
Abstract
The mangrove fern genus Acrostichum grows in the extremely unstable marine intertidal zone under harsh conditions, such as high salt concentrations, tidal rhythms and long-term climate changes. To explore the phylogenetic relationships and molecular mechanisms underlying adaptations in this genus, we sequenced the transcriptomes of two species of Acrostichum, A. aureum and A. speciosum, as well as a species in the sister genus, Ceratopteris thalictroides. We obtained 47,517, 36,420 and 60,823 unigenes for the three ferns, of which 24.39–45.63% were annotated using public databases. The estimated divergence time revealed that Acrostichum adapted to the coastal region during the late Cretaceous, whereas the two mangrove ferns from the Indo West-Pacific (IWP) area diverged more recently. Two methods (the modified branch-site model and the Kh method) were used to identify several positively selected genes, which may contribute to differential adaptation of the two Acrostichum species to different light and salt conditions. Our study provides abundant transcriptome data and new insights into the evolution and adaptations of mangrove ferns in the inhospitable intertidal zone.
Similar content being viewed by others


Introduction
The species of fern genus Acrostichum L. (Pteridaceae) are important components of mangrove community. They grow in the unstable marine intertidal zone, which is characterized by harsh conditions for plant growth, such as high salinity, tidal fluctuations and long-term climate changes1,2. Therefore, these species are referred as “mangrove ferns”2. This genus includes three species: A. danaeifolium Langsd. & Fisch., A. aureum L. and A. speciosum Willd2. A. danaeifolium and A. speciosum are restricted within the Atlantic East-Pacific (AEP) area and Indo West-Pacific (IWP) area, respectively, whereas A. aureum is the only species of mangroves that is widely distributed in both areas3. The three species of Acrostichum are all diploid, and A. aureum and A. danaeifolium have chromosome numbers of 2n = 604,5 (Supplementary Information and Supplementary Fig. S1).
In the IWP area, A. aureum and A. speciosum often occur sympatrically but occupy different habitats with respect to light and salinity6. A. aureum is an upstream fern usually found in open, light-abundant habitats that are strongly influenced by fresh water, especially in mangrove forests that have been disturbed by human activities7, whereas A. speciosum is usually found in the shady mangrove understory, which is frequently flooded by tides8. A. speciosum appears to have greater salt tolerance than A. aureum9, which is corroborated by the differing Na+ and Cl− levels in the roots and leaves of A. aureum and A. speciosum10. The differential adaptations of the two species to different light conditions are also reflected by their frond textures: A. aureum has thickly coriaceous fronds with a broadly rounded end, whereas A. speciosum has papery fronds with a pointed tip6. Although these plants prefer different environments, the two species can hybridize when their habitats overlap, especially in disturbed habitats6. However, only F1 hybrids have been found in the wild, suggesting strong postzygotic isolation between these species6. Acrostichum displays markedly differential adaptations to heterogeneous habitats, thus offering an excellent system in which to study adaptive evolution. For example, identifying positively selected genes in the genomes of Acrostichum species could contribute to our understanding of molecular mechanisms of adaptive evolution11,12. Additionally, Acrostichum is the only fern genus that grows in the intertidal zone, occupying a special position in ferns. By reconstructing the phylogeny of this genus and estimating divergence times, we can provide new insight on the origins of the genus.
To resolve these evolutionary questions, large amounts of molecular resources, such as whole-genome sequences and transcriptome data, are needed. Because fern species usually have large chromosome numbers and genome sizes, whole-genome sequencing is difficult; thus, complete genome sequences are not available for ferns, including Acrostichum13. RNA-seq is a relatively convenient choice because a large number of sequences can be obtained at low cost13. A number of ferns have been studied using this strategy, such as the bracken fern Pteridium aquilinum14, the fresh-water fern Ceratopteris richardii15, the Japanese climbing fern Lygodium japonicum16, as well as the fern species in the 1,000 Plants (1 KP) project17.
In this study, we sequenced, de novo assembled and annotated the transcriptomes of two mangrove fern species of Acrostichum (A. aureum and A. speciosum) and one species of its non-mangrove sister genus, Ceratopteris thalictroides. By combining the published chloroplast sequences and genomic data, we sought to 1) resolve the phylogenetic relationships in Acrostichum; 2) estimate the divergence times between Acrostichum and its sister genus Ceratopteris, as well as within the Acrostichum genus; and 3) detect candidate genes that are under positive selection in mangrove ferns.
Results
Transcriptome assembly and annotations
We obtained 22–26 million raw reads for the three fern species, from which 17–21 million clean reads were retrieved after quality control (Table 1). These reads were de novo assembled into 53,831, 41,661 and 69,929 contigs for A. aureum, A. speciosum and C. thalictroides, respectively, using Trinity18, a de novo transcriptome assembler (Table 1). After the redundancies were removed, 47,517, 36,420 and 60,823 contigs with N50 values of 1,136 bp, 1,687 bp and 787 bp, respectively, were treated as unigenes in the downstream analyses (Table 1). These unigenes were deposited in the NCBI GenBank under accession numbers GEEI00000000 (A. aureum), GEEJ00000000 (A. speciosum) and GEEK00000000 (C. thalictroides). The length distribution showed that 36.8–56.1% of the unigenes were longer than 500 bp (Supplementary Fig. S2). The GC content of A. aureum was 46.33%, which was slightly higher than that of A. speciosum (45.80%) and C. thalictroides (44.08%).
The functional annotations were performed based on similarity to the SwissProt protein database and NCBI non-redundancy protein database. A total of 35.89–58.35% unigenes returned a BLASTX hit above the e-value cut-off of 10−6 from these two databases (Table 2). Two annotation programs, Blast2GO19 and GOanna of Agbase20, were used for the functional annotations and Gene Ontology (GO) term retrievals. For A. aureum, A. speciosum and C. thalictroides, 12,100 (25.46%), 10,353 (28.43%) and 14,834 (24.39%) unigenes were annotated, respectively. The GO terms were assigned using Blast2GO (Table 2), and the distribution of level-2 GO terms was plotted (Fig. 1). A total of 4,501, 3,642 and 6,143 unigenes were assigned to 124, 122 and 127 KEGG pathways, respectively, for the three species (Table 2). A total of 42.05% of A. aureum, 45.63% of A. speciosum and 38.95% of C. thalictroides unigenes were matched to the annotation results from Agbase, a genomic database that contains functional annotations of agricultural species (Table 2). Detailed information on the functional annotations as well as the identified transcription factors and simple sequence repeats (SSRs) is provided in the Supplementary Information (Supplementary text, Supplementary Figs S3–S5 and Supplementary Tables S1–S10).

GO term (level 2) distribution for the transcriptomes of A. aureum, A. speciosum and C. thalictroides.
In total, 12,100, 10,353 and 14,834 unigenes for A. aureum, A. speciosum and C. thalictroides, respectively, were assigned to at least one GO term and grouped into three main GO categories and 51 GO terms.
Phylogenetic analysis based on chloroplast genes and transcriptome data
We concatenated the four chloroplast genes (atpA, atpB, rbcl and rps4) from six species (three species of Acrostichum, two species of Ceratopteris and the out-group species Pteridium aquilinum) to reconstruct the phylogenetic tree, and our results showed that each node of the tree was highly supported (Fig. 2a). The three species of Acrostichum formed a monophyletic clade, with A. danaeifolium diverging first and A. aureum and A. speciosum representing sister species. The divergence time between Acrostichum and Ceratopteris was estimated at approximately 93.8 Mya (Fig. 2b). Within Acrostichum, A. danaeifolium diverged from the other two species approximately 34.1 Mya, whereas the divergence between A. aureum and A. speciosum was much more recent (2.2 Mya).

Phylogenetic tree and divergence time estimation based on the sequences of four chloroplast genes.
(a) Phylogenetic tree. Branch lengths are marked on each branch. Numbers near each node are the bootstrap values. (b) Divergence time is based on the sequences of four chloroplast genes. The brown box indicates the earliest fossil record of Acrostichum (Maastrichtian in the late Cretaceous23, approximately 66.0–72.1 Mya). The blue bars represent the 95% confidential intervals. Q, Quaternary; P, Pliocene; Pl, Pleistocene.
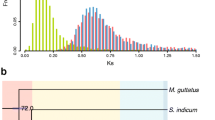
A total of 18,500 gene families were generated from the genomic and transcriptomic data of six species (A. aureum, A. speciosum, C. thalictroides, P. aquilinum, Lygodium japonicum and Selaginella moellendorffii) using OrthoMCL software21. From these data, 1,364 single-copy orthologs were chosen for phylogeny reconstruction and divergence-time estimation (Supplementary Table S11). Phylogenetic analyses were consistent with earlier studies of the phylogeny of Pteridaceae22, which suggested that Ceratopteris and Acrostichum last shared a common ancestor 88.1 million years ago in the late Cretaceous. The sister species A. aureum and A. speciosum diverged approximately 5.1 Mya. In addition, we calculated the Ks (synonymous substitution rate) of each ortholog identified before. These values reflected the relationships of Acrostichum and Ceratopteris. The peak of the distribution of the pairwise Ks values between A. aureum and A. speciosum was approximately 0.02 (Fig. 3b), suggesting a minor divergence between these two species; whereas, the peaks of the distribution of pairwise Ks values between each of the two Acrostichum species and C. thalictroides were approximately 0.7 (Fig. 3c), indicating a large divergence between the sister genera Acrostichum and Ceratopteris.

Divergence time estimated based on transcriptome data.
(a) Divergence estimation based on single-copy orthologs. The brown box indicates the earliest fossil record of Acrostichum. (b) Ks distribution between A. aureum and A. speciosum. (c) Ks distribution between Acrostichum and Ceratopteris.
Combined with the earliest fossil record of Acrostichum (Maastrichtian in the late Cretaceous23, 66.0–72.1 Mya), these results suggest that this genus might have diverged from its sister genus during the late Cretaceous. The divergence of the AEP fern A. danaeifolium at approximately 34.1 Mya might have triggered by the Eocene/Oligocene climatic crisis24.
Putative positively selected genes (PSGs) detected with the branch-site model and the Kh method
Genes under positive selection are often identified using the ratio of the nonsynonymous substitution rate to the synonymous substitution rate (Ka/Ks)25. A Ka/Ks value that is significantly larger or smaller than 1 is interpreted as evidence of positive/purifying selection, and a Ka/Ks equivalent to 1 indicates neutral evolution25. However, this method is stringent, and positive selection often acts on a few sites of a gene over a short interval26 and can be counteracted by negative selection at the remaining sites27. Therefore, two additional methods, the modified branch-site model26 and the Kh test27, were used in this study to detect candidate PSGs.
A total of 3,164 orthologs generated from the transcriptomes of four species (A. aureum, A. speciosum, C. thalictroides and C. richardii) were used to identify candidate PSGs using the modified branch-site model. We detected 27 and 31 PSGs in the branches of A. aureum and A. speciosum with p-values < 0.05 based on a Chi-square test. After the Benjamini-Hochberg correction28 was applied, only six and three genes for A. aureum and A. speciosum, respectively, remained. Because the Benjamini-Hochberg correction is a stringent correction used to reduce the false-positive rate, and may remove true PSGs, we retained all candidate PSGs from before the Benjamini-Hochberg correction for the functional annotation. These PSGs were involved in metabolic processes, RNA or DNA binding and specific enzymatic reactions that play a role in responses to light and salt stresses (see Discussion and Supplementary Tables S12–S13 for detailed information).
Using the Kh method, 7,379 orthologs from A. aureum and A. speciosum were obtained. We observed 16,183 amino acid changes between these two species, and 15,181 of which were elementary amino acid changes (changed by 1 bp). The top 10–12 classes had 3,961–5,232 elementary amino acid changes that accounted for 25–32% of the total changes. The ratio of Ki*/Ks (Ki* is the cumulative rate of the first i classes of amino acid substitution.) versus the i-th class of amino acid changes is plotted in Fig. 4a. The value of K10*/Ks was almost twice that of Ka/Ks, which is consistent with the “twofold approximation” pattern that has been observed in yeast, primates and rodents27. This twofold pattern was also supported by 53 supergenes that combined 100 orthologs with similar Ka values and presented a regression line slop of 1.76 (Fig. 4b). We defined K10* as Kh, which refers to a class of highly exchangeable substitutions, after the method of Tang and Wu27. The standard Kh/Ks > 1 may be a reasonable standard similar to Ka/Ks > 1 for use in identifying genes under positive selection27. In total, 227 genes presented Ka/Ks > 1, but only one was significant, whereas the Kh/Ks values of 15 genes were significantly greater than 1 (Table 3, Supplementary Table S14 and Supplementary Fig. S6). Three genes were involved in responses to light, and the others had functions in binding, kinase activity, metabolism, etc.(Table 3 and Supplementary Table S14).

Results of Ki*/Ks.
(a) Ki*/Ks versus i. i denotes the universal ranking of each amino acid change and range from the most exchangeable pairs (i = 1) to the least exchangeable pairs (i = 75)67. K75*/Ks is equivalent to Ka/Ks. K10*/Ks is almost twice the value of Ka/Ks. (b) Scatter plot of Kh/Ks versus Ka/Ks for 53 supergenes between A. aureum and A. speciosum. Each supergene contains 100 orthologs with similar Ka values.
Discussion
Previous studies of Acrostichum have focused on its physiology, morphology and ecology7,8, and only one recent study has reported natural hybridization between two Acrostichum species in the IWP region6. To date, there have been no published genomic data for Acrostichum, and the EST sequences in the NCBI database are too limited to address evolutionary questions, such as the origin of the genus and the identification of genes under positive selection. The transcriptome data for Acrostichum that were developed in this study provide new resources for mangrove ferns.
In this study, we used chloroplast genes and thousands of orthologous genes from transcriptomic/genomic data to estimate the divergence time between Acrostichum and its sister genus, and between species within Acrostichum. Based on the results from the two datasets, the Acrostichum genus diverged from the closely related Ceratopteris approximately 88.1 Mya and the AEP fern A. danaeifolium diverged from the other IWP ferns approximately 34.1 Mya. The earliest known Acrostichum fossil is a permineralized aerial stem with petioles and roots that was identified in the Deccan Intertrappean beds of India23. This fossil is dated to the Maastrichtian in the late Cretaceous, which is approximately 66.0–72.1 million years before present. Aerenchyma tissue is a morphological feature considered to be an adaptation to aquatic life in both Ceratopteris and Acrostichum29, and has been found in the roots of the Acrostichum fossil, which suggests a coastal palaeoenvironment23. Fossils of coastal palms, mangroves and marsh plants have also been found in this region or nearby23, indicating that the ancestor of Acrostichum had grown in and adapted to the coastal region by at least the late Cretaceous. Mangroves were pantropic by the Eocene and appeared to have originated during the Paleocene24. Our results revealed that Acrostichum is one of the oldest members of the mangrove ecosystem and dates to the late Cretaceous along with the mangrove palm Nypa30,31. It is reported that there are 10,560 extant fern species which belong to 215 genera32, and this large species richness may result from a burst of fern diversification in the Cenozoic (from 66 million years ago to present day)33. However, Acrostichum had only three extant species although it diverged from its sister genus since 88.1 Mya, which may imply that the intertidal zone might be an extremely inhospitable environment for plants to survive. A. danaeifolium, the species restricted to the AEP area, diverged approximately 34.1 Mya (in the late Oligocene) and was traced back to the Eocene/Oligocene climate crisis24.
For the divergence time between A. aureum and A. speciosum, the estimate based on the chloroplast genes (2.2 Mya) is more recent than the one based on transcriptome data (5.1 Mya). This discrepancy in estimates of divergence time may be caused by ancient chloroplast capture through interspecific introgression and hybridization. In the early stage of their divergence, the interspecific hybridization and subsequent backcrossing for several generations would create a new combination of mainly one species’ nuclear genome and completely another species’ chloroplast genome, due to maternal inheritance and absence of recombination in chloroplast DNA34. This ancient chloroplast capture would influence and reduce the divergence of chloroplast genome; thus, divergence time estimates based on chloroplast genes would be more recent. In addition, only four genes were used to estimate the divergence time; therefore, variations between the sister species may not sufficiently reflect their divergence. Additional chloroplast genes should be used in future studies to increase the accuracy of divergence estimate.
A. aureum and A. speciosum diverged from each other very recently in the IWP area and prefer different habitats with respect to salt and light conditions. To reveal the molecular mechanisms underlying these adaptations, we used two methods to detect candidate PSGs. The modified branch-site model identifies PSGs based on a likelihood ratio test of models for the foreground lineage under selection and without selection. This approach has been widely used for genomic data and exploits the advances in genome sequencing technology that have been made in recent years35,36. Many previous works showed that different amino acid pairs have different exchangeability37,38; therefore, amino acid pairs with high exchangeability could be more sensitive indicators of positive selection signals that are hidden by purifying selection. Tang and Wu developed a new method using the Kh statistic, which is the cumulative rate of nonsynonymous substitutions for the 10 most exchangeable classes, instead of the Ka statistic27. The value of Kh is approximately twice that of Ka in mangrove ferns, and a similar pattern is observed in yeast and animals27. A ratio of Kh/Ks significantly greater than 1 is a potential new criterion for detecting positive selection27.
Certain genes detected by the modified branch-site model were related to salt and light stress responses. For example, the SKIP (SNW/SKI-interacting protein) gene was identified under positive selection when A. aureum and A. speciosum were set as the foreground branch. This gene could improve the abiotic stress resistance via the regulation of abscisic acid signal transduction39, contribute to cytokinin-regulated leaf initiation40, and participate in photomorphogenesis by regulating the signaling of cell cycle41. The 27 PSGs in the lineage of A. aureum include AtBAG4 (Bcl-2-associated athanogene 4), an anti-apoptotic gene that significantly enhances the salt tolerance of rice42; adenylate kinase (ADK), a kinases of the SnRK1-ADK complexes that participates managing biotic and abiotic stresses and maintaining energy homeostasis43; and phosphomannomutase (PMM), which is required for the GDP-mannose biosynthesis, ascorbic acid biosynthesis and N-glycosylation and plays an important role in temperature adaptability44,45. Of the 31 PSGs in the A. speciosum lineage, two are involved in the response to light. Photosystem II core phosphatase (PBCP) is important for effective dephosphorylation of the core subunits of photosystem II and may influence the state transitions between photosystem I and photosystem II46. Ribosomal protein L10B (RPL10B) may participate in the responses to different stresses, especially to ultraviolet B47 (see Supplementary Tables S12–S13 for detailed information on each PSG).
Among the 15 positively selected genes identified using the Kh method, one gene was annotated as a phototropic-responsive NPH3 family protein, which function in signal transduction of phototropic response48,49. A. aureum is often found in open places with full light, while A. speciosum usually grows under mangrove forests. As the two Acrostichum species have different preferences for light, this gene may contribute to their differential adaptations to different light conditions. In addition, two genes encoded as DHNA-CoA (1,4-dihydroxy-2-naphthoyl-CoA) thioesterase may contribute to the biosynthesis of phylloquinone (vitamin K1)50, an electron acceptor of the electron transport chain in Photosystem I.
PSGs did not overlap between the two methods, which may be related to the different assumptions of the methods. The modified branch-site model uses the likelihood ratio test to detect the PSGs on a given branch, whereas the Kh method examines highly exchanged amino acid pairs for evidence of positive selection in order to reduce the influence of purifying selection. The Kh method can identify genes under selection but cannot determine the direction of selection. Although each method found a subset of PSGs, all of the candidate PSGs identified by the two methods were related to responses to light, including an elector acceptor and proteins involved in photosystem state transitions, phototropic responses and UV stress responses. Based on the recent divergence between A. aureum and A. speciosum and their different preferences for light, these PSGs might be important for the adaptation and speciation of the two mangrove ferns.
In summary, we sequenced the transcriptomes for two species of the mangrove fern genus Acrostichum and one species of its sister genus Ceratopteris, providing new genomic resources for both ferns and mangroves. Phylogenetic reconstruction and divergence time estimation based on both transcriptome data and chloroplast genes revealed that Acrostichum adapted to the coastal region during the late Cretaceous, whereas the two mangrove ferns in the Indo West-Pacific (IWP) region diverged recently. Positively selected genes, such as SKIP gene, NPH3 family protein, etc., were detected by the modified branch-site model and the Kh method, which may contribute to differential adaptations of Acrostichum species to different intertidal habitats.
Methods
Sampling, RNA extraction and sequencing
Samples of A. aureum and A. speciosum were collected from Nansha, Guangzhou, Guangdong (22°48′34.57″N, 113°34′56.38″E) and Qinglan Harbour, Wenchang, Hainan (19°37′33.11″N, 110°47′33.94″E), respectively. C. thalictroides was cultivated in the greenhouse of Sun Yat-sen University (Supplementary Table S15). Young leaves of each species were harvested to extract total RNA by the modified CTAB method51. cDNA library construction and sequencing were conducted by the Beijing Genome Institute (BGI, Shenzhen, China). Paired-end reads were obtained using the Illumina HiSeq2000 sequencing platform (Illumina, San Diego, USA). After we filtered the sequence adaptors, we deposited all raw reads into the NCBI short read archive (SRA) repository under accession numbers SRR1822234 (A. aureum), SRR1822235 (A. speciosum) and SRR1822236 (C. thalictroides).
Data filtering, de novo assembly and functional annotation
The raw reads were first trimmed using the DynamicTrim program of the SolexaQA package52 at a quality threshold of 20. We then filtered reads less than 50 bp long using the LengthSort program of the same package. The clean reads of A. aureum, A. speciosum and C. thalictroides were de novo assembled into contigs using the short read assembly program Trinity18 under the default settings except ‘min_kmer-cov = 2’. Then, the programs TGICL53 and CDHIT54 were used to remove redundant contigs under the default parameters. We mapped the clean reads to these contigs and calculated the mean coverage for each contig. Contigs with an average depth of less than two were discarded, and the remaining contigs were treated as unigenes in the subsequent analyses.
To determine the functional categories of the transcripts of three fern species, a BLASTX search was performed against the NCBI non-redundant (NR) protein database and SwissProt database55 (http://web.expasy.org/docs/swiss-prot_guideline.html) with an e-value cut-off of 10−6. The results of the NR BLASTX hits were processed with Blast2GO software19 (v.3.0.9 PRO) to assign functional annotations and retrieve the GO terms. The distribution of the level-2 GO terms for the three categories, biological processes, molecular functions and cellular components, was plotted in WEGO56. We conducted a pathway analysis against the Kyoto Encyclopedia of Genes and Genomes (KEGG) database using Blast2GO. The three transcriptomes were annotated with the SwissProt database using GOanna of Agbase20 (http://www.agbase.msstate.edu/cgi-bin/tools/GOanna.cgi) with a cut-off e-value of 10−6.
Phylogenetic analyses and divergence time estimation
To infer the phylogenetic relationships and divergence times within the Acrostichum genus, four chloroplast genes (atpA, atpB, rbcl and rps4) from six species (three species of Acrostichum, two species of Ceratopteris and the out-group species Pteridium aquilinum) were used. The chloroplast sequences of Acrostichum danaeifolium, Ceratopteris richardii and Pteridium aquilinum were downloaded from the NCBI GenBank (Supplementary Table S16), and the corresponding genes of A. aureum, A. speciosum and C. thalictroides were obtained from transcriptome data using BLASTN with a cut-off e-value of 10−6. Chloroplast genes were aligned by MUSCLE57 and concatenated into one supergene. Before the phylogeny was reconstructed, an appropriate nucleotide-substitution model was selected from 88 substitution models using the jModelTest2 program58. The phylogenetic tree was built using PhyML59 with the best-fit model (GTR + G) and 1,000 replicates of the bootstrap analysis. The divergence time of each node was calculated by MCMCTree with the PAML 4.8 package60 using ‘seq like (usedata = 1)’, ‘HKY85 + gamma (model = 4; alpha = 0.5)’ and ‘independent rates (clock = 2)’. The time constraint between Pteridium and the ancestor of Acrostichum and Ceratopteris was set at 160–170 Mya according to the results of Schuettpelz et al.33.
A phylogenetic analysis was also performed on the orthologous genes that were generated from the genome or transcriptome data of six species, including A. aureum, A. speciosum, C. thalictroides, P. aquilinum, Lygodium japonicum and Selaginella moellendorffii. The transcriptome data of P. aquilinum, L. japonicum and S. moellendorffii were downloaded from Der et al.14, the Lygodium japonicum Transcriptome Database (http://bioinf.mind.meiji.ac.jp/kanikusa/) and Phytozome61,62, respectively. For each species pair, an all-versus-all sequence similarity search was conducted on the protein sequence using BLASTP with an e-value cut-off of 10−10 and an identity threshold of 40%. The BLASTP results were imported into OrthoMCL software21 for orthologous group clustering under the default settings. The protein sequences of the single-copy orthologs were aligned with MUSCLE57 and then converted to nucleotides with Pal2nal63. Alignments longer than 150 bp were retained for the phylogeny reconstruction and dating. JModeltest2, PhyML and MCMCTree were applied for model selection, phylogenetic tree reconstruction and divergence time calculation as described above. We employed two additional time constraints when dating the divergence time between S. moellendorffii and the true ferns (400–420 Mya)64,65 and between Schizaeoid ferns and the core leptosporangiates (260–270 Mya)33. The Ks values for the orthologs were also estimated using the KaKs-Calculator66 with the YN model to examine the distance between these species. The alignments used in the phylogenetic analyses were deposited in TreeBASE (http://purl.org/phylo/treebase/phylows/study/TB2:S19541).
Identification of candidate positively selected genes (PSGs)
We applied two methods to identify putative PSGs in Acrostichum: the improved branch-site model26 implemented in codeml of the PAML 4.8 package60 and the Kh method developed by Tang and Wu27.
The modified branch-site model was used to identify PSGs along the branches of A. aureum and A. speciosum based on the 3,164 single-copy orthologs of four species (A. aureum, A. speciosum, C. thalictroides and C. richardii). The transcriptome data of C. richardii were downloaded from Bushart et al.15. A. aureum and A. speciosum were set as the foreground branch separately, and then a likelihood ratio test was performed to compare the null model (no signal of positive selection) to the alternative model (positive selection on certain codons)26. The ancestral branch of A. aureum and A. speciosum was not included in this study. The Benjamini-Hochberg correction28 with a false-discovery rate of 5% was used for multiple testing. We also annotated these genes based on the homologues of Arabidopsis in The Arabidopsis Information Resource (TAIR, https://www.arabidopsis.org/).
The orthologs between A. aureum and A. speciosum were assessed using OrthoMCL software21 and aligned as described above. The universal evolutionary index (EI(i), i = 1–75) of Tang et al.67 ranked the 75 elementary amino acid changes where the codon differed by 1 bp from the most exchangeable (i = 1) to the least exchangeable class (i = 75). To determine whether Acrostichum has the “twofold approximation” pattern reported in Tang and Wu27, we first concatenated all genes into one supergene to calculate the overall values of the cumulative rate of the first i types of amino acid changes (Ki*), synonymous substitution rates (Ks) and nonsynonymous substitution rates (Ka). We counted the observed synonymous substitutions (Ns) and total synonymous sites (Ls) of pairwise alignments to calculate the Ks value. Then, the observed substitutions and total sites of each class of elementary amino acid change (Ni and Li, respectively, i = 1, 2, …, 75) were counted to calculate the Ki value. The ratio of the transition rate to the transversion rate (kappa) was estimated from fourfold degenerate sites using baseml in the Paml 4.8 package. The values of Ks, Ki (the nonsynonymous substitution rate of the ith-type amino acid change) and Ki* were calculated using the method of Jukes and Cantor68 for multiple hit corrections. The K75* value was equivalent to Ka in this calculation. To detect this twofold pattern in genes with different Ka values, 5,304 genes with Ka > 0 were sorted in descending order, and then every 100 genes were concatenated to conduct the same estimations. To identify positively selected gene, the Ka, Ks and Ki* values of each ortholog were estimated using the same method. According to Tang and Wu27, K10* was defined as Kh, the class of high-exchangeable substitutions, and a threshold of Kh/Ks significantly greater than 1 was used as the criterion for isolating PSGs. Fisher’s exact test implemented in R was used to test for significance. We removed genes with Ka > 0.05, Ks > 0.08 or Ks < 0.005 and then treated the remaining genes with Kh/Ks > 1 and p-value < 0.05 as candidate PSGs.
Additional Information
Accession codes: All raw reads were deposited in the NCBI short read archive (SRA) repository under accession numbers SRR1822234 (A. aureum), SRR1822235 (A. speciosum) and SRR1822236 (C. thalictroides). The unigenes of three species were deposited in NCBI GenBank under the accession numbers of GEEI00000000 (A. aureum), GEEJ00000000 (A. speciosum) and GEEK00000000 (C. thalictroides).
How to cite this article: Zhang, Z. et al. Transcriptome analyses provide insights into the phylogeny and adaptive evolution of the mangrove fern genus Acrostichum. Sci. Rep. 6, 35634; doi: 10.1038/srep35634 (2016).
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Duke, N. C. Mangrove Floristics and Biogeography. In Tropical Mangrove Ecosystems (eds Rovertson, A. I. & Alongi, D. M. ) 63–100 (American Geophysical Union, 1992).
Tomlinson, P. The botany of mangroves. 312–317 (Cambridge University Press, Cambridge, 1986).
Duke, N., Ball, M. & Ellison, J. Factors influencing biodiversity and distributional gradients in mangroves. Global Ecology & Biogeography Letters 7, 27–47 (1998).
Lovis, J. D. Evolutionary patterns and processes in ferns. Adv. Bot. Res. 4, 229–415 (1978).
Marcon, A. B., Barros, I. C. L. & Guerra, M. A karyotype comparison between two closely related species of Acrostichum. Am. Fern J. 93, 116–125 (2003).
Zhang, R. et al. Molecular evidence for natural hybridization in the mangrove fern genus Acrostichum. BMC Plant Biol. 13, 74 (2013).
Medina, E., Cuevas, E., Popp, M. & Lugo, A. E. Soil salinity, sun exposure, and growth of Acrostichum aureum, the mangrove fern. Botanical Gazette 151, 41–49 (1990).
Lloyd, R. M. Reproductive biology and gametophyte morphology of New World populations of Acrostichum aureum. Am. Fern J., 99–110 (1980).
Mehltreter, K. Phenology and habitat specificity of tropical ferns. In Biology and evolution of ferns and lycophytes (eds Ranker, R. A. & Haufer, C. H. ) 201–221 (Cambridge University, 2008).
Jiang, Q. Comparisons of Element Distribution Characteristics and Salt Tolerance Between True Mangroves and Mangroves Associates, Xiamen University, (2007).
Yang, L., Wang, Y., Zhang, Z. & He, S. Comprehensive transcriptome analysis reveals accelerated genic evolution in a Tibet fish, Gymnodiptychus pachycheilus. Genome Biol. Evol. 7, 251–261 (2015).
Wang, Y. et al. Evidence for adaptation to the Tibetan Plateau inferred from Tibetan loach transcriptomes. Genome Biol. Evol. 7, 2970–2982 (2015).
Barker, M. S. & Wolf, P. G. Unfurling fern biology in the genomics age. Bioscience 60, 177–185 (2010).
Der, J. P., Barker, M. S., Wickett, N. J., dePamphilis, C. W. & Wolf, P. G. De novo characterization of the gametophyte transcriptome in bracken fern, Pteridium aquilinum. BMC Genomics 12, 99 (2011).
Bushart, T. J. et al. RNA-seq analysis identifies potential modulators of gravity response in spores of Ceratopteris (Parkeriaceae): evidence for modulation by calcium pumps and apyrase activity. Am. J. Bot. 100, 161–174 (2013).
Aya, K. et al. De novo transcriptome assembly of a fern, Lygodium japonicum, and a web resource database, Ljtrans DB. Plant and Cell Physiology 56, e5 (2015).
Matasci, N. et al. Data access for the 1,000 Plants (1KP) project. GigaScience 3, 1 (2014).
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644–652 (2011).
Conesa, A. et al. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21, 3674–3676 (2005).
McCarthy, F. et al. AgBase: a functional genomics resource for agriculture. BMC Genomics 7, 229 (2006).
Li, L., Stoeckert, C. J., Jr. & Roos, D. S. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–2189 (2003).
Schuettpelz, E., Schneider, H., Huiet, L., Windham, M. D. & Pryer, K. M. A molecular phylogeny of the fern family Pteridaceae: assessing overall relationships and the affinities of previously unsampled genera. Mol Phylogenet Evol 44, 1172–1185 (2007).
Bonde, S. & Kumaran, K. A permineralized species of mangrove fern Acrostichum L. from Deccan Intertrappean beds of India. Rev. Palaeobot. Palynol. 120, 285–299 (2002).
Plaziat, J.-C., Cavagnetto, C., Koeniguer, J.-C. & Baltzer, F. History and biogeography of the mangrove ecosystem, based on a critical reassessment of the paleontological record. Wetlands Ecol. Manage. 9, 161–180 (2001).
Nei, M. & Kumar, S. Molecular evolution and phylogenetics. (Oxford University Press, 2000).
Zhang, J., Nielsen, R. & Yang, Z. Evaluation of an improved branch-site likelihood method for detecting positive selection at the molecular level. Mol. Biol. Evol. 22, 2472–2479 (2005).
Tang, H. & Wu, C.-I. A new method for estimating nonsynonymous substitutions and its applications to detecting positive selection. Mol. Biol. Evol. 23, 372–379 (2006).
Benjamini, Y. & Hochberg, Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. Journal of the Royal Statistical Society. Series B (Methodological) 57, 289–300 (1995).
Schneider, H. Vergleichende Wurzelanatomie der Farne, Shaker Verlag, Aachen, Germany, (1996).
Gee, C. T. The mangrove palm Nypa in the geologic past of the New World. Wetlands Ecol. Manage. 9, 181–203 (2001).
He, Z. et al. De Novo Assembly of Coding Sequences of the Mangrove Palm (Nypa fruticans) Using RNA-Seq and Discovery of Whole-Genome Duplications in the Ancestor of Palms. PloS one 10, e0145385 (2015).
Christenhusz, M. J. & Byng, J. W. The number of known plants species in the world and its annual increase. Phytotaxa 261, 201–217 (2016).
Schuettpelz, E. & Pryer, K. M. Evidence for a Cenozoic radiation of ferns in an angiosperm-dominated canopy. Proc. Natl. Acad. Sci. USA 106, 11200–11205 (2009).
Raamsdonk, L. W. V., Smiech, M. P. & Sandbrink, J. M. Introgression explains incongruence between nuclear and chloroplast DNA-based phylogenies in Allium section Cepa. Bot. J. Linn. Soc. 123, 91–108 (1997).
Li, M. et al. Genomic analyses identify distinct patterns of selection in domesticated pigs and Tibetan wild boars. Nat. Genet. 45, 1431–1438 (2013).
Yan, G. et al. Genome sequencing and comparison of two nonhuman primate animal models, the cynomolgus and Chinese rhesus macaques. Nat. Biotechnol. 29, 1019–1023 (2011).
Grantham, R. Amino acid difference formula to help explain protein evolution. Science 185, 862–864 (1974).
Wyckoff, G. J., Wang, W. & Wu, C.-I. Rapid evolution of male reproductive genes in the descent of man. Nature 403, 304–309 (2000).
Lim, G. H. et al. A putative novel transcription factor, AtSKIP, is involved in abscisic acid signalling and confers salt and osmotic tolerance in Arabidopsis. New Phytol. 185, 103–113 (2010).
Zhang, X. et al. AtSKIP functions as a mediator between cytokinin and light signaling pathway in Arabidopsis thaliana. Plant Cell Rep. 33, 401–409 (2014).
Zhang, X., Ju, H.-W., Huang, P., Chung, J.-S. & Kim, C. S. Functional identification of AtSKIP as a regulator of the cell cycle signaling pathway in Arabidopsis thaliana. J. Plant Biol. 55, 481–488 (2012).
Hoang, T. M. et al. Development of salinity tolerance in rice by constitutive-overexpression of genes involved in the regulation of programmed cell death. Frontiers in plant science 6 (2015).
Mohannath, G. et al. A Complex Containing SNF1-Related Kinase (SnRK1) and Adenosine Kinase in Arabidopsis. PLoS ONE 9, e87592 (2014).
Hoeberichts, F. A. et al. A Temperature-sensitive mutation in the Arabidopsis thaliana phosphomannomutase gene disrupts protein glycosylation and triggers cell death. J. Biol. Chem. 283, 5708–5718 (2008).
Qian, W. et al. Molecular and functional analysis of phosphomannomutase (PMM) from higher plants and genetic evidence for the involvement of PMM in ascorbic acid biosynthesis in Arabidopsis and Nicotiana benthamiana. Plant J. 49, 399–413 (2007).
Samol, I. et al. Identification of a photosystem II phosphatase involved in light acclimation in Arabidopsis. The Plant Cell Online 24, 2596–2609 (2012).
Ferreyra, M. L. F., Casadevall, R., Luciani, M. D., Pezza, A. & Casati, P. New evidence for differential roles of l10 ribosomal proteins from Arabidopsis. Plant Physiol. 163, 378–391 (2013).
Motchoulski, A. & Liscum, E. Arabidopsis NPH3: a NPH1 photoreceptor-interacting protein essential for phototropism. Science 286, 961–964 (1999).
Sakai, T. NPH3 and RPT2: signal transducers in phototropin signaling pathways. In Light sensing in plants (ed Wada, M., Shimazaki, K. & Iino, M. ) Ch. 20, 179–184 (Springer, 2005).
Widhalm, J. R. et al. Phylloquinone (vitamin K1) biosynthesis in plants: two peroxisomal thioesterases of lactobacillales origin hydrolyze 1, 4-dihydroxy-2-naphthoyl-coa. The Plant Journal 71, 205–215 (2012).
Yang, G., Zhou, R., Tang, T. & Shi, S. Simple and efficient isolation of high-quality total RNA from Hibiscus tiliaceus, a mangrove associate and its relatives. Prep. Biochem. Biotechnol. 38, 257–264 (2008).
Cox, M. P., Peterson, D. A. & Biggs, P. J. SolexaQA: At-a-glance quality assessment of Illumina second-generation sequencing data. BMC Bioinformatics 11, 485 (2010).
Pertea, G. et al. TIGR Gene Indices clustering tools (TGICL): a software system for fast clustering of large EST datasets. Bioinformatics 19, 651–652 (2003).
Fu, L., Niu, B., Zhu, Z., Wu, S. & Li, W. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28, 3150–3152 (2012).
Boeckmann, B. et al. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res. 31, 365–370 (2003).
Ye, J. et al. WEGO: a web tool for plotting GO annotations. Nucleic Acids Res. 34, W293–W297 (2006).
Edgar, R. C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 (2004).
Darriba, D., Taboada, G. L., Doallo, R. & Posada, D. jModelTest 2: more models, new heuristics and parallel computing. Nat. Methods 9, 772–772 (2012).
Guindon, S. et al. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 59, 307–321 (2010).
Yang, Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591 (2007).
Banks, J. A. et al. The Selaginella genome identifies genetic changes associated with the evolution of vascular plants. Science 332, 960–963 (2011).
Goodstein, D. M. et al. Phytozome: a comparative platform for green plant genomics. Nucleic Acids Res. 40, D1178–D1186 (2012).
Suyama, M., Torrents, D. & Bork, P. PAL2NAL: robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 34, W609–W612 (2006).
Banks, J. A. Selaginella and 400 million years of separation. Annu. Rev. Plant Biol. 60, 223–238 (2009).
Kenrick, P. & Crane, P. R. The origin and early diversification of land plants. A cladistic study. Vol. 560 (Smithsonian Institute Press Washington DC, 1997).
Zhang, Z. et al. KaKs_Calculator: Calculating Ka and Ks Through Model Selection and Model Averaging. Genomics, Proteomics & Bioinformatics 4, 259–263 (2006).
Tang, H., Wyckoff, G. J., Lu, J. & Wu, C.-I. A universal evolutionary index for amino acid changes. Mol. Biol. Evol. 21, 1548–1556 (2004).
Jukes, T. H. & Cantor, C. R. Evolution of protein molecules. In Mammalian protein metabolism (ed Munro, H. N. ) 21–132 (Academic Press, New York, 1969).
Acknowledgements
We appreciate the useful comments from Dr. Harald Schneider and Dr. Sunil Kumar Sahu. This study was financially supported by the National Natural Science Foundation of China (grant numbers 41130208 and 91331202), the 985 Project (grant number 33000-18821105), the Science Foundation of State Key Laboratory of Biocontrol (grant number SKLBC16A35), the Fundamental Research Funds for the Central Universities (grant number 16lgjc39) and the Chang Hungta Science Foundation of Sun Yat-sen University.
Author information
Authors and Affiliations
Contributions
S.S., R.Z. and Z.Z. designed the project. Z.Z., S.X., Y.Y. and C.Z. contributed to sampling. Z.Z. performed the experiments and drafted the manuscript. Z.Z., Z.H., S.X., X.L., W.G. and Y.Y. analysed and interpreted the data. Z.H., R.Z. and S.S. revised the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Zhang, Z., He, Z., Xu, S. et al. Transcriptome analyses provide insights into the phylogeny and adaptive evolution of the mangrove fern genus Acrostichum. Sci Rep 6, 35634 (2016). https://doi.org/10.1038/srep35634
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep35634
- Springer Nature Limited