
Abstract
Waardenburg syndrome (WS) characterized by sensorineural hearing loss and pigmentary abnormalities is genetically heterogeneous and phenotypically variable. This study investigated the molecular etiology and genotype-phenotype correlation of WS in 36 Chinese Han deaf probands and 16 additional family members that were clinically diagnosed with WS type I (WS1, n = 8) and type II (WS2, n = 42). Mutation screening of six WS-associated genes detected PAX3 mutations in 6 (86%) of the 7 WS1 probands. Among the 29 WS2 probands, 13 (45%) and 10 (34%) were identified with SOX10 and MITF mutations, respectively. Nineteen of the 26 detected mutations were novel. In WS2 probands whose parental DNA samples were available, de novo mutations were frequently seen for SOX10 mutations (7/8) but not for MITF mutations (0/5, P = 0.005). Excessive freckle, a common feature of WS2 in Chinese Hans, was frequent in WS2 probands with MITF mutations (7/10) but not in those with SOX10 mutations (0/13, P = 4.9 × 10−4). Our results showed that mutations in SOX10 and MITF are two major causes for deafness associated with WS2. These two subtypes of WS2 can be distinguished by the high de novo rate of the SOX10 mutations and the excessive freckle phenotype exclusively associated with the MITF mutations.
Similar content being viewed by others


Introduction
Waardenburg Syndrome (WS) is relatively common among syndromic deafness, with an estimated prevalence of 1 in 42000 in the general population and 1–3% among the congenitally deaf1. It is mainly characterized by sensorineural deafness and various types of pigmentary abnormalities including heterochromic iridis, patchy de-pigmentation of the skin and premature graying of the hair. Based on the additional symptoms, WS can be further categorized into WS type I with dystrophia canthorum (WS1, OMIM193500), WS type II without additional symptoms (WS2, OMIM 193510, 600193, 606662, 608890 and 611584), WS type III with dystrophia canthorum and upper limb anomalies (WS3, also called Klein-Waardenburg syndrome, OMIM148820) and WS type IV with aganglionic megacolon (WS4, also called Shah-Waardenburg Syndrome or Waardenburg-Hirschsprung Disease, OMIM 277580, 613265 and 613266). In many cases, the clinical phenotypes of WS are incompletely penetrant. Hearing loss, for example, was estimated to occur in 60% for WS1, 90% for WS2, but only 5% for WS42,3. Variable phenotypic expression can be observed both interfamilially and intrafamilially, suggesting of interplay between genetic modifiers and environmental factors.
To date, six causative genes have been identified for WS, including PAX3 encoding the paired box 3 transcription factor, MITF encoding the microphthalmia-associated transcription factor, SOX10 encoding the SRY (sex determining region Y) box 10 transcription factor, SNAI2 encoding the snail homolog 2 transcription factor, EDN3 encoding the endothelin-3 and EDNRB encoding the endothelin receptor type B. Most cases of WS1 and some moderate cases of WS3 are caused by heterozygous mutations in PAX3, while homozygous or compound heterozygous mutations of PAX3 have been identified in some severe cases of WS34. In WS2, heterozygous mutations in MITF and SOX10 are estimated to account for 15% of cases each, while heterozygous mutations in EDNRB and homozygous mutations in SNAI2 haven been identified in less than 5%5. Two additional WS2 loci, WS2B (OMIM 600193) and WS2C (OMIM 606662), have been reported without pathogenic genes identified6. In WS4, Approximately 50% of cases are due to heterozygous mutations in SOX10 and 20–30% to homozygous or heterozygous mutations in EDNRB and EDN3.
WS1 and WS2 were two major subtypes of WS associated with hearing loss. Unlike the former, WS2 is genetically heterogeneous and so far the genotype-phenotype correlation of the WS2 genes remains unclear. In this study, 50 Chinese Han deaf patients with WS type I and II were clinically characterized and genetically screened for mutations in PAX3, MITF, SOX10, SNAI, EDN3 and EDNRB. Our results showed that heterozygous mutations in MITF and SOX10, two major causes of WS2, have distinguishable de novo rates and clinical features.
Results
Clinical characteristics of the WS patients
The clinical features of the WS probands were summarized in Table 1. All 36 probands have sensorineural, severe-to-profound hearing loss and at least one type of pigmentary abnormalities including heterochromic iridis (n = 34), excessive freckle (n = 7), patchy de-pigmentation of the skin (n = 6) and premature graying of the hair (n = 6). No musculoskeletal anomaly or intestinal aganglionosis was observed in any of the probands. Based on the W-indexes, 7 probands were diagnosed with WS1 (W-indexes > 2.10) and 29 with WS2 (W-indexes <1.90).
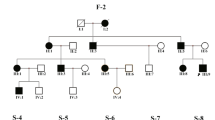
Intrafamilial varieties of the WS-associated phenotypes can be observed in all of the nine families with additional affected members (Fig. 1, marked by asterisks). Among them, heterochromic iridis is the most consistent intrafamilial phenotype (4/7), followed by excessive freckle (3/5), hearing loss (2/9), patchy de-pigmentation of the skin (0/2) and premature graying of the hair (0/6).

Pedigrees of the WS patients showing their phenotypes and genotypes.
Individuals with a number assigned participated in the current study. Phenotypes of the rest of the family members were based on relative’s description. The probands were pointed by arrows. Families with additional affected members who participated in the current study were marked by asterisks. (A) WS1 families with and without PAX3 mutations. Note in family W24, the apparent non-segregation of the p.T31S variant (marked by quotation marks) indicated a likely gross deletion in PAX3 in individual W24-3 and W24-1. (B) WS2 families with SOX10 mutations. The de novo mutations were underlined. (C) WS2 families with MITF mutations. (D) WS2 families without mutation identified.
Mutations identified in the WS patients
The mutations identified in the WS patients were summarized in Table 2 and labeled in Fig. 1. In 7 WS1 families, heterozygous mutations in PAX3 were identified in 5 of them. In addition, apparent haplotype non-segregation of a PAX3 p.T31S variant in Family W24 suggested the presence of a gross deletion in PAX3 in W24-1 and W24-3 (Fig. 1A). In 29 WS2 families, heterozygous mutations in SOX10 and MITF were identified in 13 and 10 of them, respectively (Fig. 1B,C). Seven of the 26 different mutations reported in this study were previously reported and 19 were novel (not seen in ExAC and 1000Genomes database). The majority (20/27) of the mutations were truncating or null mutations including nonsense mutations (n = 9), frameshifting indels (n = 8) and splicing site mutations (n = 2) and gross deletion (n = 1). They were predicted to lead to null alleles, prematurely stopped protein products or nonsense-mediate decay of the mRNA. The 4 missense mutations identified in this study all changed an evolutionarily conserved amino acid (Phylop scores >4.6) and were predicted to be disease-causing by all six computational tools Mutation Taster, Polyphen-2, MetaSVM, PROVEAN, SIFT and CADD (Table 3). The rest of the two mutations were a non-frameshifting insertion resulting in extra 7 amino acids in SOX10 and a non-stop mutation resulting in extra 51 amino acids in the C-terminus of MITF. None of the 26 mutations were seen in 300 ethnically matched normal hearing controls.
De novo rates of the SOX10 and MITF mutations in WS2 probands
Parental blood DNA samples were available for 13 WS2 probands with SOX10 (n = 8) and MITF (n = 5) mutations. For 7 of the 8 WS2 probands with SOX10 mutations, the corresponding mutations were not detected in either of the parents, suggesting that the mutations occurred de novo (Fig. 1B, de novo mutations were underlined). In contrast, for all 5 WS2 probands with MITF mutations, the corresponding mutations can be detected in one of the parents (Fig. 1C). The difference of de novo rates between SOX10 and MITF mutations was statistically significant (P = 0.005).
Genotype-phenotype correlation of the SOX10 and MITF mutations in WS2
The pigmentary phenotypes of the WS2 probands with SOX10 (n = 13) and MITF (n = 10) mutations were compared in Table 4. While the percentages of WS2 probands with heterochromic iridis, premature graying of the hair or patchy de-pigmentation of the skin were not significantly different between those with SOX10 and MITF mutations (P > 0.05), excessive freckle (as shown in previous reports7,8), a common WS2 phenotype in Chinese Han patients, was frequent in WS2 probands with MITF mutations (7/10) but absent in those with SOX10 mutations (0/13, P = 4.9 × 10−4). Logistic regression analysis showed that this excessive freckle phenotype was not affected by other confounding factors such as gender or age of the patients (P = 0.8129 and 0.1559, respectively, Supplementary Table S1).
Discussion
In this study, we aimed to explore the molecular etiology and genotype-phenotype correlation in deaf patients associated with WS. Among 36 probands, 7 and 29 can be classified as WS1 and WS2, respectively, based on their clinical phenotype (Table 1) and molecular diagnosis (Table 2), showing that WS1 and WS2 were two major WS subtypes associated with hearing loss. While mutations in PAX3 were the major cause for WS1 (6/7), the molecular etiology of WS2 was heterogeneous and attributable to two major causative genes SOX10 (13/29) and MITF (10/29).
By parental genotyping, we revealed an interesting inheritance pattern in WS2, as de novo mutations were frequently found in WS2 probands with SOX10 mutations (7/8) but not in those with MITF mutations (0/5, P = 0.005). Notably, no evidence suggested that the seven de novo mutations in SOX10 were from mutation hot-spots, as they occurred at different nucleotide positions and six of them were not previously reported (Table 2). The high de novo rates of SOX10 mutations in WS2 may need special attention during the course of genetic diagnosis and counseling, as it can be initially mistaken as recessive inheritance prior to testing or be interpreted with over-estimated recurrent risk without further parental testing.
Consistent with its pathogenic role in WS2, MITF encodes a basic helix-loop-helix, leucine zipper transcription factor that plays a critical function in survival and differentiation of melanocytes that produce melanin pigments9. SOX10, the SRY-related transcription factor, binds to MITF promoter and directly activates MITF’s expression10. Mutations in those two genes, therefore, are likely involved in the pathogenesis of WS2 through the same pathway and produce similar clinical phenotypes. Based on previous reports, the clinical features of WS2 were indeed indistinguishable between that resulted from SOX10 and MITF mutations11. In this study, we compared the pigmentary abnormalities between WS2 probands with SOX10 and MITF mutations (Table 4). Both previous reports and the present study showed that excessive freckle was frequently observed in Chinese Han WS patients7. Interestingly, this special subtype of cutaneous pigmentary disturbance appeared to be unique for WS2 probands with MITF mutations (7/10) but was absent in those with SOX10 mutations (0/13, P = 4.9 × 10−4). To our knowledge, this is the first report showing the clinical differences between WS2 patients with SOX10 and MITF mutations.
In conclusion, our study revealed that WS2 due to SOX10 and MITF mutations have discrepant de novo mutation rates and distinguishable clinical feature in excessive freckle. These new findings may facilitate precise diagnosis and genetic counseling of the heterogeneous WS2.
Methods and Materials
Patients
Thirty-six Chinese Han deaf probands clinically diagnosed with WS were recruited through their visit to the Department of Otolaryngology-Head and Neck Surgery, Xinhua Hospital, Shanghai, China. Fourteen of them reported family history of WS features including hearing impairment, heterochromia iridis, premature graying of the hair, excessive freckle and patchy de-pigmentation of the skin. A total of 16 additional affected members from 9 families (marked by asterisks in Fig. 1) were subsequently recruited into this study. All patients or guardians gave written, informed consent to participate in this study. This study was approved by the Ethics Committee of Xinhua Hospital, Shanghai Jiaotong University School of Medicine and was in compliance with the Declaration of Helsinki.
Clinical evaluations
Comprehensive auditory, ophthalmologic, dermatologic and neurological examinations were performed on all subjects. Auditory evaluations included otoscope examination, tympanometry, pure-tone audiometry (PTA) and/or auditory brainstem response (ABR, used for subjects with very young age). Degree of hearing impairment was calculated as the average of the hearing levels at 0.5, 1.0, 2.0 and 4.0 KHz for the better ear. The severity of hearing impairment was defined as mild (20–40 dB), moderate (41–70 dB), severe (71–95 dB) and profound (>95 dB). Special attention was given to the pigmentary abnormalities of iris, skin and hair as well as the developmental defects including dystopia canthorum, limb abnormalities and intestinal aganglionosis. W-index, the biometric index of dystopia canthorum was measured as previously described1. The patients were categorized into subtypes of WS according to the criteria proposed by the WS consortium1.
Mutation analysis
Genomic DNA was extracted from peripheral blood samples using the Blood DNA kit (TIANGEN Biotech, Beijing, China). Mutation screening of PAX3, MITF, SOX10, SNAI2, EDN3 and EDNRB was performed by polymerase chain reaction (PCR) amplification and sequencing of all exons and flanking splicing sites. Possible pathogenic effects of the missense mutations were evaluated by computational tools including Mutation Taster, Polyphen-2, MetaSVM, PROVEAN (cut-off score <−1.3), SIFT (cut-off score <0.05) and CADD (cut-off score >20).
Statistical analysis
Fisher’s exact test was used to compare: 1) percentages of WS2 probands carrying de novo mutations between those with MITF and SOX10 mutations; 2) percentages of WS2 probands exhibiting various pigmentary phenotypes between those with MITF and SOX10 mutations (The P-value thresholds of significance were set as 0.05/4 = 0.0125 for Bonferroni correction of multiple testing in the latter). P-values were presented as the result of the two-tailed analysis. Logistic regression analysis was performed to test the potential confounding effects of gender and age on the excessive freckle phenotype.
Additional Information
How to cite this article: Sun, L. et al. Molecular etiology and genotype-phenotype correlation of Chinese Han deaf patients with type I and type II Waardenburg Syndrome. Sci. Rep. 6, 35498; doi: 10.1038/srep35498 (2016).
References
Read, A. P. & Newton, V. E. Waardenburg syndrome. Journal of medical genetics 34, 656–665 (1997).
Newton, V. Hearing loss and Waardenburg’s syndrome: implications for genetic counselling. J Laryngol Otol 104, 97–103 (1990).
Puffenberger, E. G. et al. A missense mutation of the endothelin-B receptor gene in multigenic Hirschsprung’s disease. Cell 79, 1257–1266 (1994).
Hoth, C. F. et al. Mutations in the paired domain of the human PAX3 gene cause Klein-Waardenburg syndrome (WS-III) as well as Waardenburg syndrome type I (WS-I). American journal of human genetics 52, 455–462 (1993).
Pingault, V. et al. Review and update of mutations causing Waardenburg syndrome. Human mutation 31, 391–406 (2010).
Selicorni, A., Guerneri, S., Ratti, A. & Pizzuti, A. Cytogenetic mapping of a novel locus for type II Waardenburg syndrome. Hum Genet 110, 64–67 (2002).
Chen, H. et al. Novel mutations of PAX3, MITF, and SOX10 genes in Chinese patients with type I or type II Waardenburg syndrome. Biochemical and biophysical research communications 397, 70–74 (2010).
Yang, T. et al. Double heterozygous mutations of MITF and PAX3 result in Waardenburg syndrome with increased penetrance in pigmentary defects. Clin Genet 83, 78–82 (2013).
Hodgkinson, C. A. et al. Mutations at the mouse microphthalmia locus are associated with defects in a gene encoding a novel basic-helix-loop-helix-zipper protein. Cell 74, 395–404 (1993).
Verastegui, C., Bille, K., Ortonne, J. P. & Ballotti, R. Regulation of the microphthalmia-associated transcription factor gene by the Waardenburg syndrome type 4 gene, SOX10. J Biol Chem 275, 30757–30760 (2000).
Toriello, H. V. Pigmentary anomalies and hearing loss. Adv Otorhinolaryngol 70, 50–55 (2011).
Acknowledgements
This research was supported by grants from National Science Foundation of China (81570930 and 81371101 to TY, 81330023 to HW), Shanghai Municipal Science and Technology Commission (14DZ1940102 to TY; 14DZ2260300 and 14DJ1400201 to HW), Shanghai Municipal Education Commission – Gaofeng Clinical Medicine Grant (20152519 to TY) and the First Affiliated Hospital of Zhengzhou University (to XL).
Author information
Authors and Affiliations
Contributions
Conceived and designed the experiments: T.Y. and H.W. Performed the experiments: L.S., X.L., X.P. and X.W. Analyzed the data: T.Y., L.S., Y.H. and X.L. Collected the samples and reviewed the phenotypes: T.Y., H.W., J.S., L.S., X.P. and X.L. Wrote the paper: T.Y. and L.S.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Sun, L., Li, X., Shi, J. et al. Molecular etiology and genotype-phenotype correlation of Chinese Han deaf patients with type I and type II Waardenburg Syndrome. Sci Rep 6, 35498 (2016). https://doi.org/10.1038/srep35498
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep35498
- Springer Nature Limited
This article is cited by
-
A novel variant of the SOX10 gene associated with Waardenburg syndrome type IV
BMC Medical Genomics (2023)
-
Quantitative assessment of low-level parental mosaicism of SNVs and CNVs in Waardenburg syndrome
Human Genetics (2023)
-
A comprehensive genotype–phenotype evaluation of eight Chinese probands with Waardenburg syndrome
BMC Medical Genomics (2022)
-
Predicting pathogenicity for novel hearing loss mutations based on genetic and protein structure approaches
Scientific Reports (2022)
-
Analysis of genotype–phenotype relationships in 90 Chinese probands with Waardenburg syndrome
Human Genetics (2022)