mitochondrial genome evolution. Molecular Phylogenetics and Evolution 69, 328–338 (2013).
Bernt, M. et al. CREx: Inferring genomic rearrangements based on common intervals. Bioinformatics 23, 2957–2958 (2007).
Cameron, S. L. Insect Mitochondrial Genomics: Implications for Evolution and Phylogeny. Annual Review of Entomology 59, 95–117, doi: 10.1146/annurev-ento-011613-162007 (2014).
Boore, J. L. Animal mitochondrial genomes. Nucleic Acids Res 27, 1767–1780 (1999).
Evangelina García, L. & Sánchez-Puerta, M. V. Comparative and evolutionary analyses of Meloidogyne spp. based on mitochondrial genome sequences. PLoS ONE 10, e0121142, doi: 10.1371/journal.pone.0121142 (2015).
Besnard, G. et al. Fast assembly of the mitochondrial genome of a plant parasitic nematode (Meloidogyne graminicola) using next generation sequencing. Comptes Rendus Biologies 337, 295–301 (2014).
Humphreys-Pereira, D. A. & Elling, A. A. Mitochondrial genomes of Meloidogyne chitwoodi and M. incognita (Nematoda: Tylenchina): Comparative analysis, gene order and phylogenetic relationships with other nematodes. Molecular & Biochemical Parasitology 194, 20–32 (2014).
Sun, L., Zhuo, K., Lin, B., Wang, H. & Liao, J. The complete mitochondrial genome of Meloidogyne graminicola (Tylenchina): A unique gene arrangement and its phylogenetic implications. PLoS ONE 9, e98558 (2014).
Sultana, T., Han, H. & Park, J. K. Comparison of complete mitochondrial genomes of pine wilt nematode Bursaphelenchus xylophilus and Bursaphelenchus mucronatus (Nematoda: Aphelenchoidea) and development of a molecular tool for species identification. Gene 520, 39–46 (2013b).
Sultana, T. et al. Comparative analysis of complete mitochondrial genome sequences confirms independent origins of plant-parasitic nematodes. Evolutionary Biology 13, 12 (2013a).
Gibson, T. et al. The mitochondrial genome of the soybean cyst nematode, Heterodera glycines . Genome 54, 565–574 (2011).
Jacob, J. E. M., Vanholme, B., Van Leeuwen, T. & Gheysen, G. A unique genetic code change in the mitochondrial genome of the parasitic nematode Radopholus similis . BMC Research Notes 2, 192 (2009).
Gibson, T., Blok, V. C. & Dowton, M. Sequence and characterization of six mito-chondrial subgenomes from Globodera rostochiensis: multipartite structure is conserved among close nematode relatives. Journal of Molecular Evolution 65, 308–315 (2007).
He, Y., Jones, J., Armstrong, M., Lamberti, F. & Moens, M. The mitochondrial genome of Xiphinema americanum sensu stricto (Nematoda: Enoplea): considerable economization in the length and structural features of encoded genes. Journal of Molecular Evolution 61, 819–833 (2005b).
Armstrong, M. R., Blok, V. C. & Phillips, M. S. A multipartite mitochondrial genomein the potato cyst nematode Globodera pallida . Genetics 154, 181–192 (2000).
Phillips, W. S., Brown, A. M. V., Howe, D. K., Peetz, A. B., Blok, V. C., Denver, D. R. & Zasada, I. A. The mitochondrial genome of Globodera ellingtonae is composed of two circles with segregated gene content and differential copy numbers. BMC Genomics 17, 706, doi: 10.1186/s12864-016-3047-x (2016).
Okimoto, R., Chamberlin, H. M., Macfarlane, J. L. & Wolstenholme, D. R. Repeated sequence sets in mitochondrial DNA molecules of root knot nematodes (Meloidogyne): nucleotide sequences, genome location and potential for host-race identification. Nucleic Acids Research 19, 1619–1626 (1991).
Gissi, C., Iannelli, F. & Pesole, G. Evolution of the mitochondrial genome of Metazoa as exemplified by comparison of congeneric species. Heredity 101, 301–320 (2008).
Amalric, F., Merkel, C., Gelfand, R. & Attardi, G. Fractionation of mitochondrial RNA from HeLa cells by high-resolution electrophoresis under strongly denaturing conditions. Journal of Molecular Biology 118, 1–25 (1978).
Gissi, C. & Pesole, G. Transcript mapping and genome annotation of ascidian mtDNA using EST data. Genome Research 13, 2203–2212 (2003).
Mohandas, N. et al. Mitochondrial genomes of Trichinella species and genotypes - a basis for diagnosis, and systematic and epidemiological explorations. International Journal for Parasitology 44, 1073–1080 (2014).
Liu, G. H. et al. Characterization of the complete mitochondrial genomes of two whipworms Trichuris ovis and Trichuris discolor (Nematoda: Trichuridae). Infection, Genetics and Evolution 12, 1635–1641 (2012).
Jühling, F., Pütz, J., Bernt, M., Florentz, C. & Stadler, P. F. Armless mitochondrial tRNAs in Enoplea (Nematoda). RNA Biology 9, 1161–1166 (2012).
Dörner, M., Altmann, M., Pääbo, S. & Mörl, M. Evidence for import of a lysyl-tRNA into marsupial mitochondria. Molecular Biology of the Cell 12, 2688–2698 (2001).
Tan, T. H., Pach, R., Crausaz, A., Ivens, A. & Schneider, A. tRNAs in Trypanosoma brucei: genomic organization, expression, and mitochondrial import. Molecular Biology of the Cell 22, 3707–3717 (2002).
Wolstenholme, D. R., Macfarlane, J. L., Okimoto, R., Clary, D. O. & Whaleithner, J. A. Bizarre tRNAs inferred from DNA sequences of mitochondrial genomes of nematode worms. Proceedings of the National Academy of Sciences USA 84, 1324–1328 (1987).
Lavrov, D. V. & Brown, W. M. Trichinella spiralis mtDNA: A nematode mitochondrial genome that encodes a putative ATP8 and normally structured tRNAs and has a gene arrangement relatable to those of coelomate metazoans. Genetics 157, 621–637 (2001).
He, Y. et al. A molecular phylogenetic approach to Longidoridae (Nematoda: Dorylaimida). Nematology 7, 111–124 (2005).
Taanman, J. W. Review: The mitochondrial genome: structure, transcription, translation and replication. Biochimica et Biophysica Acta 1410, 103–123 (1999).
Lewis, S. C. et al. A rolling circle replication mechanism produces multimeric lariats of mitochondrial DNA in Caenorhabditis elegans . PLoS Genetics 11, e1004985 (2015).
Arunkumar, K. P. & Nagaraju, J. Unusually long palindromes are abundant in mitochondrial control regions of insects and nematodes. PLoS One 1, e110, doi: 10.1371/journal.pone.0000110 (2006).
Kim, J. et al. Mitochondrial genomes advance phylogenetic hypotheses for Tylenchina (Nematoda: Chromadorea). Zoologica Scripta 44, 446–462 (2015).
Gutiérrez-Gutiérrez, C., Palomares Rius, J. E., Cantalapiedra-Navarrete, C., Landa, B. B. & Castillo, P. Prevalence, polyphasic identification, and molecular phylogeny of dagger and needle nematodes infesting vineyards in southern Spain. European Journal of Plant Pathology 129, 427–453 (2011b).
Subbotin, S. A. et al. Characterisation of populations of Longidorus orientalis Loof, 1982 (Nematoda: Dorylaimida) from date palm (Phoenix dactylifera L.) in the USA and other countries and incongruence of phylogenies inferred from ITS1 rRNA and coxI genes. Nematology 17, 459–477 (2015).
Gutiérrez-Gutiérrez, C. et al. Genetic structure of Xiphinema pachtaicum and X. index populations based on mitochondrial DNA variation. Phytopathology 101, 1168–1175 (2011a).
Gutiérrez-Gutiérrez, C., Cantalapiedra-Navarrete, C., Montes-Borrego, M., Palomares-Rius, J. E. & Castillo, P. Molecular phylogeny of the nematode genus Longidorus (Nematoda: Longidoridae) with description of three new species. Zoological Journal of the Linnean Society 167, 473–500 (2013).
Archidona-Yuste, A., Navas-Cortés, J. A., Cantalapiedra-Navarrete, C., Palomares-Rius, J. E. & Castillo, P. Cryptic diversity and species delimitation in the Xiphinema americanum-group complex (Nematoda: Longidoridae) as inferred from morphometrics and molecular markers. Zoological Journal of the Linnean Society 176, 231–265 (2016a).
Archidona-Yuste, A., Navas-Cortés, J. A., Cantalapiedra-Navarrete, C., Palomares-Rius, J. E. & Castillo, P. Unravelling the biodiversity and molecular phylogeny of needle nematodes of the genus Longidorus (Nematoda: Longidoridae) in olive and a description of six new species. PLoS ONE 11, e0147689 (2016b).
Coolen, W. A. In Root-knot nematodes (Meloidogyne species). Systematics, biology and control (eds Lamberti, F. & Taylor, C. E. ) 317–329 (Academic Press, 1979).
Gutiérrez-Gutiérrez, C. et al. Phylogeny, diversity, and species delimitation in some species of the Xiphinema americanum-group complex (Nematoda: Longidoridae), as inferred from nuclear and mitochondrial DNA sequences and morphology. European Journal of Plant Pathology 134, 561–597 (2012).
Palomares-Rius, J. E., Subbotin, S. A., Landa, B. B., Vovlas, N. & Castillo, P. Description and molecular characterisation of Paralongidorus litoralis sp. n. and P. paramaximus Heyns, 1965 (Nematoda: Longidoridae) from Spain. Nematology 10, 87–101 (2008).
Subbotin, S., Halford, P. D., Warry, A. & Perry, R. N. Variations in ribosomal DNA sequences and phylogeny of Globodera parasitising solanaceous plants. Nematology 2, 591–604 (2000).
Lazarova, S. S., Malloch, G., Oliveira, C. M. G., Hübschen, J. & Neilson, R. Ribosomal and mitochondrial DNA analyses of Xiphinema americanum-group populations. Journal of Nematology 38, 404–410 (2006).
Untergrasser, A. et al. Primer3 - new capabilities and interfaces. Nucleic Acids Research 40, e115 (2012).
Rutherford, K. et al. Artemis: sequence visualization and annotation. Bioinformatics 16, 944–945 (2000).
Bernt, M. et al. MITOS: Improved de novo Metazoan Mitochondrial Genome Annotation. Molecular Phylogenetics and Evolution 69, 313–319 (2013).
Laslett, D. & Canback, B. ARWEN: a program to detect tRNA genes in metazoan mitochondrial nucleotide sequences. Bioinformatics 24, 172–175 (2008).
Tamura, K., Stecher, G., Peterson, D., Filipski, A. & Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Molecular Biology and Evolution 30, 2725–2729 (2013).
Thompson, J. D., Higgins, D. G. & Gibson, T. J. CLUSTAL W: improving the sensitiv-ity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Research 22, 4673–4680 (1994).
Talavera, G. & Castresana, J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Systematic Biology 56, 564–577 (2007).
Abascal, F., Zardoya, R. & Posada, D. ProtTest: selection of best-fit models of protein evolution. Bioinformatics 21, 2104–2105 (2005).
Maddison, W. P. & Maddison, D. R. Mesquite: a modular system for evolutionary analysis. Version 3.04 http://mesquiteproject.org (2015).
Darriba, D., Taboada, G. L., Doallo, R. & Posada, D. jModelTest 2: more models, new heuristics and parallel computing. Nature Methods 9, 772 (2012).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment 542 software version 7: improvements in performance and usability. Molecular Biology and Evolution 30, 772–780 (2013).
Stamatakis, A. RAxML Version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313 (2014).
Ronquist, F. & Huelsenbeck, J. P. MRBAYES 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19, 1572–1574 (2003).
Page, R. D. TreeView: an application to display phylogenetic trees on personal computers. Computer Applications in the Biosciences 12, 357–358 (1996).
Acknowledgements
The authors thank B. Landa (IAS-CSIC) and M. Metsis (Tallin University) for valuable suggestions and proofreading the manuscript, and J. Martín-Barbarroja (IAS-CSIC) and G. León Ropero (IAS-CSIC) for the excellent technical assistance. This research was supported by grants P12-AGR 1486 and AGR-136 from ‘Consejeria de Economia, Innvovacion y Ciencia’ from Junta de Andalucia, and Union Europea, Fondo Europeo de Desarrollo regional, ‘Una manera de hacer Europa’, and grant 219262 ArimNET_ERANET FP7 2012–2015 Project PESTOLIVE ‘Contribution of olive history for the management of soilborne parasites in the Mediterranean basin’ from Instituto Nacional de Investigacion y Tecnologia Agraria y Alimentaria (INIA). The James Hutton Institute receives financial support from the Scottish Government, Rural and Environment Science and Analyical Services Division.
Author information
Authors and Affiliations
Contributions
Conceived and designed the experiments: J.E.P.-R., P.C., C.C.N., A.A.-Y. and V.B. Performed the experiments: J.E.P.-R. and C.C.N. Analyzed the data: J.E.P.-R., C.C.N. and P.C. Contributed reagents/materials/analysis tools: J.E.P.-R., C.C.N. and A.A.-Y. Wrote the paper: J.E.P.-R., P.C., A.A.-Y., C.C.N. and V.B.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Palomares-Rius, J., Cantalapiedra-Navarrete, C., Archidona-Yuste, A. et al. Mitochondrial genome diversity in dagger and needle nematodes (Nematoda: Longidoridae). Sci Rep 7, 41813 (2017). https://doi.org/10.1038/srep41813
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep41813
- Springer Nature Limited
This article is cited by
-
Complete mitochondrial genome of Metathelazia capsulata (Pneumospiruridae) and comparison with other Spiruromorpha species
Parasitology Research (2024)
-
Aonchotheca (Nematoda: Capillariidae) is validated as a separated genus from Capillaria by both mitochondrial and nuclear ribosomal DNA
Parasites & Vectors (2022)
-
Inverted base composition skews and discontinuous mitochondrial genome architecture evolution in the Enoplea (Nematoda)
BMC Genomics (2022)
-
The complete mitochondrial genome of the Columbia lance nematode, Hoplolaimus columbus, a major agricultural pathogen in North America
Parasites & Vectors (2020)
-
An integrative taxonomic study of the needle nematode complex Longidorus goodeyi Hooper, 1961 (Nematoda: Longidoridae) with description of a new species.
European Journal of Plant Pathology (2020)