
Abstract
Identification of loci or genes under natural selection is important for both understanding the genetic basis of local adaptation and practical applications, and genome scans provide a powerful means for such identification purposes. In this study, genome-wide simple sequence repeats markers (SSRs) were used to scan for molecular footprints of divergent selection in Eucalyptus grandis, a hardwood species occurring widely in costal areas from 32° S to 16° S in Australia. High population diversity levels and weak population structure were detected with putatively neutral genomic SSRs. Using three FST outlier detection methods, a total of 58 outlying SSRs were collectively identified as loci under divergent selection against three non-correlated climatic variables, namely, mean annual temperature, isothermality and annual precipitation. Using a spatial analysis method, nine significant associations were revealed between FST outlier allele frequencies and climatic variables, involving seven alleles from five SSR loci. Of the five significant SSRs, two (EUCeSSR1044 and Embra394) contained alleles of putative genes with known functional importance for response to climatic factors. Our study presents critical information on the population diversity and structure of the important woody species E. grandis and provides insight into the adaptive responses of perennial trees to climatic variations.
Similar content being viewed by others


Introduction
Natural populations may survive climate change through migration and local adaptation1,2. In sessile plants, while migration involves propagule (i.e. seed and plant fragment) dispersal and population establishment in new locations2, local adaptation represents higher fitness of local than nonlocal populations resulting from divergent selection among environments3. As adaptation is often inherent in and interplayed with migration2, migration can not be viewed simply as an alternative to occurrence of local adaptation4. With the occurrence of rapid climate change, the migration rates observed commonly (e.g. 20–40 km per century) are far below the rates projected (e.g. 300–500 km per century) to track future climate shifts1, and local adaptation to future climatic conditions will likely be necessary for long-term in situ population persistence5.
Divergent natural selection, as exerted usually by environmental gradients, is the major driving force of local adaptation. Such selection can result in gene frequency changes among populations within a species, ultimately leading to variation in both phenotypic traits and genetic structure even in the presence of gene flow6,7. Identifying the effects of loci or genes under natural selection is important for elucidating the genetic basis of adaptation to different environments and also for practical applications in biodiversity conservation and selective breeding8,9. Using a candidate gene or genome scan approach, genomic loci associated with natural selection could be identified through linkage disequilibrium (LD) and quantitative trait locus mapping of adaptive traits, detection of between-population differentiation (FST) outlier loci and analysis of alleles that are correlated with environmental variables8,10.
Genome scans provide a powerful means in population genetics studies to detect outlier loci putatively under selection11,12 and identify alleles associated with environmental variables13. In contrast to a candidate gene approach, in which a limited set of candidate genes potentially influencing the adaptive trait are used, genome scans investigate a large number of loci throughout the genome even in the absence of phenotypic information14. To date, genome scans have been extensively employed for detecting loci or genes under selection in model organisms (e.g. Zea mays L.15), but its potential still remains to be explored for most non-model species10,16. Moreover, while most genome scan studies on natural selection have used high-throughput array-based single nucleotide polymorphism markers, in which the finite number of markers (at least in non-model species) and the marker nature of bi-allelism can limit the genotyping of all variations at a genomic region, few studies have attempted to use multi-allelic markers, such as microsatellites (or simple sequence repeats, SSR).
The broadleaved tree genus Eucalyptus L’Hér. (family Myrtaceae) encompasses more than 780 species and subspecies native to the Australian continent and surrounding islands17. It is highly diverse and displays significant adaptability and phenotypic plasticity18. With the projection of pronounced temperature warming and rainfall decline in Australia, eucalypts are particularly interesting for climate change studies, considering their poor dispersal capability and limited gene flow19. Also, Eucalyptus species and hybrids constitute the most widely planted hardwood trees in the world (plantations >20 million ha, http://git-forestry.com/Global_Eucalyptus_Map.htm) and are therefore important for the global forest-related industries. Within Eucalyptus, E. grandis Hill ex Maiden (flooded gum or rose gum) is one of the most important species in terms of breeding and genomics efforts18 and has been selected as the second tree species (only after Populus trichocarpa Torr. & Gray) for whole genome sequencing20. E. grandis is a diploid species with 2n = 22 chromosomes and 640 Mb genome size20. It is distributed naturally mainly in coastal areas from northern New South Wales (32° S in latitude) to southern Queensland (26° S) in Australia, with two outlier patches in central (22° S) and northern (16−18° S) Queensland. Its natural forests are mostly on low lands and hills ranging from sea level to ~600 m in altitude21. Of note, E. grandis displays significant variation in adaptive phenotypic traits, such as growth and frost tolerance (e.g. Rockwood & Meskimen22). A relatively low level of population differentiation has been revealed in E. grandis by isozyme markers (GST = 0.12)23, suggesting a weak population structure. Thus far, genomic locus-association analyses have detected loci with significant effects on economic traits in E. grandis and other Eucalyptus species18. However, there have been no association studies specifically aiming at local adaptation in E. grandis. In addition, even in Eucalyptus, only two studies have recently reported candidate loci for diversifying selection in natural populations of E. gomphocephala DC.24 and E. tricarpa (L.A.S. Johnson) L.A.S. Johnson & K.D. Hill25.
In this study, we used 110 SSR markers distributed across the genome sequence of E. grandis, including 45 genomic SSR (gSSR) and 65 expressed sequence tag (EST) derived SSR (EST-SSR) markers, to search for molecular footprints of divergent selection in E. grandis natural populations. The genetic diversity and population structure at putatively neutral gSSR loci were first assessed for 16 E. grandis populations across the species range. The FST outlier SSRs were then tested against the 110 SSRs to identify loci potentially involved in divergent natural selection. SSR alleles associated with climatic variables were finally investigated to reveal gene polymorphisms contributing to local adaptation to temperature, isothermality and/or precipitation.
Materials and Methods
Population sampling and DNA extraction
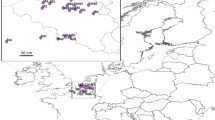
A total of 159 individuals were sampled from 16 provenances across the range of E. grandis (Fig. 1 and Table 1), each provenance representing one population and each individual originating from an open-pollinated mother tree in the native forests. The sample size ranged from nine to twelve individuals per population (Table 1). The mother trees were at least 100 m apart to minimise the risk of collecting seed from closely related trees (R Arnold, Australian Tree Seed Centre, Australia, pers. comm.). The original provenance seedlots had been provided by Australian Tree Seed Centre (Canberra, Australia). The leaf samples were stored at −80 °C prior to DNA extraction. Leaf samples were collected in July 2011 from a 2-year-old E. grandis provenance-family trial located at Zhaoqing City (112°27′ E, 23°03′ N), Guangdong Province, China.

Geographic distribution of the 16 Eucalyptus grandis populations studied.
The map was generated using software ArcGIS 10.0 (http://www.esri.com/software/arcgis/). Full description of the populations can be found in Table 1. SF, state forest; ACT, Australian Capital Territory.
Leaf tissues (~300 mg) were powder homogenized in a MM400 mixer mill (Retsch GmbH, Haan, Germany), and genomic DNA was subsequently extracted using a modified CTAB method26. DNA concentration and quality were determined by agarose gel electrophoresis and a Nanodrop 2000 spectrophotometer (Thermo Fisher Scientific Inc., Waltham, MA, USA).
Microsatellite markers and their genotyping
In total, 110 SSR markers distributed across the 11 main scaffolds and one small scaffold (Supplementary Table S1) were used in this study, including 45 gSSRs27,28 and 65 EST-SSRs29,30,31. Polymerase chain reactions (PCRs) were performed in a 10 μL volume on a DNA Engine (Bio-Rad Laboratories, Hercules, CA, USA) following a florescent-dUTP based protocol32, with MgCl2 concentration and melting temperature (Tm) depending on marker. The PCR products (1 μL) were diluted 1:10.5 with loading buffer (9.34 μL deionized formamide and 0.16 μL GeneScan 500LIZ) and then detected on an ABI 3130xl genetic analyzer using GeneMapper 4.1 software (Applied Biosystems, Foster City, CA, USA). Sixteen individuals, each randomly selected from one population, were PCR repeated to test the marker reproducibility.
Marker polymorphism, population diversity and structure
For each locus, number of alleles (NA), observed heterozygosity (HO), expected heterozygosity (HE) and fixation index (F) were estimated with GENALEX 6.4.133. Polymorphic information content (PIC) was computed with PowerMarker 3.2534. Null allele frequencies (NAFs) were assessed following 50,000 bootstrap resamples in FREENA35. Hardy-Weinberg equilibrium (HWE, with manual Bonferroni correction for P values) was tested over all populations with GENEPOP 4.236. The software FSTAT 2.9.3.2 (http://www2.unil.ch/popgen/softwares/fstat.htm) was used to investigate per-locus inbreeding coefficients of individuals relative to the total population (FIT) and to the sub-population (FIS), FST and allelic richness (AR, based on the minimal sample size of nine individuals) as well as between-locus LD (with Bonferroni correction).
Only the genomic SSRs that neither departed significantly from HWE (P < 0.01) nor showed FST outlying values in lositan analysis on 16 populations (implemented as stated below) were included (31 markers, see Results) in subsequent population diversity and structure analyses. For each population, average number of alleles per locus (ANA), HO, HE, F and AR were estimated similarly as above. Significance in diversity differences between populations or population groups was tested using the ‘Compare among groups of samples’ function with ‘two sided’ option and 1,000 permutations in fstat 2.9.3.2 (http://www2.unil.ch/popgen/softwares/fstat.htm). Nei’s genetic distance between populations was calculated for construction of an unweighted pair group method with arithmetic mean (UPGMA) dendrogram in POWERMARKER 3.2534. The package GENALEX 6.4.133 was used under default settings to conduct analyses of principal coordinates (PCoA), overall between-population differentiations (FST) and molecular variances (AMOVA). The Bayesian clustering program STRUCTURE 2.3.437 was performed to assign individuals to a number (K) of genetically homogeneous clusters assuming an admixture model and correlated allele frequencies between populations. For each of K values (1−16), the Markov Chain Monte Carlo (MCMC) sampling was replicated with 10 iterations38 each following 100,000 burn-ins and 100,000 MCMC repetitions. The optimal K value was determined by the highest ΔK method39 in STRUCTURE HARVESTER 0.640 and by a complementary assessment of the change in lnP(K) slope with increase in each K value.
Climatic data and partitioning of populations
Values of 19 climatic variables during the years 1950–2000 were obtained for each original population location from a standard set of climate grids (http://www.worldclim.org/) at a spatial resolution of 30 arc-seconds41. Correlations between the climatic variables were computed using R function COR.TEST, and only three non-correlated variables (mean annual temperature, isothermality and annual precipitation, see Results) were included in subsequent association analysis of candidate loci.
The 16 populations were partitioned into homogeneous groups using a k-means analysis on each of the non-correlated climatic variables42, as implemented similarly in black spruce [Picea mariana (Mill.) B.S.P.]43.
Detection of FST outlier loci
Three FST outlier detection methods were used to test all the 110 SSR loci for evidence of natural selection among the population groups of each climatic partition. The first was a summary-statistics method44 implemented in LOSITAN45. The outlying values of FST were identified from a plot of FST vs. expected heterozygosity. Following the infinite allele mutation model, the distribution of FST vs. expected heterozygosity was calculated in LOSITAN with 100,000 simulation replicates under the option of neutral mean FST. Markers outside the 95% and 5% confidence intervals of the distribution were considered as candidates (outliers) for positive (divergent) and balancing selections, respectively. False discovery rate (FDR) was set to 0.01.
The second was also a summary-statistics method44 modified for hierarchically structured populations46 as implemented in ARLEQUIN 3.547. As the hierarchical structure was considered, coalescent simulations (500,000) were implemented under a hierarchical island model to obtain more realistic null distribution for FST statistics and therefore avoid possible false positives. Significant loci at the 95% or 5% confidence level were recognized as candidates for positive (divergent) selection or balancing selection, respectively.
The third was a hierarchical Bayesian modeling method modified from Beaumont and Balding48 and implemented in BAYESCAN 2.149. Three runs were performed in BAYESCAN with standard parameters, except 100 prior odds for the neutral model. FDR was set to 0.01. A Bayesian factor and its logarithm value were generated for each marker and thereafter classified into five grades according to Jeffrey’s scale of evidence of selection (http://cmpg.unibe.ch/software/bayescan/index.html), namely, barely worth mentioning, substantial, strong, very strong and decisive evidences, in which the last three grades were considered as candidates for divergent selection.
Detection of candidate SSR loci associated with environmental variables
The FST outlier loci detected above were corroborated using a spatial analysis method (SAM)13. The associations between allele frequencies and climatic variables were tested using the likelihood ratio (G) and Wald tests13. With the Bonferroni correction, the significance was declared at 99.99% confidence level, and a conservative alpha of 0.01 was adopted in the Wald test to reduce false positives. Moreover, significant associations were further evaluated in OFFICE EXCEL 2010 (Microsoft Corp., Redmond, WA, USA) to correct allele frequency autocorrelation using univariate linear regressions between group allele frequencies and environmental variables.
Results
Marker polymorphism, population diversity and structure
In total, 1,857 alleles were identified at the 110 polymorphic SSR loci. Single-locus parameters exhibited striking differences among loci (Supplementary Table S2), with NA ranging from three to 52 (mean 16.9), HO from 0.209 to 1.000 (mean 0.623), HE from 0.200 to 0.890 (mean 0.706), AR from 1.434 to 3.835 (mean 3.027) and NAF from zero to 0.278 (mean 0.075). No pair of loci showed significant LD in two or more populations (P < 0.05), suggesting the independent segregation of the marker loci used. Twenty-eight loci deviated significantly from HWE (P < 0.01), including 12 gSSRs and 16 EST-SSRs (Supplementary Table S2). The 12 non-HWE gSSRs, along with gSSR loci (eight, including six non-HWE loci; Supplementary Fig. S1 and Table S2) that exhibited FST outlying values for the 16 populations, were excluded from subsequent population diversity and structure analyses.
High levels of putatively neutral (gSSR) diversity were revealed in E. grandis populations (Table 2), e.g. HE ranging from 0.706 to 0.809 (mean 0.774) and AR from 4.295 to 5.300 (mean 4.929). Specifically, the five northern populations (codes Pic, Cop, MS, Fin and Cre, mean HO = 0.711) were significantly less diverse as compared to the remaining (southern) populations (mean HO = 0.758, P = 0.007). The fixation index (F) per population suggested little evidence of inbreeding, with a range between zero and 0.127 (mean 0.037). The overall FST values were generally low as demonstrated by between-population comparisons (mean FST = 0.037; Supplementary Table S3) and AMOVA analysis (among-population variation percentage being 3.7%, P < 0.001; Supplementary Table S4), indicating a weak population structure.
Similar patterns of neutral population structure were observed in PCoA analysis, UPGMA dendrogram and STRUCTURE analysis (Fig. 2). For PCoA analysis (Fig. 2a), the first coordinate accounted for 33.2% of the variation and separated clearly the northern and southern populations. Further, the UPGMA dendrogram (Fig. 2b) based on Nei’s genetic distance confirmed the clustering of northern vs. southern populations, coinciding well with geographic distribution. Furthermore, the STRUCTURE analysis (Fig. 2c) illustrated the differentiation between the northern and southern populations, providing evidence for the presence of two genetically distinct clusters (i.e. K = 2).

Genetic structure of 16 Eucalyptus grandis populations based on 31 putatively neutral gSSR loci.
Full description of the populations can be found in Table 1. (a) Principal coordinates analysis (PCoA). (b) Unweighted pair group method with arithmetic mean (UPGMA) dendrogram. (c) Individual proportion and population membership to each of the clusters inferred in STRUCTURE analysis (K = 2).
Climatic data and partitioning of populations
Of the 19 climatic variables obtained (http://www.worldclim.org/), only three were not correlated with each other, namely, mean annual temperature, isothermality and annual precipitation. The k-means analysis partitioned optimally the 16 populations into six, four and four groups in mean annual temperature, isothermality and annual precipitation, respectively (Table 1). Each group comprised two or more populations, with the only exception of one population in group 4 for isothermality partition.
FST outlier loci
A total of 58 FST outlier loci were identified collectively using LOSITAN, ARLEQUIN and BAYESCAN against the three non-correlated climatic factors (Table 3), including 35 (31.8%, 24 positive and 11 balancing selections; Table 3), 22 (20.0%, 10 positive and 12 balancing selections; Table 3) and 20 (18.2%, 13 positive and seven balancing selections; Table 3) outliers for mean annual temperature, isothermality and annual precipitation, respectively. Four outliers were detected in all three climatic partitions, namely, EUCeSSR0755, EUCeSSR1070, Embra394 and EUCeSSR0893. In addition, the rates of EST-SSR outliers (20.0–33.8%) for all the climatic partitions were significantly greater (P = 0.006) than those of genomic SSRs (15.6–28.9%; Table 3).
LOSITAN, ARLEQUIN and BAYESCAN detected consistently the largest, medium and least numbers, respectively, of FST outlying loci for all the three climatic partitions (Table 3 and Supplementary Fig. S2). There were 11 (10.0%), 10 (9.1%) and five (4.5%, Table 3) outliers identified simultaneously in LOSITAN and ARLEQUIN analyses for mean annual temperature, isothermality and annual precipitation, respectively, while only one outlier locus EUCeSSR0893 (0.9%; Table 3) identified in both LOSITAN and ARLEQUIN was detected with BAYESCAN.
Loci and alleles associated with climatic variables
A total of nine significant associations were detected between FST outlier allele frequencies and climatic variables, involving seven alleles from five loci (Table 4). Regression analysis indicated that three of the associations were significantly negatively linear (Fig. 3) while the remaining six were non-linear (Supplementary Fig. S3). Five, two and two alleles were significantly associated with mean annual temperature, isothermality and annual precipitation, respectively. All alleles exhibited significant association with only one climatic factor, with the exceptional allele EUCeSSR0755-276 bp associated with all the three climatic factors. In addition, two alleles (266 and 276 bp) of the same locus EUCeSSR0755 were significantly associated with the same climatic factor, mean annual temperature.

Linear regression for three significant associations between FST outlier allele frequencies and climatic variables.
Each dot represents a group of homogeneous populations in K-means climatic partition. (a) The 120 bp allele of locus Embra180 associated with mean annual temperature. (b,c) The 276 bp allele of EUCeSSR0755 associated with mean annual temperature and isothermality, respectively.
Discussion
High genetic diversity and low population structure in E. grandis
Despite the large geographical (latitudinal) range, E. grandis populations show high levels of neutral genetic diversity (mean HE = 0.774; Table 2), consistent with SSR-derived population diversity in other eucalypt species, e.g. HE being 0.82 in E. globulus Labill.50, 0.75 in E. gomphocephala24 and 0.739 in E. urophylla S.T. Blake51. Northern populations tend to be significantly less diverse than the southern populations, similar to the observations based on chloroplast DNA sequences52. It is possible that the relatively isolated northern populations have been colonized from the south52 as genetic diversity is generally higher in source populations than in younger populations derived from them51. Moreover, as E. grandis may cross with co-occurring close relatives such as E. saligna Smith, E. robusta Smith and E. resinifera Smith21, gene flow from the related species may have contributed to the high genetic diversity, in the southern populations in particular52. Additionally, all populations analysed here were sampled from a field trial, in which selection might have taken place during earlier seed germination, seedling establishment and juvenile survival stages, and the neutral genetic diversity could be thus more or less affected. Nevertheless, the overall high levels of genetic diversity may reflect the maintenance of large population size in E. grandis.
The low levels of population differentiation (mean FST = 0.037) are less than the mean of widespread eucalypt species, e.g. mean FST of 0.062 and 0.055 at restriction fragment length polymorphism (RFLP) and SSR loci, respectively53. These results confirm also earlier findings of limited population structure in E. grandis based on isozyme markers23. Generally speaking, forest tree species including eucalypts have weak population structure54. Such weak population structure, in light of the large geographical disjunctions in the natural range of E. grandis (e.g. populations MS vs. Fin and Cre vs. Kin) that could contribute as a barrier to gene flow and induce genetic differentiation, may reflect recent emergence of the disjunctions from a more continuous distribution due to climate change52.
In this study, besides the regions within E. grandis distribution range sampled earlier by Jones et al.52, an additional isolated region (populations Fin and Cre) in central coastal Queensland was included, providing a more comprehensive picture of the population diversity and structure. These results can have important implications for conservation and utilization of the species. The high levels of population diversity and certain genetic differentiation among populations detected herein could imply the great potential of further genetic resource exploitation. The populations featured with high diversity and/or rich private alleles, particularly those under potential threat by predicted climate change, should be considered in future conservation programs. For the purpose of association mapping studies, the population subdivision information will be of practical importance.
Detection of FST outlier loci
Similar to the k-means partition of populations according to climatic variables implemented in genome scans with black spruce43, the 16 populations of E. grandis analysed were partitioned into homogeneous groups to identify FST outliers related with genetic divergence under specific environmental factors. Such climatic partitions may offer better odds to identify genes potentially involved in adaptation to a specific climate factor43, and those outliers specific only to a climatic factor could provide evident support in this respect. Based on the climatic partition of population groups, 35, 22 and 20 outlier loci (Table 3) were identified collectively for mean annual temperature, isothemality and annual precipitation, respectively. Of these, eight, eight and five loci presented higher genetic differentiation (FST) values in at least two outlier detection methods than expected in the respective climatic variables, suggesting that they could have been involved in divergent selection among homogeneous population groups. In contrast, 11, 12 and seven loci presented lower FST values than expected in the respective variables, signaling balancing selection within population groups and/or homogeneous selection among groups. In addition, quantitative trait differentiation coefficients (QST) are 0.33 and 0.28 in 7-year-old tree height and diameter at breast height, respectively, for a range-wide provenance/family trial of E. grandis (Trial E110-155; J Luo, China Eucalypt Research Centre, China, pers. comm., assuming an overall coefficient of relatedness of 0.4 amongst open-pollinated siblings), being much larger than the mean FST (0.037; Supplementary Table S3) and thus indicating the role of divergent selection on quantitative traits56 (our field trial was not used for estimating QST due to abnormal growth caused by infection of gall wasp, Leptocybe invasa Fisher and La Salle).
The rates of FST outlier loci detected totally with all the three methods (18.2–31.8% depending on climatic partition; Table 3) are parallel to those reported in several forest trees, e.g. 16.7% (3/18) at genomic SSR and EST-SSR markers in E. gomphocephala24, 22–53% at gene-linked loci in Quercus robur L. and Q. petraea (Matt.) Liebl.57 and 31.3% (10/32) at EST-SSRs in Castanopsis fargesii Franchet58. However, such rates are generally greater than those reported by other studies in plants and animals, e.g. 12.0% (3/25) at SSRs in eelgrass (Zosteria marina L.)59, 11.4 % (5/44) at genomic SSRs and EST-SSRs in Q. robur and Q. ellipsoidalis E.J. Hill60 and 0.3% (18/6145) at restriction site associated DNAs (RADs) in kokanee salmon (Oncorhynchus nerka Walbaum)61. Given that only one FST outlier (0.9%; Table 3) was detected with the more conservative Bayesian method of BAYESCAN, the high proportion of outliers reported here is likely attributable to the utilization of relaxed summary-statistics approaches conducted in LOSITAN and ARLEQUIN. As a consequence, false positive outliers may exist, which remain still a challenge to be separated from real outliers59. Moreover, besides some common outliers (5–11 depending on climatic partition; Table 3) between LOSITAN and ARLEQUIN analyses, higher proportions of outliers were consistently detected by LOSITAN for the three climatic partitions as compared with those of ARLEQUIN, but it is impossible to figure out which detection method is more powerful. Furthermore, given the multiplicity of natural habitats of E. grandis and other eucalypt species, the FST outliers identified herein can be used as candidates for further genome scans against extended population samples.
Significant loci associated with climatic variables
Five (8.6%) of the FST outlier loci showed significant associations between allelic frequencies and climatic variations (Table 4). However, the remaining FST outliers should not be considered as false positives as they may be involved in adaptation on only a fraction of the climatic gradient or in part of adaptation complexes resulting from epistatic interactions43. Of the five significant loci, two (EUCeSSR1044 and Embra394) were homologous to known gene or predicted protein, but three (Embra180, EUCeSSR0755 and EUCeSSR0849) did not have significant matches or known functions when they were BlastX searched against the NCBI non-redundant protein database (http://blast.ncbi.nlm.nih.gov/Blast.cgi).
The locus EUCeSSR1044, associated significantly with annual precipitation, was functionally annotated as C3HC4 type RING finger protein. Similarly, two C3HC4 RING finger genes were identified to be associated with local precipitation in black spruce43. In eukaryotes, C3HC4 type RING finger proteins act as E3 ubiquitin ligases, targeting numerous intracellular regulators in a wide range of physiological processes62,63, including abscisic acid (ABA) mediated drought stress responses in plants64,65. The plant hormone ABA can induce stomatal closure to mitigate transpirational water loss and is the master modulator of drought responses64. Changes in C3HC4 RING finger gene expression have been reported in response to drought stress in many plants, such as Arabidopsis thaliana (L.) Heynh.64,65 and Populus species66−68. Thus, it is not surprising to find significant associations between C3HC4 type RING finger protein and annual precipitation as critically low precipitations can cause drought stress.
The marker Embra394, significantly associated with mean annual temperature, was functionally annotated as thionin-like protein 2. The thionin-like proteins have been well documented in plants to be involved in protection against pathogens, including bacteria and fungi69. Furthermore, in wheat (Triticum aestivum L.), thionin-like genes can express differently in response to heat stress70 or be involved in pathogen resistance induced by low temperatures71. As similarities in adaptation exist among phylogenetically remote plants43, thionin-like proteins may take part in temperature responses in Eucalyptus.
The remaining three loci were of unknown function, including EUCeSSR0755 which was associated with all three climatic variables and EUCeSSR0849 which was related to both mean annual temperature and isothermality (Table 4). These EST-derived loci may represent candidate genes for physiological response to environmental variation. Alternatively, they may be artefacts of genetic hitchhiking11, in which neutral markers are linked to a single gene under positive selection.
Implications for adaptation genomics in perennial trees
The relatively large number of FST outlier loci (58 in total, 52.7%; Table 3) may indicate that adaptive genetic variation is a genome-wide phenomenon72, in which multiple loci of small adaptive effect are spread across the genome. Similarly, relatively large numbers of FST outliers were revealed by genome scan in some other broadleaved trees (e.g. Po. alba L.73) and conifers (e.g. black spruce43).
Divergent natural selection can change allele frequencies and thereby increase the adaptive trait value of a population and the number of individuals with the fitness traits, leading to local adaptation6,7. Here, seven alleles were revealed to be significantly associated with the three climatic variables in E. grandis, indicating the importance of these climate factors as selective agents. Most of the significant alleles were associated with mean annual temperature followed by isothermality and annual precipitation, suggesting the critical role of mean annual temperature in determining adaptive responses. Many studies have demonstrated the influence of these climatic agents on selection and adaptation in forest trees, such as black spruce43, E. gomphocephala24, C. fargesii58 and Pi. albies (L.) Karst.74. With the occurrence of globally rapid climatic change, in temperature in particular, these results could inform the adaptive responses of perennial trees to the environment. The existence of divergent natural selection, in addition to high genetic diversity, implies the great potential for evolution of tree populations in adaptive traits in the face of climate change6. The populations that harbour the highest frequency of favourable alleles underlying the adaptive traits will be the most adaptive, and vice versa, under a climatic scenario of pronounced warming temperature and declining rainfall19.
Only one or two alleles of each significantly associated locus showed population variation with climatic variables, which may indicate that the significant alleles alter protein function in a way that the remaining alleles do not24. Given that none of the five significant loci have been directly investigated for their functional roles in perennial trees, these loci and their variation patterns described here will provide a foundation on which further functional characterisation work can rely.
In conclusion, the widespread woody species E. grandis provides an unusual opportunity to study local population adaptation in perennial trees. Although a relatively low density of multi-allelic loci were used for genome scans (only 110 SSRs spanning the E. grandis genome), we found evident footprints of divergent selection at a suite of loci. Two loci that showed significant associations with climatic variables represent alleles of putative genes with known functional importance for response to climatic factors. In addition, high genetic diversity levels and weak population structure were detected in E. grandis natural populations. These results have implications for understanding the genomic basis of adaptation to climates in perennial woody trees as well as for conservation and utilization of the important hardwood tree E. grandis. Further genetic association and expression studies would be required to confirm the functional role of putative genes. Also, next-generation sequencing technologies, including those for genotyping of multi-allelic markers (e.g. short tandem repeats75), will help to investigate fine-scale genome-wide patterns of natural selection and local adaptation.
Additional Information
How to cite this article: Song, Z. et al. Genome scans for divergent selection in natural populations of the widespread hardwood species Eucalyptus grandis (Myrtaceae) using microsatellites. Sci. Rep. 6, 34941; doi: 10.1038/srep34941 (2016).
References
Davis, M. B. & Shaw, R. G. Range shifts and adaptive responses to Quaternary climate change. Science 292, 673–679 (2001).
Jump, A. S. & Peñuelas, J. Running to stand still: adaptation and the response of plants to rapid climate change. Ecol. Lett. 8, 1010–1020 (2005).
Aitken, S. N. & Whitlock, M. C. Assisted gene flow to facilitate local adaptation to climate change. Ann. Rev. Ecol. Evol. Syst. 44, 367–388 (2013).
Jackson, S. T. & Overpeck, J. T. Responses of plant populations and communities to environmental changes of the late Quaternary. Paleobiology 26, 194–220 (2000).
Anderson, J. T., Inouye, D. W., McKinney, A. M., Colautti, R. I. & Mitchell-Olds, T. Phenotypic plasticity and adaptive evolution contribute to advancing flowering phenology in response to climate change. Proc. Royal Soc. B: Biol. Sci. 279, 3843–3852 (2012).
Savolainen, O., Pyhäjärvi, T. & Knürr, T. Gene flow and local adaptation in trees. Ann. Rev. Ecol. Evol. Syst. 38, 595–619 (2007).
Thurman, T. J. & Barrett, R. D. H. The genetic consequences of selection in natural populations. Mol. Ecol. 25, 1429–1448 (2016).
Savolainen, O., Lascoux, M. & Merilä, J. Ecological genomics of local adaptation. Nat. Rev. Genet. 14, 807–820 (2013).
Luikart, G., England, P. R., Tallmon, D., Jordan, S. & Taberlet, P. The power and promise of population genomics: from genotyping to genome typing. Nat. Rev. Genet. 4, 981–994 (2003).
Pardo-Diaz, C., Salazar, C. & Jiggins, C. D. Towards the identification of the loci of adaptive evolution. Method. Ecol. Evol. 6, 445–464 (2015).
Schlötterer, C. Hitchhiking mapping – functional genomics from the population genetics perspective. Trends Genet. 19, 32–38 (2003).
Teshima, K. M., Coop, G. & Przeworski, M. How reliable are empirical genomic scans for selective sweeps? Genome Res. 16, 702–712 (2006).
Joost, S. et al. A spatial analysis method (SAM) to detect candidate loci for selection: towards a landscape genomics approach to adaptation. Mol. Ecol. 16, 3955–3969 (2007).
Stinchcombe, J. R. & Hoekstra, H. E. Combining population genomics and quantitative genetics: finding the genes underlying ecologically important traits. Heredity 100, 158–170 (2008).
Wright, S. I., Bi, I. V., Schroeder, S. G., Yamasaki, M. & Doebley, J. F. The effects of artificial selection on the maize genome. Science 308, 1310–1314 (2005).
Bragg, J. G., Supple, M. A., Andrew, R. L. & Borevitz, J. O. Genomic variation across landscapes: insights and applications. New Phytol. 207, 953–957 (2015).
Slee, A. V., Brooker, M. I. H., Duffy, S. M. & West, J. G. EUCLID – Eucalypts of Australia, 3rd edn. CD (CSIRO Publishing, 2006).
Grattapaglia, D. et al. Progress in Myrtaceae genetics and genomics: Eucalyptus as the pivotal genus. Tree Genet. Genomes 8, 463–508 (2012).
Booth, T. H. et al. Native forests and climate change: Lessons from eucalypts. For. Ecol. Manag. 347, 18–29 (2015).
Myburg, A. A. et al. The genome of Eucalyptus grandis. Nature 510, 356–362 (2014).
FAO. Eucalypts for Planting. 422–424 (Food and Agriculture Organization of the United Nations, 1979).
Rockwood, D. L. & Meskimen, G. F. Comparison of Eucalyptus grandis provenances and seed orchards in a frost frequent environment. South Afr. For. J. 159, 51–59 (1991).
Burgess, I. P. & Bell, J. C. Comparative morphology and allozyme frequencies of Eucalyptus grandis Hill ex Maiden and E. saligna Sm. Aust. For. Res. 13, 133–149 (1983).
Bradbury, D., Smithson, A. & Krauss, S. L. Signatures of diversifying selection at EST-SSR loci and association with climate in natural Eucalyptus populations. Mol. Ecol. 22, 5112–5129 (2013).
Steane, D. A. et al. Genome-wide scans detect adaptation to aridity in a widespread forest tree species. Mol. Ecol. 23, 2500–2513 (2014).
Gan, S. et al. Moderate-density molecular maps of Eucalyptus urophylla S. T. Blake and E. tereticornis Smith genomes based on RAPD markers. Genetica 118, 59–67 (2003).
Brondani, R. P. V., Williams, E. R., Brondani, C. & Grattapaglia, D. A microsatellite-based consensus linkage map for species of Eucalyptus and a novel set of 230 microsatellite markers for the genus. BMC Plant Biol. 6, 20 (2006).
He, X., Li, F., Shi, J. & Gan, S. Seven genomic SSR markers revealed in Eucalyptus by re-sequencing of DNA sequences from GenBank. Silvae Genet. 60, 92–94 (2011).
Zhou, C. et al. Comparison between direct sequencing and pool-cloning-based sequencing of PCR products in EST-SSR marker development in Eucalyptus. Mol. Plant Breed. 8, e1 (2010).
He, X. et al. Development of 198 novel EST-derived microsatellites in Eucalyptus (Myrtaceae). Am. J. Bot. 99, e134–e148 (2012).
Zhou, C. et al. Development of 240 novel EST-SSRs in Eucalyptus L’Hérit. Mol. Breed. 33, 221–225 (2014).
Li, F. & Gan, S. An optimised protocol for fluorescent-dUTP based SSR genotyping and its application to genetic mapping in Eucalyptus. Silvae Genet. 60, 18–25 (2011).
Peakall, R. & Smouse, P. GenAlEx 6: genetic analysis in Excel. Population genetic software for teaching and research. Mol. Ecol. Notes 6, 288–295 (2006).
Liu, K. & Muse, S. V. PowerMarker: an integrated analysis environment for genetic marker analysis. Bioinformatics 21, 2128–2129 (2005).
Chapuis, M. P. & Estoup, A. Microsatellite null alleles and estimation of population differentiation. Mol. Biol. Evol. 24, 621–631 (2007).
Rousset, F. Genepop'007: a complete reimplementation of the Genepop software for Windows and Linux. Mol. Ecol. Resour. 8, 103–106 (2008).
Falush, D., Stephens, M. & Pritchard, J. K. Inference of population structure: extensions to linked loci and correlated allele frequencies. Genetics 164, 1567–1587 (2003).
Gilbert, K. J. et al. Recommendations for utilizing and reporting population genetic analyses: the reproducibility of genetic clustering using the program STRUCTURE. Mol. Ecol. 21, 4925–4930 (2012).
Evanno, G., Regnaut, S. & Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol. Ecol. 14, 2611–2620 (2005).
Earl, D. A. & vonHoldt, B. M. STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 4, 359–361 (2012).
Hijmans, R. J., Camerson, S. E., Parra, J. L., Jones, P. G. & Jarvis, A. Very high resolution interpolated climate surfaces for global land areas. Int. J. Climatol. 25, 1965–1978 (2005).
Legendre, P. & Legendre, L. Numerical Ecology, 2nd edn (Elsevier, 1998).
Prunier, J., Laroche, J., Beaulieu, J. & Bousquet, J. Scanning the genome for gene SNPs related to climate adaptation and estimating selection at the molecular level in boreal black spruce. Mol. Ecol. 20, 1702–1716 (2011).
Beaumont, M. A. & Nichols, R. A. Evaluating loci for use in the genetic analysis of population structure. Proc. Royal Soc. B: Biol. Sci. 263, 1619–1626 (1996).
Antao, T., Lopes, A., Lopes, R. J., Beja-Pereira, A. & Luikart, G. LOSITAN: a workbench to detect molecular adaptation based on a Fst-outlier method. BMC Bioinformatics 9, 323 (2008).
Excoffier, L., Hofer, T. & Foll, M. Detecting loci under selection in a hierarchically structured population. Heredity 103, 285–298 (2009).
Excoffier, L. & Lischer, H. E. L. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 10, 564–567 (2010).
Beaumont, M. A. & Balding, D. J. Identifying adaptive genetic divergence among populations from genome scans. Mol. Ecol. 13, 969–980 (2004).
Foll, M. & Gaggiotti, O. A. genome-scan method to identify selected loci appropriate for both dominant and codominant markers: a Bayesian perspective. Genetics 180, 977–993 (2008).
Steane, D. A., Conod, N., Jones, R. C., Vaillancourt, R. E. & Potts, B. M. A comparative analysis of population structure of a forest tree, Eucalyptus globulus (Myrtaceae), using microsatellite markers and quantitative traits. Tree Genet. Genomes 2, 30–38 (2006).
Payn, K. G., Dvorak, W. S., Janse, B. J. H. & Myburg, A. A. Microsatellite diversity and genetic structure of the commercially important tropical tree species Eucalyptus urophylla, endemic to seven islands in eastern Indonesia. Tree Genet. Genomes 4, 519–530 (2008).
Jones, M. E., Shepherd, M., Henry, R. J. & Delves, A. Chloroplast DNA variation and population structure in the widespread forest tree. Eucalyptus grandis. Conserv. Genet. 7, 691–703 (2006).
Byrne, M. Phylogeny, diversity and evolution of eucalypts. In Plant Genome Biodiversity and Evolution, Part E, Phanerogams – Angiosperms (eds Sharma, A. K. & Sharma, A. ). 303–346 (Science Publishers, 2008).
Petit, R. & Hampe, A. Some evolutionary consequences of being a tree. Ann. Rev. Ecol. Evol. Syst. 37, 187–214 (2006).
Luo, J. et al. Genetic variation and age-age correlations of Eucalyptus grandis at Dongmen Forest Farm in southern China. Aust. For. 73, 67–80 (2010).
Whitlock, M. C. & Guillaume, F. Testing for spatially divergent selection: comparing QST to FST . Genetics 183, 1055–1063 (2009).
Scotti-Saintagne, C. et al. Genome scanning for interspecific differentiation between two closely related oak species [Quercus robur L. and Q. petraea (Matt.) Liebl.]. Genetics 168, 1615–1626 (2004).
Li, C., Sun, Y., Huang, H. W. & Cannon, C. H. Footprints of divergent selection in natural populations of Castanopsis fargesii (Fagaceae). Heredity 113, 533–541 (2014).
Oetjen, K. & Reusch, T. B. H. Genome scans detect consistent divergent selection among subtidal vs. intertidal populations of the marine angiosperm Zostera marina. Mol. Ecol. 16, 5156–5167 (2007).
Lind-Riehl, J. F., Sullivan, A. R. & Gailing, O. Evidence for selection on a CONSTANS-like gene between two red oak species. Ann. Bot. 113, 967–975 (2014).
Lemay, M. A. & Russello, M. A. Genetic evidence for ecological divergence in kokanee salmon. Mol. Ecol. 24, 798–811 (2015).
Smalle, J. & Vierstra, R. D. The ubiquitin 26S proteasome proteolytic pathway. Annu. Rev. Plant Biol. 55, 555–590 (2004).
Wu, W., Cheng, Z., Liu, M., Yang, X. & Qiu, D. C3HC4-type RING finger protein NbZFP1 is involved in growth and fruit development in Nicotiana benthamiana. PLoS One 9, e99352 (2014).
Cho, S. K., Ryu, M. Y., Seo, D. H., Kang, B. G. & Kim, W. T. The Arabidopsis RING E3 ubiquitin ligase AtAIRP2 plays combinatory roles with AtAIRP1 in abscisic acid-mediated drought stress responses. Plant Physiol. 157, 2240–2257 (2011).
Yang, L. et al. Arabidopsis C3HC4-RING finger E3 ubiquitin ligase AtAIRP4 positively regulates stress-responsive abscisic acid signaling. J. Integr. Plant Biol. 58, 67–80 (2016).
Street, N. R. et al. The genetics and genomics of the drought response in Populus. Plant J. 48, 321–341 (2006).
Wilkins, O., Waldron, L., Nahal, H., Provart, N. J. & Campbell, M. M. Genotype and time of day shape the Populus drought response. Plant J. 60, 703–715 (2009).
Cohen, D. et al. Comparative transcriptomics of drought responses in Populus: a meta-analysis of genome-wide expression profiling in mature leaves and root apices across two genotypes. BMC Genomics 11, 630 (2010).
Stec, B. Plant thionins – the structural perspective. Cell. Mol. Life Sci. 63, 1370–1385 (2006).
Qin, D. et al. Heat stress-responsive transcriptome analysis in heat susceptible and tolerant wheat (Triticum aestivum L.) by using Wheat Genome Array. BMC Genomics 9, 432 (2008).
Koike, M., Okamoto, T., Tsuda, S. & Imai, R. A novel plant defensin-like gene of winter wheat is specially induced during cold acclimation. Biochem. Bioph. Res. Co. 298, 46–53 (2002).
Neale, D. B. & Kremer, A. Forest tree genomics: growing resources and applications. Nat. Rev. Genet. 12, 111–122 (2011).
Stölting, K. N. et al. Genome-wide patterns of differentiation and spatially varying selection between postglacial recolonization lineages of Populus alba (Salicaceae), a widespread forest tree. New Phytol. 207, 723–734 (2015).
Di Pierro, E. A. et al. Climate-related adaptive genetic variation and population structure in natural stands of Norway spruce in the South-Eastern Alps. Tree Genet. Genomes 12, 16 (2016).
Carlson, K. D. et al. MIPSTR: a method for multiplex genotyping of germline and somatic STR variation across many individuals. Genome Res. 25, 750–761 (2015).
Acknowledgements
We thank Yonghuai Huang for assistance with leaf sampling, Xiaoli Yu for help in the laboratory and Yangkun Li for assistance with data analyses. We also thank Jiangming Yao (Guangxi University) for downloading the climatic data and Jianzhong Luo (China Eucalypt Research Centre) for providing the QST data as well as Yanlong Jia and Liang Kou (CAS) for production of Figure 1. We gratefully acknowledge the valuable comments and suggestions of J. F. Scheepens and an anonymous reviewer that helped to improve our manuscript. This work was financially supported by the Ministry of Science and Technology of China (2013AA102705), the National Natural Science Foundation of China (31270702) and Guangdong Provincial Department of Science and Technology (2014B020202013).
Author information
Authors and Affiliations
Contributions
S.G. and X.M. conceived and designed the project. M.Z., F.L., X.M., H.H. and S.G. collected the samples. Z.S., M.Z., Q.W., C.Z., M.L. and J.L. performed molecular labwork and scored the markers. S.G., X.M., Z.S. and M.Z. analysed the data and wrote the manuscript with assistance from all other authors. All authors read and approved final manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Song, Z., Zhang, M., Li, F. et al. Genome scans for divergent selection in natural populations of the widespread hardwood species Eucalyptus grandis (Myrtaceae) using microsatellites. Sci Rep 6, 34941 (2016). https://doi.org/10.1038/srep34941
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep34941
- Springer Nature Limited
This article is cited by
-
Genetic diversity and population structure in Cary’s Beardtongue Penstemon caryi (Plantaginaceae), a rare plant endemic to the eastern Rocky Mountains of Wyoming and Montana
Conservation Genetics (2019)
-
The first identification of genomic loci in plants associated with resistance to galling insects: a case study in Eucalyptus L'Hér. (Myrtaceae)
Scientific Reports (2018)
-
A novel set of 223 EST-SSR markers in Casuarina L. ex Adans.: polymorphisms, cross-species transferability, and utility for commercial clone genotyping
Tree Genetics & Genomes (2018)