
Abstract
Adults of the lady beetle species Harmonia axyridis (Pallas) are bred artificially en masse for classic biological control, which requires egg-laying by the H. axyridis ovary. Development-related genes may impact the growth of the H. axyridis adult ovary but have not been reported. Here, we used integrative time-series RNA-seq analysis of the ovary in H. axyridis adults to detect development-related genes. A total of 28,558 unigenes were functionally annotated using seven types of databases to obtain an annotated unigene database for ovaries in H. axyridis adults. We also analysed differentially expressed genes (DEGs) between samples. Based on a combination of the results of this bioinformatics analysis with literature reports and gene expression level changes in four different stages, we focused on the development of oocyte reproductive stem cell and yolk formation process and identified 26 genes with high similarity to development-related genes. 20 DEGs were randomly chosen for quantitative real-time PCR (qRT-PCR) to validate the accuracy of the RNA-seq results. This study establishes a robust pipeline for the discovery of key genes using high-throughput sequencing and the identification of a class of development-related genes for characterization.
Similar content being viewed by others


Introduction
The multi-coloured Asian lady beetle Harmonia axyridis (Pallas) is a well-known and highly efficient predator of aphids, and other insect pests of fruit trees, flowers, and vegetables1,2,3. Extensive research has focused on aspects of H. axyridis, such as genetics or evolutionary responses to population dynamics4, biological control4, life history5, biology6, and potential non-target impacts4. As an important part of previous research, H. axyridis has been extensively released for classic biological control worldwide4 (e.g., eastern Asia, Europe, Africa, and South and North America) with many significant benefits4,7. The ovary is an import reproductive organ in H. axyridis and typically associated with the egg-laying ability of H. axyridis adults. Therefore, the ovary contributes to the prosperity of the population and influences mass artificial breeding for classic biological control8. Ovary development can be divided into the following four development stages according to the presence of egg rooms and egg yolk deposition: the undifferentiated stage (Stage 1), ovarian developmental growth (Stage 2), the yolk sedimentary period (Stage 3) and early maturity for maternity (Stage 4)8. Previous studies have been performed at the phenotypic level, and few studies focused on understanding the key biological processes based on the H. axyridis transcriptome, especially among the ovary developmental genes. To understand the molecular mechanisms underlying ovary development, improve the artificial reproductive capacity of H. axyridis from the perspective of genes for biological control, and lay a foundation for other basic studies, it is indispensable to detect development-related genes in the ovarie of H. axyridis adult.
RNA-seq is a convenient and accurate technology that has been adopted to study gene function in many eukaryotes9. Following its rapid development, RNA-seq has been used to detect the genome sequences of many species rapidly with relatively high accuracy, extension and cost performance, resulting in great breakthroughs in biological research10. The research applications include basic research, clinical diagnosis and drug development11. RNA-seq provides a far more precise measurement of transcript levels and their isoforms than other methods12. Vera (2008) first reported the Melitaea cinxia. L transcriptome sequence obtained with RNA-seq, which was considered as paradigm in insects based on the new generation of transcriptome sequencing technology13. Gibbons (2009) sequenced Anopheles gambiae. L and Aedes aegypti. L, which confirmed that the short-read sequencing platform was feasible in non-model insect transcriptome studies14. Malone and Oliver (2011) adopted Illumina to perform RNA-seq experiments with Drosophila pseudoobscura. L and confirmed that this sequencing technology was reliable for gene expression research15.
In our study, we applied RNA-seq to obtain ovary transcriptomes from H. axyridis adults in Stages (1–4) (S1–S4), and performed an in depth analysis of these data to detect development-related genes. Additionally, we performed quantitative reverse transcription (qRT-PCR) analysis to validate the RNA-seq results. This transcriptome-wide study of mRNAs in the H. axyridis ovary will facilitate the study of the functions of development-related genes and provide potential RNAi targets to accelerate the breeding of H. axyridis for use in biological control.
Results
Transcriptome sequencing and read assembly
After Illumina Solexa deep sequencing, 21.5 Gb of clean data were obtained from the transcriptome sequencing of the H. axyridis adult ovary. To obtain an overview of the ovary expression profile at the different developmental stages in H. axyridis adults, four expressed gene libraries (S1: 22,364, 281; S2: 18, 460,991; S 3: 20,636,344; and S4: 24,531,820) containing clean reads were obtained for analysis. All base calling rates were greater than 92% (Table 1). A large number of reads was distributed at approximately 500 bp, with lengths ranging from 300 to 2000 bp. After assembly, 64,231 transcripts and 47,550 unigenes were obtained; the N50s of the transcripts and unigenes were 1448 and 1282 bp, respectively (Table 2), indicating high integrity assembly.
Unigene annotation
For annotation of the unigenes, BLAST software (version 2.2.26)16 was used to compare the unigene sequences with the Clusters of Orthologous Groups (COG)17, Gene Ontology (GO)18, Kyoto Encyclopedia of Genes and Genomes (KEGG)19, euKaryotic Orthologous Groups (KOG)20, Protein family (Pfam)21, Swiss-Prot22, and non-redundant (nr) databases23 to annotate the unigenes. Functions were predicted and classified with the COG classification system, resulting in the annotation of 7296 unigenes (Table 3). Additionally, 12,985 unigenes were annotated by the GO classification system. The pathway involvement of 8144 unigenes identified in this study was predicted based on a comparison with the KEGG database. A total of 17,235, 16,822, 16,863, and 28,186 unigenes were analysed with the KOG, Pfam, Swiss-Prot, and nr databases, respectively. Overall, 28,558 unigenes were annotated by selecting parameters with an E value ≤1e-5 (Table 3), to obtain an unigene annotation database for ovaries in H. axyridis adults (Supplementary Dataset File 1).
Differentially expressed genes (DEGs) analysis
To detect specific target genes that affected the developmental process, we performed a series of genome-wide expression profiling comparisons to examine gene activity changes between S1 and the other three stages. An Fragments per kilobase of exon model per million mapped reads (FPKM) fold change >2 and a false discovery rate (FDR) <0.05 were used as the thresholds to identify significant differences in gene expression.
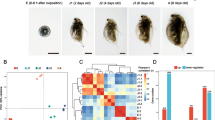
Comparing S2 with S1, 962 genes were differentially expressed, including 134 up-regulated genes and 828 down-regulated genes (Table 4). In S3 compared with S1, the expression of 524 genes was up-regulated and the expression of 810 genes was down-regulated (Table 4). When the gene expression levels were compared between S1 and S4, 4994 genes were up-regulated and 4646 genes were down-regulated, for a total number of 9640 genes (Table 4). These results revealed that the number of DEGs increased as development proceeded (Fig. 1). All of the obtained DEGs were combined into one DEG database (Supplementary Dataset File 2).

Scatter diagrams representing genome-wide expression profiling comparisons between S1 and the other three stage.
Note: Each black point represents a non-significant difference in gene expression, and each red point indicates a significant differentially expressed gene. Abscissa: log10 (Counts); Ordinate: log2 (FC) expression difference multiple values between two gene samples. a: S1vs. S2, b: S1vs. S3, c:S1vs. S4.
Identification and sequence analysis of development-related genes in a time series from the H. axyridis adult ovary
We combined the unigene annotations from the unigene annotation databases obtained above (Supplementary Dataset File 1), literature reports24,25,26,27,28,29,30,31, and transcription level changes in the four different stages (i.e., preference was given to genes that were up- or down- regulated continuously from S1 to S4) to identify genes associated with the formation of the polar plasm, the signal transduction mechanism in the ovary microenvironment and yolk formation, which define the development of the insect ovary31. In total, we identified 26 unigenes with high similarity to development-related genes (Table 5). All of them can be annotated in nr and divided into the following eight groups based on specific molecular functions: one gene related to polar formation, two genes related to bone morphogenetic protein function related, one gene related to DE-cadherin function related, two genes related to insulin related function, one gene related to ecdysone signalling pathways, 11 genes related to Notch signalling pathway, five genes related to Jak-STAT signalling pathway, and threegenes related to vitellogenin function. One gene related to polar formation was predicted to be full-length and to contribute to embryonic polarity protein dorsal-like isoform X1. Two putative sequences associated with bone morphogenetic protein functions were also predicted to be full-length. Their nucleotide lengths ranged from 3, 186 bp to 4, 758 bp, which corresponded to protein lengths of 517 aa to 821 aa, and the genes contained annotations for the bone morphogenetic protein receptor. TRINITY_DN17181_c3_g1 was the only gene related to DE-cadherin functions after screening and was annotated as a DE cadherin-like protein. Two genes related to insulin function were identified; neither gene was full length, and the genes contained annotations for the insulin receptor substrate. One gene was annotated as an ecdysone-induced protein included in the ecdysone signalling pathway. Of the 11genes predicted to be related to the Notch signalling pathway, six genes were full-length. All homologous functions from the nr database are shown in Table 5. In this study, we identified five genes related to the Jak-STAT signalling pathway; four of these genes were full-length, and all of the genes were predicted to possess different homologous functions. Three genes were related to vitellogenin functions, which plays an important role in yolk formation. TRINITY_DN20508_c0_g4 was annotated as the vitellogenin receptor. TRINITY_DN20502_c0_g1 and TRINITY_DN20736_c0_g1 were annotated as vitellogenin. In total, 18 unigenes in the H. axyridis adult ovary could be blasted against Tribolium castaneum Herbst, which accounted for 69.2% of all identified unigenes with high similarity to development-related genes. The others genes blasted to Anoplophora glabripennis Motschulsky (15.4%), Agrilus planipennisFairmaire (11.5%) and Polistes canadensisL (3.8%) (Table 5). Heat maps of the development-related genes identified in the ovaries of H. axyridis adults during development indicated that the same gene had different expression levels in different stages (Fig. 2).

Clustering of development-related genes.
The colour represents the gene expression levels in the samples in different columns. Different columns represent different samples, and different lines represent different genes.
Functional annotation clustering of development-related genes
Following GO annotation, the 26 development-related gene were classified into 35 different groups belonging to three main categories (“biological process”, “cellular component”, and “molecular function”) (Fig. 3). “Biological regulation” (14 genes), “single-organism process” (14 genes), and “cellular process” (12 genes) were the top three abundant categories in “biological process”, whereas “membrane” (13 genes),“membrane part” (9 genes), “cell” (8 genes), “cell part” (8 genes) and “organelle” (7 genes) were the top five categories in “cellular component”. Gene number involved in “binding” (11 genes) was the dominant in the “molecular function” category (Fig. 3) (Supplementary information Table 1). A total of 21 development-related genes were annotated by the KEGG classification system, including 13 genes related to signal transduction, four genes for ageing or endocrine and metabolic diseases, two genes for development, and one gene for carbohydrate metabolism or glycan biosynthesis and metabolism (Fig. 4) (Supplementary information Table 2).

GO annotation classification of development-related genes converted to unigenes annotation database of ovaries in H. axyridis adult.
The abscissa is the GO classification, the ordinate left is the gene number percentage, and the right is the number of genes.

KEGG pathway annotation classification of development-related genes converted to unigenes annotation database of ovaries in H. axyridis adult.
The abscissa is the gene number, and the ordinate left is the KEGG classification.
qRT-PCR validation of the DEGs results
To validate our DEG results, 20 randomly selected genes were analysed by qRT-PCR. Primer pairs for qRT-PCR were designed based on the nucleotide sequences of the transcriptome results. β-Actin was chosen as the internal control. The expression changes of these genes in S1, S2, S3 and S4 ovaries are shown in Fig. 5. Expression patterns similar to the DEG results were obtained. In combination with the previous study, the accuracy of the DEG results was verified8 (Fig. 5).

The correlation between mRNA expression levels using qPCR and mRNA sequencing.
The blue bar denotes expression values based on qPCR, and the red line denotes expression values based on mRNA sequencing.
Discussion
The ovary is a reproductive response organ that plays a crucial role in population breeding and undergoes dynamic and molecular changes. The development mechanism of the ovary has been studied in some insects. Liu (2015) studied the function of the vitellogenin gene in Chrysopa septempunctata. Wesmael32. Based on the sequence alignment with the results in this study, the TRINITY_DN20736_c0_g1 sequence shows 46.52% similarity with the vitellogenin gene reported in the study of Liu (2015)32. This finding suggests that these two genes have a certain similarity. Xie (2015) screened two genes related to polar cell formation; however, their sequence similarity was 6.96% and 14.82% with TRINITY_DN20651_c0_g14 identified in this study, indicating that these genes are not similar31. Based on the RNA-seq results, approximately 405 cellular-related genes were identified during ovary development in Bombyx mori. L in the fifth instar and early pupal stages, thus providing new insights into our understanding of the molecular mechanism of ovary development and oogenesis in the silkworm33. However, ovarian development related to natural enemy insects has mostly remained at the anatomic level11. H. axyridis is an important natural enemy. The ovary of H. axyridis has obvious spatial and temporal characteristics, and all four developmental stages are regulated by developmental gene expression4. Consequently, a comprehensive understanding of the molecular mechanisms underlying the regulation of ovary growth and development is very important for improving the reproductive capacity of H. axyridis.
Previous reports suggest that the formation and differentiation of germline stem cells (GSCs) directly determine the insect ovipositor34. GSCs develop from primordial germ cells (PGCs), which also maintain the balance of differentiation in the microenvironment35. In this series of physiological activities, the formation of polar plasm decides the development of the PGCs31. In this study, TRINITY_DN20651_c0_g14 was annotated as embryonic polarity protein dorsal-like isoform X1 and might have participated in the formation of polar cells in the ovaries, similar to XP_015172818.1 in Polistes canadensisL (Table 5). This process might represent the basic conditions of cellular differentiation36 and organismal zygote development37. The signal transduction mechanism maintains the stability of the microenvironment38. Six groups of genes related to signal transduction were identified. Among them, bone morphogenetic protein function-related genes are dominant in maintaining GSC quality31. Two of these genes were annotated for the bone morphogenetic protein receptor, similar to the report of Yamashita39 (Table 5). TRINITY_DN17181_c3_g1 was predicted to be a DE cadherin-like proteinrelated to GSC activity29. TRINITY_DN19612_c0_g1 and TRINITY_DN19612_c0_g9 are related to the insulin receptor, which affects insulin signalling pathways and controls the microenvironment via upstream regulation, thereby affecting GSC development31. TRINITY_DN20112_c0_g2 was the only gene related to the ecdysone signalling pathways and plays an important role in maintaining the stability of the microenvironment and GSC differentiation31. In the organization of many species, insulin signalling pathways and ecdysone signalling pathways correlate with one another40. The 11 genes related to the Notch signalling pathway may regulate the cell cap. Deficiency of the Notch signalling pathway might lead to a decrease in GSCs26. Five genes were included in the Jak-STAT signalling pathway, which can affect the bone morphogenetic protein25. Three genes were related to vitellogenin, which provides nutrition to the ovary31 and thus is indispensable for embryonic development41.
TRINITY_DN20502_c0_g1 and TRINITY_DN20736_c0_g1 were predicted to be vitellogenin, but the similarity between these genes was only 38.73%, indicating that they are different genes. The functional genes identified in this study are closely associated with ovarian development. The 18 (69.2%) development-related genes identified in this study could be blasted against Tribolium castaneum Herbst, indicating high homology between them, and four (15.4%), three (11.5%), and one (3.8%) development-related genes could be blasted against Anoplophora glabripennis. Motschulsky, Agrilus planipennis. Fairmaire and Polistes Canadensis. L, respectively, revealing evolutionary relationships among these species. Screening for functional genes could provide a foundation for research on developmental mechanisms in the H. axyridis adult ovary.
This study adopted functional transcriptome analysis with deep sequencing and the identification of genes after focusing on key ovary development processes, providing a new method to identify genes related to ovary development. Egg production might be stimulated in H. axyridis adults by interfering with development-related genes using the information obtained in this study. In a future study, a series of functional validation experiments will be performed to evaluate the development-related genes identified in this study.
Materials and Methods
Experimental insects
The H. axyridis used in this study were collected from the suburbs of Beijing and reared in the laboratory at 26±1 °C and 60–70% RH under a 16L:8D photoperiod for the experiments42. The insects were fed Acyrthosiphon pisum Harris reared on Pisum sativum. L. (Millborn Seeds, Zhewan 1, Beijing, China) in the laboratory. Ovary samples were collected from 100 H. axyridis adults on each of four typical dates, including the first day after emergence (Stage 1), the third day after emergence (Stage 2), the fourth day after emergence (Stage 3), and the seventh day after emergence (Stage 4)8.
Total RNA preparation
Total RNA was extracted from each stage on the first, third, fourth, and seventh days after insect emergence using TRIzol (Reagent, Thermo Fisher Scientific, Waltham, USA) according to the manufacturer’s instructions43. Each RNA sample was incubated with RNase-free DNase I (Reagent, Thermo Fisher Scientific, Waltham, USA) for 30 minutes at 37 °C43. The total amount of RNA in each sample was approximately 30 μg. The RNA samples were isolated and purified from the RNA pool with an Oligotex mRNA Midi kit (Reagent, Qiagen, Dusseldorf, Germany). A NanoDrop ND-1000 spectrophotometer (Specialized equipment, Thermo Fisher Scientific, Waltham, USA) was used to measure the absorbance at 260/280 nm (A260/A280) to determine the quality and quantity of the purified RNA. The integrity of the RNA was verified by electrophoresis on a 1.5% (w/v) agarose gel.
cDNA library construction and Illumina Solexa sequencing
A supported oligo ligation detection (SOLiD) Whole Transcriptome Analysis kit (Reagent, Thermo Fisher Scientific, Waltham, USA) was used to build a random fragment sequencing library following the manufacturer’s standard procedure44. The purified mRNA was fragmented with an interrupt reagent in a thermomixer and used as the template to synthesize first-strand cDNA. RNase H (Reagent, Thermo Fisher Scientific, Waltham, USA), DNA polymerase I (Reagent, Promega, Madison, USA), and dNTPs (Reagent, Thermo Fisher Scientific, Waltham, USA) were used to synthesize the second-strand cDNA. For ligation to sequence adapters, the cDNA was purified and repaired, and A bases were added to the 3′-end. Fragments of the appropriate size were selected and amplified by polymerase chain reaction (PCR) to construct the final sequencing library. After testing the quality control using an ABI Step One Plus Real-Time PCR System (Specialized equipment, Applied Biosystems, Foster, USA) and an Agilent 2100 Bioanalyzer (Specialized equipment, Agilent, Palo Alto, USA), the library was sequenced using the Illumina HiSeq 4000 platform (Specialized equipment, Illumina, SanDiego, USA)44.
Quality control of sequencing data
The raw Solexa sequencing data were processed using in-house Perl scripts to remove low-quality reads, reads containing poly-Ns and reads containing adapters to obtain clean data44. Concurrently, the Q20, Q30, and GC contents were calculated for the clean data. Clean data of high quality were used for the following analyses44.
De novo transcriptome assembly and quantification of gene expression levels
The trimmed reads were de novo assembled using trinityrnaseq_r20140717 with the default setting45. Bowtie v2.23 was used to map the clean reads to the Trinity assembly46. Then, RSEM47 was used to estimate the gene and isoform expression levels by calculating the FPKM of each gene and isoform (which considers the effect of the sequencing depth and gene length of the reads counted simultaneously)48.
Unigene annotation
The gene functions were annotated using the COG (http://www.ncbi.nlm.nih.gov/COG/)17, GO (http://www.geneontology.org/)18, KEGG (http://www.genome.jp/kegg/)19, KOG (http://www.ncbi.nlm.nih.gov/KOG/)20, Pfam (http://pfam.xfam.org/)21, Swiss-Prot (http://www.uniprot.org/)22, and nr databases (ftp://ftp.ncbi.nih.gov/blast/db/)23. The annotations of the unigenes were com-bined to create a database of annotated unigenes in the ovaries of adult H. axyridis.
DEGs analysis
Prior to differential gene expression analysis, for each sequenced library, the read counts were adjusted using the edgeR program package through one scaling normalized factor49. The DEGSeq R package (1.12.0) (https://bioconductor.org/packages/release/bioc/html/DESeq.html) was used to analyse differential expression under two conditions50. The Benjamini & Hochberg method was used to correct the P-values. Differential expression was considered significant at a P-value of 0.005 and log2 (fold change) of 151 A series of genome-wide expression profiling comparisons were performed to examine gene activity changes between S1 and the other three stages. All obtained DEGs were combined into one DEG database.
Identification and sequence analysis of development-related genes
To obtain a comprehensive perspective on the molecular basis of ovary development, we focused on the formation of the polar plasm, the signal transduction mechanism in the ovary microenvironment and yolk formation31. The analysis was based on the database of annotated unigenes in the ovaries of adult H. axyridis and the DEG database obtained in this study, literature reports24,25,26,27,28,29,30,31, and changes in gene expression levels in the four different stages to identify genes related to ovary development.
Development-related genes functional annotation clustering. Development-related genes were converted to unigenes annotation database of ovaries in H. axyridis adult and underwent functional enrichment analysis within GO and KEGG pathway terms with DAVID v6.7 (http://david.abcc.ncifcrf.gov/). The value was considered as significant when it was <0.05 for GO terms and KEGG pathways52.
qRT-PCR validation of the DEGs results
A total of 20 randomly selected mRNAs were tested by qRT-PCR (Table 6). Total RNA was extracted following the RNA-seq method. Using nuclease-free water, the concentration of each RNA sample was adjusted to 1 μg/μl, and 6 μl of total RNA from each sample was used as the template to synthesize first-strand cDNA in a reverse transcription system using a first-strand synthesis kit (Reagent, Thermo Fisher Scientific, Waltham, USA) following the manufacturer’s instructions. The β-actin gene was chosen as an internal control in all qRT-PCR experiments. Twenty pairs of primers were designed to validate the RNA-seq data (Table 6). Analysis of a particular gene was performed three times under identical conditions in a 25-μl volume in the Roche Light Cycler 480 system (Reagent, Roche, Basel, Switzerland)53. The E (Efficiency)-method from Roche Applied Science was used to analyse the relative quantification of all target genes53. The expression levels of the target genes were normalized by comparison with the β-actin reference gene53.
Additional Information
How to cite this article: Du, W. and Zeng, F. Identification of Development-Related Genes in the Ovaries of Adult Harmonia axyridis (Pallas) Lady Beetles Using a Time-Series Analysis by RNA-seq. Sci. Rep. 6, 39109; doi: 10.1038/srep39109 (2016).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Wheeler, A. G. & Hoebeke, E. R. Coccinella novemnotata in northeastern North America: historical occurrence and current status (Coleoptera: Coccinellidae). Proc Entomol, Soc Wash. 97, 701–716 (1995).
Gordon, R. D. The Coccinellidae (Coleoptera) of America north of Mexico. J N Y Entomol Soc. 93, 95 (1985).
Koch, R. L., Hutchison, W. D., Venette, R. C. & Heimpel, G. E. Susceptibility of immature monarch butterfly, danaus plexippus, (lepidoptera: nymphalidae: danainae), to predation by harmonia axyridis, (coleoptera: coccinellidae). Biolcontrol. 28, 265–270 (2003).
Koch, R. L. The multicolored asian lady beetle, harmonia axyridis: a review of its biology, uses in biological control, and non-target impacts. J Insect Sci. 3, 32 (2003).
Stathas, G. J., Eliopoulos, P. A., Kontodimas, D. C. & Giannopapas, J. Parameters of reproductive activity in females of Harmonia axyridis (Coleoptera: Coccinellidae). Eur J Entomol. 98, 547–549 (2001).
Fauske, G. M., Tinerella, P. P. & Rider, D. A. A list of the lady beetles(Coleoptera: Coccinellidae) of North Dakota with new records from North Dakota and Minnesota. J Kansas Entomol Soc. 76, 38–46 (2003).
Koch, R. L., Venette, R. C. & Hutchison, W. D. Invasions by Harmonia axyridis (Pallas) (Coleoptera: Cocci-nellidae) in the Western Hemisphere: implications for South America. Neotrop Entomol. 35, 421–434 (2006).
Chen, J. et al. Ovarian development and oogenesis of Harmonia axyridis Pallas. J Plant Pro. 42, 237–243 (2015).
Valenzuela, M. V., Chavez, M. J. & Gallardo, E. C. RNA-seq analysis evidences multiple gene responses in Caligus rogercresseyi exposed to the anti-salmon lice drug azamethiphos. Aquaculture. 446, 156–166 (2015).
Yue, G. D. et al. The application of high-throughput sequencing technology in plant and animal research. Scientia Sinica Vitae. 42, 107–124 (2012).
Liu, H. L., Zheng, L. M., Liu, Q. Q. & Quan, F. S. Studies on the transcriptomes of non-model organisms. hereditas. 35, 955–970 (2013).
Wang, Z. & Gerstein, M. M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 10, 57–63 (2009).
Cristobal, J. V. E. R. A. et al. Rapid transcriptome characterization for a nonmodel organism using 454 pyrosequencing. Mol Ecol. 17, 1636–1647 (2008).
Gibbons, J. G. et al. Benchmarking next-generation transcriptome sequencing for functional and evolutionary genomics. Mol Biol Evol. 26, 2731–2744 (2009).
Malone, J. H. & Oliver, B. Microarrays, deep sequencing and the true measure of the transcriptome. BMC Biology. 9, 34 (2011).
Altschul, S. F. et al. Gapped BLAST and PSI BLAST: A New Generation of Protein Database Search Programs. Nucleic Acids Res. 25, 3389–3402 (1997).
Tatusov, R. L., Galperin, M. Y. & Natale, D. A. The COG database: a tool for genome scale analysis of protein functions and evolution. Nucleic Acids Res. 28, 33–36 (2000).
Ashburner, M. et al. Gene ontology: tool for the unification of biology. Nat Genet. 25, 25–29 (2000).
Kanehisa, M., Goto, S., Kawashima, S., Okuno, Y. & Hattori, M. The kegg resource for deciphering the genome. Nucleic Acids Res. 32, 277–280 (2004).
Koonin, E. V. et al. A comprehensive evolutionary classification of proteins encoded in complete eukaryotic genomes. Genome Biology. 5, 60 (2004).
Finn, R. D. Pfam: the protein families database. Nucleic Acids Res. 42, D222–230 (2014).
Apweiler, R. et al. UniProt: the Universal Protein knowledge base. Nucleic Acids Res. 32, 115–119 (2004).
Deng, Y. Y. et al. Integrated nr Database in Protein Annotation System and Its Localization. Computer Engineering. 32, 71–74 (2006).
Harris, R. E. & Ashe, H. L. Cease and desist: modulating short-range dpp signalling in the stem-cell niche. Embo Reports. 12, 519–526 (2011).
López, O. L., Fernández, M. A. & González, R. A. Jak/stat signalling in niche support cells regulates dpp transcription to control germline stem cell maintenance in the drosophila ovary. Development. 135, 533–540 (2008).
Song, X., Call, G. B. & Kirilly D. Notch signaling controls germline stem cell niche formation in the Drosophila ovary. DEVELOPMENT. 134, 1071–1080 (2007).
Uhlenhaut, N. H. & Treier, M. Forkhead transcription factors in ovarian function. Reproduction. 142, 489–495 (2011).
Hsu, H. J. & Drummond-Barbosa, D. Insulin signals control the competence of the drosophila, female germline stem cell niche to respond to notch ligands. Developmental Biology. 350, 290–300 (2011).
Jin, Z. et al. Differentiation-defective stem cells outcompete normal stem cells for niche occupancy in the drosophila, ovary. Cell Stem Cell. 2, 39–49 (2008).
Ables, E. T. & Drummond B. D. The steroid hormone ecdysone functions with intrinsic chromatin remodeling factors to control female germline stem cells in drosophila. Cell Stem Cell. 7, 581–592 (2010).
Xie, Y. F. The influence of piRNA pathway related gene tudor and trehalase gene on ovary development in Bactrocera dorsalis, PhD thesis. Southwest university of china (June 2015).
Liu, C., Mao, J. & Zeng, F. Chrysopa septempunctata (Neuroptera: Chrysopidae) Vitellogenin Functions Through Effects on Egg Production and Hatching. J Econ Entomol. 108, 2780–2788 (2015).
Yin, Y. J., Cao, G. L., Huang, M. L., Xue, R. Y. & Gong, C. L. Characterization of cellular process related genes during ovary development of bombyx mori in the fifth instar and early pupal stage based on rna-seq. Science Of Sericulture. 39, 1078–1089 (2013).
Zhao, R., Yang, X., Li, X. & Xi, R. Age-related changes of germline stem cell activity, niche signaling activity and egg production in drosophila. Aging Cell. 7, 344–354 (2008).
Extavour, C. G. & Akam, M. Mechanisms of germ cell specification across the metazoans: epigenesis and preformation. Development. 130, 5869–5884 (2003).
Siegrist, S. E. & Doe, C. Q. Microtubule-induced cortical cell polarity. Gene Dev. 21, 483–96 (2007).
Pruyne, D., Legesse, M. A., Gao, L., Dong, Y. & Bretscher, A. Mechanisms of polarized growth and organelle segregation in yeast. Annu Rev Cell Dev Biol. 20, 559–91 (2004).
Kaibuchi, K. Neuronal polarity: from extracellular signals to intracellular mechanisms. Nat Rev Neurosci. 8, 194–205 (2007).
Yamashita, H., Dijke, P. T., Heldin, C. H. & Miyazono, K. Bone morphogenetic protein receptors. Bone. 19, 569–574 (1996).
Yan, L. et al. Transcriptional regulation of the insulin signaling pathway genes by starvation and 20-hydroxyecdysone in the bombyx, fat body. J Insect Physiol. 56, 1436–1444 (2010).
Tufail, M., Nagaba, Y., Elgendy, A. M. & Takeda, M. Regulation of vitellogenin genes in insects. Entomol sci. 17, 269–282 (2014).
Sighinolfi, L. et al. Biological and biochemical characteristics for quality control of harmonia axyridis (pallas) (coleoptera, coccinellidae) reared on a liver-based diet. Archinsect Biochem. 68, 26–39 (2008).
Firmino, A. A. et al. Transcriptome analysis in cotton boll weevil (Anthonomus grandis) and RNA interference in insect pests. Plos One. 8, e85079–e85079 (2013).
Lang, D. L. et al. Rna-seq analysis of yak ovary: improving yak gene structure information and mining reproduction-related genes. Sci China Life Sci. 57, 925–935 (2014).
Manfred, G. et al. Trinity: reconstructing a full-length transcriptome without a genome from RNA-Seq data. Nat Biotechnol. 29, 644–652 (2011).
Robledo, D. et al. Integrative transcriptome, genome and quantitative trait loci resources identify single nucleotide polymorphisms in candidate genes for growth traits in turbot. Int J Mol Sci. 17, 243 (2016).
Li, B. & Colin, N. D. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. Bmc Bioinformatics. 12, 323 (2011).
Trapnell, C. et al. Transcript assembly and quantification by rna-seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 28, 511–515 (2015).
Robinson, M. D., Mccarthy, D. J. & Smyth, G. K. Edger: a bioconductor package for differential expression analysis of digital gene expression datasmotn. Bioinformatics. 26, 139–140 (2010).
Wang, L., Feng, Z., Wang, X., Wang, X. & Zhang, X. Degseq: an r package for identifying differentially expressed genes from rna-seq data. Bioinformatics. 26, 136–138 (2010).
Zhang, D. et al. The cysteine protease cep1, a key executor involved in tapetal programmed cell death, regulates pollen development in arabidopsis. Plant Cell. 26, 2939–2961 (2014).
Huang, D. W., Sherman, B. T. & Lempicki, R. A. Systematic and integrative analysis of large gene lists using DAVID bioinformaticsresources. Nat Protoc. 4, 44–57 (2009).
Tellmann, G. & Geulen, O. Light Cycler 480 Real-Time PCR System: Innovative Solutions for Relative Quantification. Biochemica. 4, 16–18 (2006).
Acknowledgements
We thank Jinsong Zhu for comments and suggestions on an earlier version of the manuscript. This study was supported by a research fund (2013CB127600) from the Ministry of Science and Technology of China.
Author information
Authors and Affiliations
Contributions
Wenxiao Du wrote the main manuscript text and prepared all figures. Fanrong Zeng revised the manuscript. All authors reviewed the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Du, W., Zeng, F. Identification of Development-Related Genes in the Ovaries of Adult Harmonia axyridis (Pallas) Lady Beetles Using a Time- Series Analysis by RNA-seq. Sci Rep 6, 39109 (2016). https://doi.org/10.1038/srep39109
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep39109
- Springer Nature Limited
This article is cited by
-
Reference gene selection for RT-qPCR analysis in Harmonia axyridis, a global invasive lady beetle
Scientific Reports (2018)