
Abstract
The PTA-NiMo/ZSM-5 catalyst impregnated with phosphotungstic acid (PTA) was designed for the transformation of Jatropha oil into benzene, toluene, and xylenes (BTX) aromatics. The produced catalyst was characterized by N2 adsorption-desorption, powder X-ray diffraction (XRD), Fourier transform infrared (FT-IR), X-ray photoelectron spectroscopy (XPS), and the temperature-programmed desorption of ammonia (NH3-TPD). The catalytic performance was evaluated by gas chromatography (GC). The liquid products were 70 wt% of the feed oil, and the majority of the liquid products were BTX. The aromatization activity of the catalyst was improved by the addition of PTA and the hierarchical process. The favorable PTA amount was 20 wt% and the yield of BTX was 59 wt% at 380 °C, 3 MPa, H2/oil (v/v) = 1000 and LHSV = 1 h−1 over the PTA20-NiMo/HZ0.5 catalyst (PTA 20 wt%) without sulfurization.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Introduction
The hydrocracking of vegetable oils (Jatropha oil, palm oil, algae oil, etc.) for use as bio-fuels or bio-chemicals is a promising solution due to the problem of depleting petroleum reserves as well as environmental concerns1,2,3. The Hydrocracking is commonly applied to convert vegetable oils into biodiesel consisted of normal alkanes and isoalkanes. In our previous work, the biodiesel was produced successfully from hydrocracking of Jatropha oil using a non-sulfide catalyst4,5,6. Another study reported that vegetable oils could be utilized in the production of bio-chemicals, especially aromatic compounds7,8,9. Aromatics, especially monocyclic aromatics such as benzene, toluene, and xylene (BTX) have industrially important applications. These compounds can be used as a lead substitute for the octane number boosting of transportation fuel, and the seal of the engine can be made to swell by aromatics to prevent gas or oil leakage7,10. For these reasons, aromatics are often added to jet fuels to guarantee its quality11. Therefore, it is important to find a new method to efficiently prepare bio-aromatics from vegetable oil.
Currently, conventional fuels are derived from petroleum through distillation processes, and aromatics are derived from catalytic reforming technology11. It was reported that ZSM-5 zeolite was suitable as a catalyst for the production of aromatics from vegetable oils7. Use of this catalyst for the processing of Jatropha oil resulted in an aromatic yield of 28 wt%8. However, this yield was still too low to effectively use.
Hierarchical zeolite preserves the zeolitic properties of its bulk analogue but exhibits greatly enhanced active-site accessibility due to a reduction in the size of the microporous domains in which the diffusion of hydrocarbons is often strongly constrained. The pore structure may prolong catalyst lifetime by enhancing the tolerance to coking during the aromatization of branched alkenes12,13,14,15,16,17. The introduction of additives such as metal or sulfur active phase is expected to promote the transfer of hydrogen atoms or help to maintain the catalysts in this form18,19,20. The yield of aromatics can be improved by additives21. Metals (Ni, Mo, Co, Fe, and Zn) can be supported on ZSM-5, and metal sulfide catalysts have been widely used for the cracking process1,20,22,23,24,25. The hydrocracking of Jatropha oil was performed by NiMo-modified hierarchical ZSM-5 to obtain the highest total aromatic yield, approximately 50 wt%, but the catalyst was presulfided9. Although the yield of the product can be increased by use of a presulfide process, it results in the environmental harmful consequences of sulfur dioxide emissions, corrosion, and sulfur residues.
In this work, a modified ZSM-5 catalyst was employed with the goal of increased BTX aromatic production. Heteropoly acids (HPAs), especially those with the Keggin type, such as phosphotungstic acid (PTA), possess unique properties, and the immobilization of Keggin-type HPAs on zeolite can improve its regenerability and activity to suppress the generation of outgrowths26,27. In our previous work, environmentally friendly PTA catalysts (Ni-PTA/Al2O3 and Ni-PTA/nano-hydroxyapatite) were prepared without sulfurization for use in a clean process to generate high yields of green diesel from Jatropha oil5,6. In this work, the hierarchical ZSM-5 impregnated with PTA or/and NiMo were developed for the production of aromatics and light oil from the hydrocracking of Jatropha oil without sulfidation. The performance and the possible reaction pathway of the mechanism with the prepared catalysts were also investigated.
Methods
Catalyst preparation and characterization
The catalysts were developed by the incipient wetness impregnation method. All used reagents were of analytical grade and were used without further purification. The MFI-type ammonium ZSM-5 zeolite (NH4-ZSM-5) with a SiO2/Al2O3 mole ratio (SAR) of 25 was supplied by the Nankai University Catalyst Co., Ltd. Ni(NO3)2·6H2O, (NH4)6Mo7O24·4H2O, H3PW12O40 (PTA), NH4NO3, NaOH, and HCl were purchased from the Sinopharm Chemical Reagent Co., Ltd.
A typical treatment procedure for the support was as described previously with some modifications28. Briefly, the NH4-ZSM-5 powder was calcined at 550 °C in air for 5 h to convert the powder from the ammonium form to its protonated form (HZSM-5), denoted as HZ. The HZ zeolite supports (10 g) were soaked in 100 mL of NaOH solution at different concentrations (0.2, 0.5, and 1.0 M) for 30 min at 65 °C and 600 rpm. The solution was then filtered off and thoroughly washed with deionized water. The desilicated zeolites were subsequently treated with 100 mL of 0.1 M aqueous HCl under identical conditions for 6 h to remove the alumina debris. The sample was washed with deionized water until reaching neutral pH, and then filtered, dried at 120 °C for 12 h and calcined at 550 °C for 5 h to obtain the hierarchical ZSM-5 zeolites. The obtained samples were transformed into a protonated form by ion-exchanging three times in 0.5 M NH4NO3 solution at 80 °C for 4 h, followed by the separation−calcination procedure as described above. The hierarchical samples were denoted as HZ0.2, HZ0.5, HZ1.0, respectively, corresponding to the NaOH concentration.
Ni (4.0 wt%) and Mo (12 wt%) were loaded onto the hierarchical ZSM-5 zeolites by incipient wetness impregnation with an aqueous solution containing Ni(NO3)2 and (NH4)6Mo7O249. Impregnated samples were dried at 120 °C for 12 h and calcined at 500 °C for 5 h, denoted as NiMo/HZ.H3PW12O40 (PTA) was also loaded on the NiMo/HZ zeolites by impregnation with an aqueous solution containing 5 to 45 wt% PTA. Impregnated samples were dried at 120 °C for 3 h and calcined at 500 °C for 5 h, denoted as PTA(x)-NiMo/HZ (where × indicates the PTA content, from 5 to 45 wt%)6.
X-ray diffraction (XRD) was performed to determine the structural properties of the samples using a Rigaku D/MAX-2500 X-ray diffractometer with Cu Kα (λ = 0.1541 nm) radiation at 40 kV and 40 mA. Each sample was scanned at a speed of 4° min−1 over a range from 5° to 80°. Analysis was carried out with Highscore plus analysis software equipped with a standard ICDD X-ray diffraction database supplied by Panalytical.
Chemical analysis of the composite was carried out using a Fourier transform infrared (FT-IR) spectrophotometer (Lambda FTIR-7600) in the range from 4000 cm−1 to 400 cm−1 at 1.5 cm−1 resolution, averaging 32 scans.
The Si/Al mole ratio (SAR) of the samples was determined by inductively coupled plasma optical emission spectrometry (ICP-OES) using an Optima-7000DV instrument equipped with photomultiplier tube detection.
The specific surface area, pore volume, and average pore diameter of catalysts were determined by N2 adsorption and desorption isotherms using the Quantachrome NOVA 2200e Surface Area and Pore Distribution Analyzer instrument at −196 °C. The samples were degassed at 300 °C for more than 6 h under high vacuum conditions. The specific surface area was calculated using the Brunauer Emmett Teller (BET) method based on the adsorption data (P/P0 range = 0.05–0.30). The t-plot method was used to discriminate microporosity and mesoporosity. The mesopore surface area and micropore volume were determined from the isotherms using the t-plot method. The mesopore volume and micropore diameter were determined from the isotherms using the density functional theory (DFT) method. In order to avoid the Tensile Strengthening Effect, the average mesopore diameter was determined from the isotherms using the Barrett Joyner Halenda (BJH) method.
X-ray photoelectron spectroscopy (XPS) data were obtained using an ESCALab 250 electron spectrometer from Thermo Scientific Corporation at 280 eV pass energy. The base pressure was about 6.5 × 10−10 mbar. The binding energies were corrected for sample charging using the C 1 s peak at 284.6 eV for adventitious carbon as a reference. The peak areas of the samples were determined by measuring the Ni 2p and Mo 3d peak areas (after linear subtraction of the background).
The acidity of catalysts was measured with a temperature programmed desorption of ammonia (NH3-TPD) using a chemisorption analyzer (TP-5080 Multi-functional automatic Adsorption Instrument, Tianjin Industrial Technology CO., Ltd) with a thermal conductivity detector. Prior to each TPD run, the catalyst (0.2 g) was heated to 500 °C for 30 min under N2 to remove all moisture. The catalyst was then cooled to 100 °C and saturated with NH3, and then flushed with N2 to remove the physically adsorbed NH3. Finally, desorption of NH3 was performed from 100 °C to 600 °C at a heating rate of 10 °C min−1.
Catalytic activity measurements
The Jatropha oil was purchased from Jiangsu Donghu Bioenergy Co., Ltd, including myristic acid (0.8%), arachic acid (0.5%), linolenic acid (0.9%), palmitoleic acid (1.2%), stearic acid (7.3%), palmitic acid (14.8%), linoleic acid (36.2%) and oleic acid (38.3%).
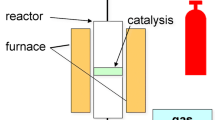
All reactions were carried out in a continuous fixed-bed flow reactor (1.2 cm I.D. and 56 cm in length) equipped with an electrically heating system from Tianjin Golden Eagle Technology Co., Ltd, (JF-2). The equipment for continuous hydrocracking consisted of a feed system, heating section, tubular reactor, condensation section, storage section, and an instrumentation and control section, as schematically depicted in Fig. 1. The reaction temperature was controlled by microcomputer, and the system pressure was maintained by a backpressure regulator. Jatropha oil was delivered from a feed pot into the reactor using a high pressure pump. All gases used were UHP grade and were controlled by a separate mass flow meter. Typically, 10 g non-sulfided catalyst was loaded into the reactor and heated under H2 to 400 °C at a ramp rate of 5 °C min-1, and then incubated at 3 MPa for 3 h to convert the metal into its non-oxide form. The main feedstock was mixed with high pressure hydrogen and entered the reactor where the feed molecules undergo the hydrocracking reactions. The reaction conditions for the experiment were as follows: temperature 380 °C, pressure 3 MPa, LHSV 1.0 h−1, and H2 to a feed ratio of 1000 (v/v). The product exited the reactor as a mixed gas liquid phase and was cooled before it entered a high pressure low temperature separator, where the gas and liquid phases separated for analysis.

Simplified schematic diagram of the reaction apparatus (diagram was drawn by Xiaosong Yang).
The reaction system achieved stability after 8 h. The gas products were analyzed online by a gas chromatograph (GC-900C) equipped with a thermal conductivity detector (TCD) and two columns, a TDX-01 packed column (3 m in length and 3 mm I.D.) for the determination of CO and CO2, and a GDX-104 packed column (3 m in length and 2 mm I.D.) for the determination of light hydrocarbons (C1–C4). The liquid products were analyzed by using a GC-900C equipped with a flameionization detector (FID) using a AT.SE-30 capillary column (30 m in length and 0.32 mm I.D.) with a film thickness of 0.5μm. The following temperature program was used: the injection port and detector temperature were set to 300 °C, the initial temperature was 50 °C for 5 min, followed by heating at 8 °C min−1 to 180 °C, then heating at 15 °C min−1 to 210 °C, and heating at 8 °C min−1 to 280 °C, with a 10 min hold at 280 °C. The individual products were identified using GC standards (purchased from Sigma-Aldrich, LLC). The triglycerides in the liquid product were determined by methyl esterification with GC analysis.
The conversion (C) of Jatropha oil was calculated as:

where C(TG) was the concentrations of triglycerides (TG) in the liquid product.
The yield (Y) of miscellaneous hydrocarbons was calculated as:

where M(HC) was the mass of the corresponding hydrocarbons (HC) in the liquid product.
Results and Discussion
Catalyst characterization
The X-ray diffraction patterns of all freshly prepared ZSM-5 catalysts are shown in Fig. 2. The patterns exhibited the characteristic diffraction peaks at 2θ = 7.89°, 8.73°, 14.82°, 23.04°, 23.86°, and 24.26°, which were indexed to the MFI zeolitic structure (JCPDS No. 42-0024). The XRD patterns of the original ZSM-5 zeolites showed that no new phases were generated and the intrinsic lattice structure of the ZSM-5 was unchanged. Thus the fine structure of zeolites was maintained in the catalyst after the desilication and impregnation process, but the relative crystallinity decreased. This result was similar to that reported previously9,29,30. After being impregnated with NiMo species, no peak was observed for the Ni or Mo compounds. The absence of any metal oxide peaks in the XRD patterns indicated good metal dispersion with small particle size on the zeolite surfaces31,32. After being impregnated with PTA, the peaks observed at 2θ = 10.52°, 25.54°, and 34.02° were attributed to PTA (JCPDS No. 50-0657), and were only in the PTA45-NiMo/HZ0.5 catalyst sample. This indicated that the PTA was highly dispersed on the PTA5-NiMo/HZ0.5 and PTA20-NiMo/HZ0.5 catalysts, but the PTA was aggregated due to excessive impregnation on the PTA45-NiMo/HZ0.5 sample. In addition, there were no other diffraction peaks for the impregnated PTA catalyst samples, indicating that the Keggin structures remained intact33.

XRD patterns of the ZSM-5 catalysts.
The FTIR spectra of the HZ and PTA20-NiMo/HZ0.5 samples collected at room temperature are presented in Fig. 3. The characteristic bands at ν = 455, 545, and 795 cm−1 were associated with the stretching vibration of [AlO4] or [SiO4] groups in the HZ framework. One band at ν = 1080 cm−1 with a shoulder at ν = 1240 cm−1, were attributed to the characteristic asymmetric stretching band of a Si–O–Si bridge34. Overall, no obvious framework change was detected for the PTA20-NiMo/HZ0.5 catalyst, consistent with the XRD result. The bands at ν = 1637 and 3440 cm−1 were assigned to the bending vibration of the adsorbed water molecules, and the bands at ν = 3440 cm−1 were attributed to hydrogen-bonded silanol groups and water molecules adsorbed in the zeolite34. After PTA impregnation, the PTA20-NiMo/HZ0.5 catalyst showed new peaks at ν = 984, 892, and 811 cm−1 that corresponded to ν(W = O), ν(W–O–W) edge, and ν(W–O–W) corner, respectively35. The distance between the bands at ν = 811 and 795 cm−1 was too close, so they were combined smoothly into a bigger peak. Therefore, the Keggin structure of PTA was preserved in the catalyst without damage36.

FTIR spectra of (a) the HZ catalyst and (b) the PTA20-NiMo/HZ0.5 catalyst. (▲W = O; ●W–O–W edge; ◆W–O–W corner).
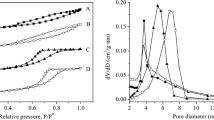
The nitrogen adsorption-desorption isotherms of the hierarchical ZSM-5 samples are shown in Fig. 4. The parent HZ zeolite presented a type I isotherm with the plateau starting at a very low relative pressure, which implied that the parent zeolite was dominated by microporous structure. After impregnation with NiMo species, the NiMo/HZ sample also presented a type I isotherm, which implied that the NiMo/HZ remained the same microporous structure. These results were consistent with the calculated micropore diameters shown in Table 1. As compared to the parent zeolite, the alkali-treated and acid-washed samples exhibited type IV isotherms typical of micro- and mesoporous materials. The isotherm with a H4-type hysteresis loop is a known fingerprint of a hierarchical porous system with size-homogeneous 1D slit channels37,38,39. The major differences in the isotherms of hierarchical samples compared to the parent zeolite were an increase in the N2 adsorption for the region 0.4 < P/P0 < 0.9, which was interpreted as the presence of capillary condensation in the intra-crystalline mesoporous spaces and external surface40. These changes should have a positive effect on the catalytic activity for the zeolite. The isotherm of the NiMo/HZ1.0 sample presented a rapid increase in absorption with P/P0 > 0.8, which was indicative of the substantial damage to the internal pores.

The nitrogen adsorption-desorption isotherms of the hierarchical ZSM-5 samples.
The textural properties of the various ZSM-5 samples are listed in Table 1. The Barrett Joyner Halenda (BJH) analysis of the pore size revealed mesopores in a size range of 2–20 nm. The results showed enhanced BET and mesopore surface areas for the hierarchical zeolites except the NiMo/HZ1.0 zeolite. This enhancement exhibited by the hierarchical zeolites was expected to have a positive effect on catalytic activity. The enhanced mesopore surface areas and the decreased micropore surface areas indicated that the loss in microporosity and the development of mesoporosity occurred as a result of the alkaline treatment and acid wash process. However, the development of mesoporosity occurred with micropore damage when the zeolites were treated at the higher NaOH concentration of 1.0 M, and the surface area and pore volume decreased drastically. This reduction of surface area and pore volume was expected to have a negative effect on catalytic activity. The surface area and pore volume of the NiMo/HZ zeolite catalysts decreased relative to the parent HZ zeolite, which was attributed to the action of the NiO and MO3 particles on the external surface to impede the access of N2 molecules and block the zeolite micropores. After PTA impregnation, it was clear that the surface area and pore volume decreased. The loss of specific surface area could be the result of the strong adsorption of PTA with a low surface area. Nevertheless, the decrease in surface area for low loading of PTA (5–20 wt%) was small, indicating that the majority of PTA was located outside of mesopores. However, when the PTA content was further increased to 45 wt%, the surface area and total volume were significantly decreased, which suggested that at this higher level, many micropores or mesopores of the zeolite were blocked. These results were in accordance with the XRD results.
The XPS spectra for the NiMo/HZ0.5 and PTA20-NiMo/HZ0.5 catalysts are shown in Fig. 5. The Ni 2p binding energy is shown in Fig. 5a. The metallic Ni in the two catalysts was 852.09 eV and 852.25 eV, respectively, and the other two peaks of each sample were assigned to Ni2+41. The Mo 3d binding energy is shown in Fig. 5b. The low-binding energy (about 233 eV) was attributed to Mo4+ in MoO2, and the high-binding energy (about 236 eV) was attributed to Mo6+ in MoO342. The shift of the peaks towards higher binding energy values in the Ni 2p but towards lower values in Mo 3d for the PTA20-NiMo/HZ0.5 catalyst might be ascribed to the strong interaction among diverse elemental species of the sample.

XPS spectra of (a) Ni 2p and (b) Mo 3 d levels for NiMo/HZ0.5 and PTA20-NiMo/HZ0.5 catalysts.
Table 2 shows that after adding PTA, the peak area ratio of Ni0/Ni2+ in the NiMo/HZ0.5 and PTA20-NiMo/HZ0.5 catalysts increased from 30.5% to 43.3%, and the Mo6+/Mo4+ ratio increased from 60.0% to 71.5%. These changes suggested that the PTA-loaded catalyst was able to promote the reduction of Ni2+ and the oxidation of Mo4+. The Mo species may donate partial electrons to the Ni oxide species and keep metal oxide species in the reduction state. The electron transfer among metal species was due to the Keggin structure of PTA, which allowing replacement of the presulfurization and maintenance of the catalysts in the active form during the hydrocracking reaction4,5. The similar phenomenon was also reported in our previous work5,6.
The acidity of the ZSM-5 samples was studied by NH3-TPD. The acid properties of the different samples are summarized in Table 3. Two desorption peaks were observed at 220 °C and 380 °C approximately corresponding to the desorption of NH3. The adsorbed NH3 molecules desorbed from the weak acid sites at low temperatures, and from the strong acid sites at high temperatures. The weak acid sites were ascribed to the acidic framework of the Si–OH–Si group, whereas the strong acid sites were ascribed to the acidic framework of the Si–OH–Al group34. The loss of the weak and strong acid sites in the HZ0.5 and NiMo/HZ0.5 samples could be because desilication and dealumination reduced the amount of framework acid sites after alkali treatment and subsequent acid washing15. The other potential explanation for the reduction in acidity is that the acid sites located on the external/mesopore surface had lower acid strength than those inside the micropore system43. Overall, compared to the parent HZ catalyst, the hierarchical samples HZ0.5 and NiMo/HZ0.5 exhibited decreased total acidity, likely due to the partial loss of framework acid sites and the formation of mesopore structure. The incorporation of NiMo appeared to affect the number of weak acid sites. The increase in weak acid sites might be related to the formation of new sites by NiMo species with weak acidic features. For the hierarchical NiMo/HZ0.5 sample, the high temperature peak become broader and was shifted toward higher temperature (370 °C to 383 °C) compared to the NiMo-free hierarchical HZ0.5. These results may indicate the better dispersion and contact of the NiMo particles with the zeolitic support for the hierarchical zeolite samples, which should have a positive effect on the catalytic activity. The ratio of weak acid sites and strong acid sites was gradually increased for the catalysts. With the addition of PTA, the number of strong acid sites of PTA20-NiMo/HZ0.5 sample increased. This result suggested that the PTA was loaded on the zeolite surface, and the strong acidic protons of PTA caused the shift. Compared with the desorption temperature peak of NiMo/HZ0.5, the peak assigned to the strong acid sites were shifted to lower temperature, but the weak acid sites were shifted to higher temperature, showing moderation of the strength of the acid sites, which weakened the coke deposition and restrained the deactivation of the catalyst. The nature of a solid acid catalyst is mainly determined by the strong acid sites on the surface44.
Analysis of the liquid products
The FTIR spectra of the standard toluene and liquid products from the reaction using HZ catalyst are shown in Fig. 6. The GC analysis charts of liquid products over the catalyst are shown in Fig. 7. The hydrocarbon distribution of the products is shown in Fig. 8.

IR of (a) standard toluene and (b) liquid products produced using the HZ catalyst (A:CHx stretching; BD: C=C stretching; C: C=C vibrations; E: aromatic rings vibrations).

The GC analysis charts of liquid products.

Hydrocarbon distribution of the liquid products analyzed by GC (wt%).
The structure was confirmed by FTIR spectra. The peaks at 2920 cm−1 and 2850 cm−1 were assigned to methylene groups45. These results were consistent with GC analysis of the liquid products produced using the HZ catalyst (Figs 7 and 8). The peak at 3050 cm−1 was due to aromatic CHx stretching, the peak at 1600 cm−1 and 1421 cm−1 were attributed to the symmetric stretching of the C=C aromatic ring45,46,47, and the peak at 1500 cm−1 was due to aromatic C=C vibrations48,49. The peak at 752 cm−1 was assigned to the vibrations of aromatic rings with a low degree of substitution45. The characteristic peaks of the liquid products were consistent with the toluene standard, which suggested that aromatics were generated via the catalytic conversion of Jatropha oil.
The hydrocracking of Jatropha oil to bio-aromatics using the ZSM-5 catalysts was investigated. GC analysis charts of liquid products over the catalyst are shown in Fig. 7. The hydrocarbon distribution of the products is shown in Fig. 8. The amount of each product group was calculated based on the product of the relative area (%) of each component as determined by the GC analysis. The products consisted of three fractions, gaseous products, liquid products and coke. The liquid products accounted for about 70 wt% of the Jatropha oil, and the majority of the liquid products were BTX and light oil. The remaining 30 wt% was a gaseous product that was mainly composed of CO, CO2, CH4, C2H4, and C3H8. The C15–C18 fraction from the deoxygenation reactions of triglyceride was barely detected in the samples except for that prepared using the HZ0.5 catalyst sample that showed low amounts. The acidity of the catalysts may have been sufficient for the deoxygenation and cracking reactions, but the acidity of the HZ0.5 catalyst was slightly lower, according to the values presented in Table 3 24. In the cracking reaction, the C15–C18 fraction was cut off and broken into (C2–C10) fractions, which provided abundant materials for the aromatization50. The hydrocarbons in the liquid products were primarily hydrocarbon fractions of C6–C9 (Fig. 8). Analysis of the aromatic fraction indicated that it primarily consisted of the most valuable C6–C8 BTX aromatics (Fig. 9).

The yields of BTX aromatics in the liquid products.
The <C6 light hydrocarbon fractions, including C2–C5 alkenes/alkanes, CO and CO2, might be direct materials for the aromatization, based on the correlation between the ratio of weak acid sites to strong acid sites and the yields of BTX or <C6 fractions (Fig. 10). The yield of BTX increased but the yield of <C6 fractions decreased, suggesting an inverse relationship. The yield of BTX aromatics was practically proportional to the ratio of weak acid sites and strong acid sites except for reactions performed using the PTA20-NiMo/HZ0.5 sample, which revealed that the weak acid sites of the catalyst might participate in the rate-limiting reaction for aromatization. For the PTA20-NiMo/HZ0.5 sample, although the weak and strong acid sites also increased, but the ratio of weak acid sites and strong acid sites decreased due to the more increment in strong acid sites.

Correlation between the ratio of weak acid sites to strong acid sites and the yields of BTX or <C6 fractions for the different catalysts.
The conversion of Jatropha oil and the yield of BTX using the catalysts (HZ, PTA20-NiMo/HZ0.5) are shown in Fig. 11. The conversion of Jatropha oil was more than 96.5% by 15 h for the two catalysts. The conversion gradually decreased from 98.3% to 84.7% between 15 h and 40 h for the PTA20-NiMo/HZ0.5 catalyst. For the HZ catalyst, the conversion quickly decreased from 96.5% to 68.1% between 15 h and 55 h. The conversions of the two catalysts exhibited little change after 55 h. The BTX yield was gradually decreased from 64.7% to 47.2% by 35 h and remained steady subsequently for the PTA20-NiMo/HZ0.5 catalyst. The BTX yields decreased from 41.6% to 26.5% by 30 h for the HZ catalyst. The results showed that the stability or catalytic activity of the prepared PTA20-NiMo/HZ0.5 catalyst were better than that of the HZ catalyst.

Conversion of Jatropha oil and BTX yields in product oil as a function of reaction time using the HZ and PTA20-NiMo/HZ0.5 catalysts.
The stability and recyclability of the best-performing catalyst (PTA20-NiMo/HZ0.5) were assessed by performing four consecutive cycles. The conversion and yields of BTX over the catalyst (8 h) upon consecutive use are shown in Fig. 12. The catalyst was thermally treated in air to simulate a typical regeneration procedure between each cycle. The conversion gradually decreased from 99.5% to 93.2% and the BTX yields from 59.5% to 50.1%. Such a minor change just shown the stability of the catalyst activity.

The conversion and yields of BTX for consecutive use of the PTA20-NiMo/HZ0.5 catalyst.
A comparison of the BTX yields performance of the ZSM-5 revealed distinct aromatization activity for the different catalytic zeolites. For the HZ and NiMo/HZ catalysts, the latter sample exhibited higher formation of BTX aromatic products. The incorporation of NiMo metal promoted the formation of gaseous hydrocarbons likely associated with the occurrence of hydrogen transfer reactions, which increased the formation of light fractions and provided more materials for aromatization (Fig. 10). Despite the decreased acidic sites on the NiMo/HZ0.5 catalyst, the ratio of weak acid sites and strong acid sites was increased and the BTX aromatic products also increased compared to the NiMo/HZ sample. The difference in performance was only attributed to the hierarchical treatment. This may be because the hierarchical NiMo/HZ0.5 catalyst exhibited higher external and mesopore surface areas, bigger pore volume, and larger pore diameter (Table 1), which broadened the reaction area and improved the diffusion limitation. In addition, the active center located at the mesopore walls of the hierarchical zeolites could catalyze reactions involving bulky molecules that were unable to enter the zeolite micropores and enhanced the reaction accessibilities51. The NH3-TPD analysis indicated better dispersion and contact with NiMo in the NiMo/HZ0.5 catalyst which may act synergistically to increase activity. As listed in Fig. 13, all samples that contained PTA exhibited very high catalytic properties for the aromatization reaction. Moreover, the PTA20-NiMo/HZ0.5 catalyst exhibited the highest BTX yield (59 wt%) compared with the NiMo/HZ0.5 catalyst. This could be because there were more acid sites on the PTA20-NiMo/HZ0.5 catalyst than the NiMo/HZ0.5 catalyst. In addition, according to the XPS analysis, the Mo species might donate partial electrons to the Ni oxide species, resulting in the transfer of electrons between Mo and Ni, which could increase the production of carbenium ions and enhance catalytic activity.

The yields of BTX aromatics for the NiMo/HZ0.5 catalysts (wt %) with various amounts of PTA.
The possible reaction pathway
The majority of products converted from Jatropha oil were CO, CO2, CH4, C2H4, C3H8 and hydrocarbons ranging from C4 to C18. In the initial stage of the hydrocracking reaction, each triglyceride molecule was cracked into three hydrocarbon molecules via three types of deoxygenation reactions (equ. 3), which were hydrodecarboxylation (HDCO), hydrodecarbonylation (HDC) and hydrodeoxygenation (HDO)52. C15 and C17 hydrocarbons were the products of lesser hydrogen consumption, direct decarboxylation and decarbonylation (carbon removal pathway), and the C16 and C18 hydrocarbons were the products of the higher hydrogen consuming process of hydrodeoxygenation (hydrogen addition pathway).
Hydrocracking reaction:

Meanwhile, the oxygen content of triglycerides was deleted as CO2, CO, and H2O. CO, CO2, CH4, C2H4, and C3H8 were the major gaseous products found in the effluent gas mixture. Methanation reactions (eqs 4 and 5) and water gas shift (equ. 7) were probable side-reactions, and these reactions can also increase hydrogen consumption or methane composition. The methane could be partly converted to ethylene (equ. 6), and might promote the methane dehydro-aromatization (MDA) reaction, which contributed to the BTX yields53,54. Two types of reactions that mainly utilized CO also occurred in the gaseous products. One was via the Fischer–Tropsch (equ. 8) reaction to generate alkylcarbenium ion. The other was via the Boudouard reaction (equ. 9) to result in the deposition of carbon on the surface of the catalyst54.
Methanation reaction:



Water gas shift:

Fischer–Tropsch reaction (FT):

Boudouard reaction:

The subsequent cracking reaction might occur as follows55. A tiny fraction of the C15-C18 alkanes could be directly cracked (equ. 10) to generate the alkylcarbenium ion. A majority of the C15-C18 alkanes were dehydrogenated on the metal sites to generate the corresponding alkenes (equ. 12), then the alkenes moved to the strong acid sites and generated alkylcarbenium ion (equ. 13), and the alkenes were then rearranged and cracked via β-scission (eqs 11 and 13). Next, the generated light alkylcarbenium ion was oligomerized, and C2-C10 alkenes were generated (equ. 13) as the principal intermediate products of the aromatization reaction50,56. Finally, the alkenes moved to the metal sites and were hydrogenated to generate C2-C10 alkanes (equ. 12), and then the alkanes joined in the cracking circulation to generate lighter alkenes.
Subsequent cracking reaction:


Dehydrogenation and rearranged reaction:


It was presumed that aromatics were formed by Diels–Alder reactions from light alkenes, which was the rate-limiting, key reaction for aromatization56. The formed alkenes came to the weak acid site to undergo the Diels–Alder cyclization reactions and produce the cycloalkanes, and then the cycloalkanes transformed into aromatics at strong acid sites by dehydrogenation-aromatization (eqs 14,15,16,17,18). Then, only a small portion of aromatics polymerized on the surface of the catalyst to form coke.
The Diels–Alder reactions and dehydrogenation-aromatization to form BTX:





Conclusions
This study presents the preparation of hierarchical PTA-NiMo/ZSM-5 catalysts with phosphotungstic acid (PTA) for the transformation of Jatropha oil into benzene, toluene, and xylenes (BTX) aromatics. The yield of BTX was 59 wt% at 380 °C, 3 MPa, H2/oil (v/v) = 1000, and LHSV = 1 h−1 for the reaction by the PTA20-NiMo/HZ0.5 catalyst (PTA 20 wt%). The optimal PTA amount was 20 wt%. The aromatization activity of the catalyst was enhanced by the moderate treatment of a hierarchical process, which was due to the increase in surface areas and improved diffusion/dispersion. The activity was further enhanced by the addition of PTA, because it increased the moderate acid sites or the electronic transformation to generate more carbenium ions to accelerate the reaction. Furthermore, the possible mechanism of BTX formation was presumed through Diels–Alder reactions and dehydrogenation-aromatization from the reaction pathway. This work presents a method to produce an efficient catalyst that does not require sulfurization, preventing environment and human health risks.
Additional Information
How to cite this article: Yang, X. et al. Hydrocracking of Jatropha Oil over non-sulfided PTA-NiMo/ZSM-5 Catalyst. Sci. Rep. 7, 41654; doi: 10.1038/srep41654 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Verma, D., Kumar, R., Rana, B. S. & Sinha, A. K. Aviation fuel production from lipids by a single-step route using hierarchical mesoporous zeolites. Energy & Environmental Science 4, 1667 (2011).
Kandaramath Hari, T., Yaakob, Z. & Binitha, N. N. Aviation biofuel from renewable resources: Routes, opportunities and challenges. Renewable and Sustainable Energy Reviews 42, 1234–1244 (2015).
Wang, W. C. & Tao, L. Bio-jet fuel conversion technologies. Renewable and Sustainable Energy Reviews 53, 801–822 (2016).
Liu, J., Liu, C., Zhou, G., Shen, S. & Rong, L. Hydrotreatment of Jatropha oil over NiMoLa/Al2O3 catalyst. Green Chemistry 14, 2499 (2012).
Fan, K., Liu, J., Yang, X. & Rong, L. Hydrocracking of Jatropha oil over Ni-H3PW12O40/nano-hydroxyapatite catalyst. International Journal of Hydrogen Energy 39, 3690–3697 (2014).
Liu, J. et al. A Non-sulfided flower-like Ni-PTA Catalyst that Enhances the Hydrotreatment Efficiency of Plant Oil to Produce Green Diesel. Scientific reports 5, 15576 (2015).
Sadrameli, S. M., Green, A. E. S. & Seames, W. Modeling representations of canola oil catalytic cracking for the production of renewable aromatic hydrocarbons. Journal of Analytical and Applied Pyrolysis 86, 1–7 (2009).
Saxena, S. K. & Viswanadham, N. Selective production of green gasoline by catalytic conversion of Jatropha oil. Fuel Processing Technology 119, 158–165 (2014).
Chen, H., Wang, Q., Zhang, X. & Wang, L. Hydroconversion of Jatropha Oil to Alternative Fuel over Hierarchical ZSM-5. Industrial & Engineering Chemistry Research 53, 19916–19924 (2014).
Corporan, E. et al. Chemical, Thermal Stability, Seal Swell, and Emissions Studies of Alternative Jet Fuels. Energy & Fuels 25, 955–966 (2011).
Chiaramonti, D., Prussi, M., Buffi, M. & Tacconi, D. Sustainable bio kerosene: Process routes and industrial demonstration activities in aviation biofuels. Applied Energy 136, 767–774 (2014).
Perez-Ramirez, J., Christensen, C. H., Egeblad, K., Christensen, C. H. & Groen, J. C. Hierarchical zeolites: enhanced utilisation of microporous crystals in catalysis by advances in materials design. Chemical Society reviews 37, 2530–2542 (2008).
Milina, M., Mitchell, S., Trinidad, Z. D., Verboekend, D. & Pérez-Ramírez, J. Decoupling porosity and compositional effects on desilicated ZSM-5 zeolites for optimal alkylation performance. Catalysis Science & Technology 2, 759 (2012).
Kim, Y. H., Lee, K. H., Nam, C.-M. & Lee, J. S. Formation of Hierarchical Pore Structures in Zn/ZSM-5 to Improve the Catalyst Stability in the Aromatization of Branched Olefins. ChemCatChem 4, 1143–1153 (2012).
Milina, M., Mitchell, S., Michels, N.-L., Kenvin, J. & Pérez-Ramírez, J. Interdependence between porosity, acidity, and catalytic performance in hierarchical ZSM-5 zeolites prepared by post-synthetic modification. Journal of Catalysis 308, 398–407 (2013).
Zhou, J. et al. Enhanced accessibility and utilization efficiency of acid sites in hierarchical MFI zeolite catalyst for effective diffusivity improvement. RSC Adv. 4, 43752–43755 (2014).
Gueudré, L., Milina, M., Mitchell, S. & Pérez-Ramírez, J. Superior Mass Transfer Properties of Technical Zeolite Bodies with Hierarchical Porosity. Advanced Functional Materials 24, 209–219 (2014).
Yakovlev, V. A. et al. Development of new catalytic systems for upgraded bio-fuels production from bio-crude-oil and biodiesel. Catalysis Today 144, 362–366 (2009).
Maia, A. J., Louis, B., Lam, Y. L. & Pereira, M. M. Ni-ZSM-5 catalysts: Detailed characterization of metal sites for proper catalyst design. Journal of Catalysis 269, 103–109 (2010).
Abdelsayed, V., Shekhawat, D. & Smith, M. W. Effect of Fe and Zn promoters on Mo/HZSM-5 catalyst for methane dehydroaromatization. Fuel 139, 401–410 (2015).
Vichaphund, S., Aht-ong, D., Sricharoenchaikul, V. & Atong, D. Production of aromatic compounds from catalytic fast pyrolysis of Jatropha residues using metal/HZSM-5 prepared by ion-exchange and impregnation methods. Renewable Energy 79, 28–37 (2015).
Liu, Y., Sotelo-Boyás, R., Murata, K., Minowa, T. & Sakanishi, K. Hydrotreatment of Vegetable Oils to Produce Bio-Hydrogenated Diesel and Liquefied Petroleum Gas Fuel over Catalysts Containing Sulfided Ni–Mo and Solid Acids. Energy & Fuels 25, 4675–4685 (2011).
Wang, H., Yan, S., Salley, S. O. & Ng, K. Y. S. Hydrocarbon Fuels Production from Hydrocracking of Soybean Oil Using Transition Metal Carbides and Nitrides Supported on ZSM-5. Industrial & Engineering Chemistry Research 51, 10066–10073 (2012).
Gong, S., Shinozaki, A. & Qian, E. W. Role of Support in Hydrotreatment of Jatropha Oil over Sulfided NiMo Catalysts. Industrial & Engineering Chemistry Research 51, 13953–13960 (2012).
Ishihara, A., Fukui, N., Nasu, H. & Hashimoto, T. Hydrocracking of soybean oil using zeolite–alumina composite supported NiMo catalysts. Fuel 134, 611–617 (2014).
Qiu, B., Yi, X., Lin, L., Fang, W. & Wan, H. The hydrocracking of n-decane over bifunctional Ni-H3PW12O40/SiO2 catalysts. Catalysis Today 131, 464–471 (2008).
Jin, H. et al. Influence of H4SiW12O40 loading on hydrocracking activity of non-sulfide Ni– H4SiW12O40/SiO2 catalysts. Fuel 89, 1953–1960 (2010).
Verboekend, D., Mitchell, S., Milina, M., Groen, J. C. & Pérez-Ramírez, J. Full Compositional Flexibility in the Preparation of Mesoporous MFI Zeolites by Desilication. The Journal of Physical Chemistry C 115, 14193–14203 (2011).
Zhang, C. et al. Aqueous-phase hydrodeoxygenation of lignin monomer eugenol: Influence of Si/Al ratio of HZSM-5 on catalytic performances. Catalysis Today 234, 145–152 (2014).
Rostamizadeh, M. & Taeb, A. Highly selective Me-ZSM-5 catalyst for methanol to propylene (MTP). Journal of Industrial and Engineering Chemistry 27, 297–306 (2015).
Laurent, M., Ricard, J.-L., Bahain, J.-J., Voinchet, P. & Rousseau, L. Datation du site Paléolithique moyen de la Butte d’Arvigny (Moissy-Cramayel, Seine-et-Marne). Comptes Rendus de l’Académie des Sciences - Series IIA - Earth and Planetary Science 330, 581–583 (2000).
Abdelsayed, V., Shekhawat, D., Poston, J. A. & Spivey, J. J. Synthesis, characterization, and catalytic activity of Rh-based lanthanum zirconate pyrochlores for higher alcohol synthesis. Catalysis Today 207, 65–73 (2013).
Kumar, A. S. H. et al. Nitration of phenol over silica supported H4PW11VO40 catalyst. Catalysis Communications 18, 37–40 (2012).
Zhang, Q. et al. Sorbitol transformation into aromatics: A comparative evaluation of Ni/HZSM-5 and Ni/Hβ. Fuel 165, 152–158 (2016).
Li, Z. et al. Synthesis and characterization of tungstophosphoric acid intercalated Ni/Al HTlc with magnetite. Journal of Materials Processing Technology 209, 2613–2619 (2009).
Kong, J. et al. Ultrasound-assisted synthesis of nanosized hierarchical ZSM-5 and its catalytic performance as the support for heteropolyacid. Journal of Porous Materials 21, 241–249 (2014).
Yin, A., Guo, X., Dai, W.-L. & Fan, K. Effect of initial precipitation temperature on the structural evolution and catalytic behavior of Cu/SiO2 catalyst in the hydrogenation of dimethyloxalate. Catalysis Communications 12, 412–416 (2011).
Wang, S., Li, X., Yin, Q., Zhu, L. & Luo, Z. Highly active and selective Cu/SiO2 catalysts prepared by the urea hydrolysis method in dimethyl oxalate hydrogenation. Catalysis Communications 12, 1246–1250 (2011).
Thommes, M. et al. Physisorption of gases, with special reference to the evaluation of surface area and pore size distribution (IUPAC Technical Report). Pure and Applied Chemistry 87, (2015).
Serrano, D. P., Aguado, J., Rodríguez, J. M. & Peral, A. Catalytic cracking of polyethylene over nanocrystalline HZSM-5: Catalyst deactivation and regeneration study. Journal of Analytical and Applied Pyrolysis 79, 456–464 (2007).
Bykova, M. V. et al. Ni-based sol–gel catalysts as promising systems for crude bio-oil upgrading: Guaiacol hydrodeoxygenation study. Applied Catalysis B: Environmental 113–114, 296–307 (2012).
Belatel, H., Al-Kandari, H., Al-Khorafi, F., Katrib, A. & Garin, F. Catalytic reactions of methylcyclohexane (MCH) on partially reduced MoO3 . Applied Catalysis A: General 275, 141–147 (2004).
Serrano, D. P. et al. Acidic and catalytic properties of hierarchical zeolites and hybrid ordered mesoporous materials assembled from MFI protozeolitic units. Journal of Catalysis 279, 366–380 (2011).
Liu, Y., Sotelo-Boyás, R., Murata, K., Minowa, T. & Sakanishi, K. Production of Bio-Hydrogenated Diesel by Hydrotreatment of High-Acid-Value Waste Cooking Oil over Ruthenium Catalyst Supported on Al-Polyoxocation-Pillared Montmorillonite. Catalysts 2, 171–190 (2012).
Wang, S., Tang, Y., Schobert, H. H., Guo, Y. n. & Su, Y. FTIR and13C NMR Investigation of Coal Component of Late Permian Coals from Southern China. Energy & Fuels 25, 5672–5677 (2011).
Tahmasebi, A., Yu, J., Han, Y. & Li, X. A study of chemical structure changes of Chinese lignite during fluidized-bed drying in nitrogen and air. Fuel Processing Technology 101, 85–93 (2012).
Nasrollahzadeh, M., Mohammad Sajadi, S., Rostami-Vartooni, A., Alizadeh, M. & Bagherzadeh, M. Green synthesis of the Pd nanoparticles supported on reduced graphene oxide using barberry fruit extract and its application as a recyclable and heterogeneous catalyst for the reduction of nitroarenes. Journal of Colloid and Interface Science 466, 360–368 (2016).
Müller, U., Rätzsch, M., Schwanninger, M., Steiner, M. & Zöbl, H. Yellowing and IR-changes of spruce wood as result of UV-irradiation. Journal of Photochemistry and Photobiology B: Biology 69, 97–105 (2003).
Colom, X., Carrillo, F., Nogués, F. & Garriga, P. Structural analysis of photodegraded wood by means of FTIR spectroscopy. Polymer Degradation and Stability 80, 543–549 (2003).
Chen, H., Wang, Q., Zhang, X. & Wang, L. Quantitative conversion of triglycerides to hydrocarbons over hierarchical ZSM-5 catalyst. Applied Catalysis B: Environmental 166–167, 327–334 (2015).
Shetti, V., Kim, J., Srivastava, R., Choi, M. & Ryoo, R. Assessment of the mesopore wall catalytic activities of MFI zeolite with mesoporous/microporous hierarchical structures. Journal of Catalysis 254, 296–303 (2008).
Wang, C. et al. One-step hydrotreatment of vegetable oil to produce high quality diesel-range alkanes. ChemSusChem 5, 1974–1983 (2012).
Fila, V., Bernauer, M., Bernauer, B. & Sobalik, Z. Effect of addition of a second metal in Mo/ZSM-5 catalyst for methane aromatization reaction under elevated pressures. Catalysis Today 256, 269–275 (2015).
Wang, H. et al. Carbon dioxide hydrogenation to aromatic hydrocarbons by using an iron/iron oxide nanocatalyst. Beilstein journal of nanotechnology 5, 760–769 (2014).
Wang, C. et al. High quality diesel-range alkanes production via a single-step hydrotreatment of vegetable oil over Ni/zeolite catalyst. Catalysis Today 234, 153–160 (2014).
Corma, A., Huber, G., Sauvanaud, L. & Oconnor, P. Processing biomass-derived oxygenates in the oil refinery: Catalytic cracking (FCC) reaction pathways and role of catalyst. Journal of Catalysis 247, 307–327 (2007).
Author information
Authors and Affiliations
Contributions
L.R. initiated the project and conceived. X.S.Y. supervised the experiments and wrote the paper. J.L. provided assistance with the analysis of the data. K.F. prepared and characterized the samples. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Yang, X., Liu, J., Fan, K. et al. Hydrocracking of Jatropha Oil over non-sulfided PTA-NiMo/ZSM-5 Catalyst. Sci Rep 7, 41654 (2017). https://doi.org/10.1038/srep41654
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep41654
- Springer Nature Limited
This article is cited by
-
Self-regeneration of Monodisperse Hierarchical Porous NiMo/Silica Catalyst Induced by NaHCO3 for Biofuel Production
Waste and Biomass Valorization (2022)
-
WO3/Al-HZSM-5 catalysts for effective catalytic conversion of biomass-derived levulinic acid to angelica lactones
Emergent Materials (2021)
-
Mechanistic correlation between water infiltration and framework hydrophilicity in MFI zeolites
Scientific Reports (2019)