
Abstract
Microalgae are regarded as the most promising biofuel candidates and extensive metabolic engineering were conducted but very few improvements were achieved. Long non-coding RNA (lncRNA) investigation and manipulation may provide new insights for this issue. LncRNAs refer to transcripts that are longer than 200 nucleotides, do not encode proteins but play important roles in eukaryotic gene regulation. However, no information of potential lncRNAs has been reported in eukaryotic alga. Recently, we performed RNA sequencing in Chlamydomonas reinhardtii, and obtained totally 3,574 putative lncRNAs. 1440 were considered as high-confidence lncRNAs, including 936 large intergenic, 310 intronic and 194 anti-sense lncRNAs. The average transcript length, ORF length and numbers of exons for lncRNAs are much less than for genes in this green alga. In contrast with human lncRNAs of which more than 98% are spliced, the percentage in C. reinhardtii is only 48.1%. In addition, we identified 367 lncRNAs responsive to sulfur deprivation, including 36 photosynthesis-related lncRNAs. This is the first time that lncRNAs were explored in the unicellular model organism C. reinhardtii. The lncRNA data could also provide new insights into C. reinhardtii hydrogen production under sulfur deprivation.
Similar content being viewed by others


Introduction
A considerable portion of the genome of eukaryotes can be transcribed to RNAs, but will not be translated to proteins. These non-coding RNAs (ncRNAs) consist of housekeeping, regulatory and functional unknown ncRNAs. Regulatory ncRNAs are usually classified as small non-coding RNAs and long non-coding RNAs (lncRNAs) according to their lengths1,2. Little was known about the function of lncRNA for a long time, but the discovery of the function of HOTAIR lifts ncRNAs to new levels3. HOTAIR is a 2.2 kb non-coding RNA, which represses transcription of the HOXD locus in trans across 40 kb4. In fact, previous studies have showed that HOX gene clusters play important roles in embryonic development. After the discovery of HOTAIR, lncRNAs began come into the spotlight. Now it is well known that lncRNAs play important roles in cell differentiation and development1,5,6, silencing gene expression in X-chromosome7, neurological diseases occurrence8, cancer progression9,10 and immune response genes mediation11,12.
In plants, lncRNAs express differentially in various organs and under different treatment conditions, which indicates lncRNAs can modulate gene activity during development and in response to external stimuli13. For instance, in Arabidopsis 6,480 intergenic transcripts were classified as long intergenic non-coding RNAs (lincRNAs). A subset of these lincRNAs express organ-specifically, whereas others are responsive to biotic and/or abiotic stresses14. Interestingly, a large number of Arabidopsis long non-coding natural antisense transcripts responded to light are dynamically correlated with histone acetylation15. Very recently, genome-wide screening and functional analysis in rice identified a set of lncRNAs that are involved in the sexual reproduction16. A 1.2 kb rice lncRNA, referred as long-day-specific male-fertility-associated RNA (LDMAR) regulates photoperiod-sensitive male sterility (PSMS), which is the essential component of hybrid rice17. In addition, there are 1,704 high-confident lncRNAs identified in maize, and the tissue-specific expression of maize lncRNAs is more significant than that of filtered genes18.
As to microorganisms, lncRNAs were proved to mediate sporulation via chromatin regulation in both budding yeast Saccharomyces cerevisiae and fission yeast Schizosaccharomyces pombe19,20. Subset of ncRNAs, natural antisense transcripts (NATs) had been genome-wide investigated in the ascomycetes S. cerevisiae, Candida albicans, Aspergillus flavus, Magnaporthe oryzae, Tuber melanosporum and S. pombe, as well as the basidiomycetes Cryptococcus neoformans, Ustilago maydis and Schizophyllum commune. The results demonstrated that a large number of NATs existed in various fungi21. In the model filamentous fungus Neurospora crassa, 939 lncRNAs have been successfully identified based on the results of RNA sequencing. Interestingly, these lncRNAs could be regulated by different environmental stimuli22.
Microalgae are regarded as the most promising biofuel candidates and extensive metabolic engineering are being conducted to reduce the biofuel production cost but very few improvements are achieved yet. Based on the regulatory functions found in other higher plants, lncRNA investigation and manipulation may provide new insights and solutions for this issue. The Chlamydomonas reinhardtii is a unicellular green alga, which is a model organism in the study of chloroplast-based photosynthesis, the structure and function of eukaryotic flagella, as well as many metabolic processes23. C. reinhardtii is also an ideal model organism to study hydrogen metabolism in photosynthetic eukaryotes24. Sulfur deprivation has been proved to be highly correlated to hydrogen photoproduction, and it leads to sustained hydrogen production in C. reinhardtii24. Transcriptome and proteome analyses indicated that sulfur deprivation affects massive pathways including sulfur metabolism, cell wall structure, photosystems, protein biosynthetic apparatus, molecular chaperones and 20 S proteasomal components25,26,27. It was previously demonstrated that hydrogen production can be regulated by an artificial non-coding RNA miRNA (amiRNA) targeting OEE2 encoded gene (a photosystem II related protein, oxygen evolving enhancer)28. This suggested a prospective way for continuous hydrogen production in green algae using regulatory ncRNAs. LncRNAs might be one of the potential solutions for above issues, which explain why we first used the sulfur-deprived C. reinhardtii cells when screening the whole genome for lncRNAs.
The complete genome of C. reinhardtii had been sequenced in ref. 23, and miRNAs had also been found in C. reinhardtii29,30,31. However, the lncRNAs still remains completely unknown in any microalga. In this study, we have carried out a genome-wide scanning using cutting edge high-throughput RNA-seq to discover and characterize the lncRNAs in C. reinhardtii. LncRNA target genes were also predicted to annotate lncRNA functions. Finally, the lncRNAs expression changes in sulfur-replete and sulfur-deprived conditions were explored as well.
Results
Genome-wide identification of lncRNAs in C. reinhardtii
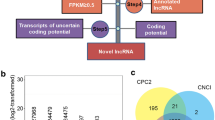
To identify lncRNAs in C. reinhardtii, we performed RNA-seq using C. reinhardtii cells cultured under sulfur-replete and sulfur-deprived conditions. We performed four samples in total, including two sulfur-replete samples and two sulfur-deprived samples. Cells were cultured in TAP + S (with sulfate 40.55 mg/L) and TAP-S (the S-salts was replaced by their chloride counterparts) medium respectively. Total RNA was isolated using RNAiso and cDNA libraries were constructed for sequencing with NEBNext® Ultra™ Directional RNA Library Prep Kit for Illumina® (NEB, USA) according to the manufacturer’s instructions, respectively. 194,241,412 and 174,811,628 clean reads were obtained from sulfur-replete (+S) and sulfur-deprived (−S) libraries, respectively (Fig. 1A). The sequences were mapped to C. reinhardtii genome retrieved from NCBI (ftp://ftp.ensemblgenomes.org/pub/plants/release-23/fasta/chlamydomonas_reinhardtii/dna/). Details of sequencing and mapping steps can be found in Supplementary Data S1 and Table S1. Identification of lncRNAs was executed according to the pipeline shown in Fig. 1. Briefly, the data were firstly filtered using five basic principles: (1) Recurrence in ≥ 3 samples or by ≥ 2 assemblers; (2) Transcript length ≥ 200, and exon number ≥ 1; (3) Minimal reads coverage ≥ 3; (4) Filter known non-lncRNA annotation; (5) Classification of candidate lncRNAs. As a result, 3,574 sequences were obtained after the sifting (Fig. 1B). To effectively distinguish protein-coding and non-coding sequences, coding potential filtering was performed subsequently according to CPC (Coding Potential Calculator) and Pfam Scan (v1.3). By this way, 2,413 and 1597 candidate lncRNAs were predicted by CPC and Pfam Scan, respectively. Finally 1,440 lncRNAs were obtained in the intersection of CPC and Pfam Scan (Fig. 1C). The sequences of all 1,440 lncRNAs identified by CPC and Pfam were listed in Supplementary Data S2.

(A) Informatics pipeline for the identification of lncRNAs in C. reinhardtii. (B) The candidate transcripts numbers of the five filtering steps. (C) Venn chart showing the numbers of candidate lncRNAs filtered by the PFAM, CPC assemblies or by both assemblies. PFAM: Pfam Scan, CPC: Coding Potential Calculator.
Validation of transcription levels of C. reinhardtii lncRNAs
To confirm the expression of C. reinhardtii lncRNAs and their response to sulfur-deprived stress, quantitative Real-Time PCR (qRT-PCR) analysis was applied to verify the results of the high-throughput RNA-seq sequencing. Total RNA extracted from the same samples as RNA-seq used for C. reinhardtii cells cultured under sulfur-replete and sulfur-deprived conditions was converted to cDNA by reverse transcription. Real-Time PCR was next employed to validate the expression levels of 21 lncRNAs selected from the RNA-seq results at random (Fig. 2). The U4 was used as the internal control for quantification31,32.

21 putative lncRNAs, including 17 lincRNAs and 4 intronic lncRNAs are selected for quantitative PCR validation. U4 was used as the reference gene. Colors and numbers under the bar charts refers to the FPKM of genes.
LncRNAs are classified into four types according to their genomic location and context33,34. In this study we detected three main types of lncRNAs in C. reinhardtii: intergenic lncRNAs (lincRNAs), intronic lncRNAs and anti-sense lncRNAs.
Totally 21 putative lncRNAs, including 17 lincRNAs and 4 intronic lncRNAs were randomly selected for quantitative PCR validation. The results demonstrated that in most cases results of qRT-PCR were consistent with those of RNA-seq. This correlation confirmed that the results of RNA-seq technique are reliable. The expression levels of 18 lncRNAs matched these of high throughput sequencing data. However, 3 of the chosen low read lncRNAs did not match the RNA-seq results. We deduce that it is likely due to the low abundance of lncRNAs and the amplification efficiency.
Characterization of C. reinhardtii lncRNAs
For the first time, characteristics and transcription patterns of C. reinhardtii lncRNAs were investigated in this study. The 1440 newly identified C. reinhardtii lncRNAs included 936 lincRNAs, 310 intronic lncRNAs and 194 anti-sense lncRNAs (Fig. 3A). LincRNAs comprise the major part of total lncRNAs (65% of the total C. reinhardtii lncRNAs). Full-length C. reinhardtii lncRNA transcripts (median length of 509 nucleotides) are longer than Arabidopsis lncRNA transcripts (median length of 285 nucleotides), but shorter than human (median length of 592 nucleotides) and rice (median length of 852 nucleotides)19,20,21,35. Interestingly, anti-sense lncRNAs with about 1200 nucleotides in length were found to be the longest transcripts among the three types of lncRNAs in C. reinhardtii. In contrast, a majority of intronic lncRNAs are shorter than 300 nucleotides (Fig. 3B). When we compared the exon number in different type of lncRNA of C. reinhardtii, 67.9% of intronic lncRNAs and 88.6% of anti-sense lncRNAs have only one exon, while lincRNAs usually have one or two exons (33.3% of lincRNAs have one exon and 40.3% of lincRNAs have two exons) (Fig. 3C). In addition, more than 20% of C. reinhardtii lincRNAs and anti-sense lncRNAs exons are shorter than 100 nucleotides, but almost 60% of intronic lncRNAs exons distribute among the regions of 200–300 nucleotides (Fig. 3D). LncRNAs evenly distributed in each chromosome and lincRNAs do not show significant chromosome location preference either (Fig. 3E).

(A) Numbers of lincRNAs, intronic lncRNAs and anti-sense lncRNAs in C. reinhardtii. (B) Transcript length distribution of lincRNAs, intronic lncRNAs and anti-sense lncRNAs. (C) The number of exons per transcript for lncRNAs. (D) Exon length distribution of lincRNAs, intronic lncRNAs and anti-sense lncRNAs. (E) Distribution of lncRNAs along each chromosome. The numbers of lincRNAs (outer circle, depicted in blue), intronic lncRNAs (middle circle, depicted in green) and anti-sense lncRNAs (inner circle, depicted in red) in physical bins of 500 kb for each chromosome.
In contrast with more than 98% human lncRNAs are spliced34, only 48.1% of spliced C. reinhardtii lncRNAs were observed in our study. Interestingly, the percentage of spliced C. reinhardtii lincRNAs (66.7%) is higher than that of rice spliced lincRNAs (46.5%)21.
The conservation of lncRNAs is considered lower than that of protein coding genes in comparisons between species. All C. reinhardtii lncRNAs were blasted against the genomes of Arabidopsis thaliana, Coccomyxa subellipsoidea, Oryza sativa, Sorghum bicolor and Volvox carteri (Fig. 4). In C. reinhardtii only 38 lncRNAs were predicted to be conserved with that of Arabidopsis, while 169 lncRNAs shared homology with Volvox carteri genome (Table 1). The entire list of all conserved lncRNAs can be found as Supplementary Data S3. The C. reinhardtii have longer conserved sequences compared with Coccomyxa subellipsoidea and Volvox carteri (Table 1), which indicated that C. reinhardtii may have higher conservation with these two species in terms of lncRNA conservation. Besides, the coverage value referred to percentage of conserved sequence regions in full length lncRNAs was also investigated to predict the most homologue specie with C. reinhardtii. LncRNAs with more than 10% or 20% coverage were summarized, and the results showed that C. reinhardtii possessed most conserved lncRNAs sequences when compared with Volvox carteri at both over 10% and over 20% coverage levels. This result suggested that C. reinhardtii lncRNAs were most conserved with V. carteri.

All the C. reinhardtii lncRNAs are blasted with the genomes of Arabidopsis thaliana (A), Coccomyxa subellipsoidea (B), Oryza sativa (C), Sorghum bicolor (D) and Volvox carteri (E). X axis: percentage of identity. Y axis: align length. E value < 1.0E-5. (F) Conserved lncRNAs numbers with more than 20% or 10% coverage regions.
Basic property comparison of lncRNAs and mRNAs
The properties such as transcript abundance, lengths, exon numbers and ORFs (Open Reading Frames) of C. reinhardtii lncRNAs and mRNAs have also been compared under the same conditions (Fig. 5). The data of FPKM (expected number of fragments per kilobase of transcript sequence per million mapped reads) represented the abundance of lncRNAs were lower than those of mRNAs in RNA-seq samples, indicting lncRNAs were less transcribed (Fig. 5A). Also, we found that the lengths of lncRNAs were usually shorter than mRNAs. For instance, the lengths of most C. reinhardtii lncRNAs were from 200 to 300 nucleotides, while most mRNAs were longer than 2,000 nucleotides (Fig. 5B). Moreover, fewer exons existed in lncRNAs than in mRNAs. For example, most lncRNAs have fewer than six exons, while mRNAs have more exons and exon numbers distribute in a wider range instead. Some mRNAs have as many as thirty exons (Fig. 5C). The C. reinhardtii lncRNAs also have shorter (60–90 nucleotides) ORFs than those of mRNAs, while most mRNAs ORFs are more than 500 nucleotides (Fig. 5D).

(A) Expression levels of lncRNAs and mRNAs. (B) Transcript length of lncRNAs and mRNAs. (C) Comparison of exon numbers of lncRNAs and mRNAs. Y axis: frequency count. (D) Comparison of ORFs length of lncRNAs and mRNAs. Y axis: frequency count.
Alternative splicing events
As one of the most reported functional bioprocesses by lncRNAs, alternative splicing events36,37 based on the RNA-seq data were investigated in C. reinhardtii. The numbers of different alternative splicing events were calculated and recognized as 12 types: (1) TSS: Alternative 5′ first exon; (2) TTS: Alternative 3′ last exon; (3) SKIP: Skipped exon; (4) XSKIP: Approximate SKIP; (5) MSKIP: Multi-exon SKIP; (6) XMSKIP: Approximate MSKIP; (7) IR: Intron retention; (8) XIR: Approximate IR; (9) MIR: Multi-IR; (10) XMIR: Approximate MIR; (11) AE: Alternative exon ends (5′, 3′, or both) and (12) XAE: Approximate AE. No difference in numbers of all kinds of alternative splicing events could be identified between the sulfur-replete and sulfur-deprived C. reinhardtii samples (see Supplementary Figure S1). As a consequence, further study is needed for the alternative splicing events case by case in C. reinhardtii.
LncRNAs responsive to sulfur deprivation
Totally 367 lncRNAs responsive to sulfur deprivation (see Supplementary Data S4), including 194 up-regulated lncRNAs and 173 down-regulated lncRNAs were identified in this study (Table 2), which were classified to 289 lincRNAs (78.7%), 30 intronic lncRNAs (8.2%) and 48 anti-sense lncRNAs (13.1%). The lncRNAs with up-regulated levels after sulfur deprivation were more than down-regulated ones in all three types of lncRNAs. The proportion of differentially expressed lncRNAs under sulfur-deprived conditions was also analyzed. For instance, 30.9% of lincRNAs changed after sulfur deprivation, and the numbers of up-regulated and down-regulated lincRNAs were very similar. However, only 9.7% intronic lncRNAs were responsive to sulfur deprivation (Fig. 6). The results showed that the lincRNAs were more responsive to sulfur deprivation, while intronic lncRNAs were less affected. Interestingly, when we looked at the chromosome preference of lncRNAs under sulfur deprivation, more than 60% lncRNAs on chromosome 2 and chromosome 7 were responsive to sulfur deprivation, respectively. In contrast, only 11.0% lncRNAs on chromosome 10 were changed under sulfur-deprived condition (Fig. 7).

Percentage of changed C. reinhardtii lncRNAs under sulfur-deprived condition.

The Y axis represents numbers of lncRNAs.
Among the 367 lncRNAs responsive to sulfur deprivation, 6 lncRNAs and 10 lncRNAs were only expressed under sulfur-replete (Table 3) and sulfur-deprived (Table 4) condition, respectively. The top 10 lncRNAs with the most up- or down-regulation were listed (Tables 5 and 6). Both the up-regulated and the down-regulated assembles had 6 lincRNAs (60%), 2 intronic lncRNAs (20%) and 2 anti-sense lncRNAs (20%). Accordingly, the proportions of intronic lncRNAs and anti-sense lncRNAs were higher in most changed top 10 lncRNAs (20%) than in all lncRNAs responsive to sulfur deprivation (9.7%).
LncRNA target prediction, annotation and enrichment analysis
LncRNAs usually act on neighboring target genes, which is known as the Cis role of lncRNAs. We searched for coding genes 100 kb upstream and downstream of lncRNAs to predict putative Cis target genes of lncRNAs, followed by analyzing functions of these coding genes to annotate lncRNAs. KOBAS software was used to analyze the statistical enrichment of differentially expressed lncRNA target genes in KEGG pathways. Based on the results of significantly differentially expressed lncRNAs analysis, 99 pathways were found responsive to sulfur deprivation. The most enriched pathways including pentose phosphate pathway, plant hormone signal transduction, protein export, glutathione metabolism, amino acid metabolism and fatty acids metabolism. The analysis result was showed in a heat map indicating the expression levels of all pathways, and the entire pathways expression level heat map can be found in Supplementary Figure S2. The different pathways included pentose phosphate pathway, RNA polymerase and degradation, protein export, plant hormone signal transduction, base excision repair, and some metabolic pathways.
LncRNAs related to photosynthesis
The differentially expressed photosynthetic proteins were investigated, and their regulating lncRNAs were located. In total, 23 photosynthesis-related mRNAs were found responsive to sulfur-deprived condition, and they were predicted to be the target genes of 36 sulfur deprivation-responsive lncRNAs. We classified lncRNA target genes into five types according to their positions in photosynthetic systems (Table 7). In detail, we were able to predict 14 lncRNAs related to Photosystem II, 10 lncRNAs related to Photosystem I, 7 lncRNAs related to photosynthetic electron transport, 3 lncRNAs related to ATP synthase and 2 lncRNAs related to cytochrome b6-f complex.
Discussion
LncRNAs play important roles in various metabolic pathways in animal, plant, and yeast. Recent studies showed that they are closely related to cancer, nervous disease and autoimmune disease8,9,10,11,12. In plants, rice lncRNAs were reported to regulate the essential component photoperiod-sensitive male sterility of hybrid rice17, which has greatly contributed to the global increase of rice productivity and solution to food problem. The importance of lncRNA has been emphasized in many species, however, still remained completely unknown until this study in C. Reinhardtii. As a unicellular eukaryotic model organism, C. reinhardtii is an ideal model for studying chloroplast-based photosynthesis, structure, assembly and function of eukaryotic flagella (cilia) inherited from the common ancestor of plants and animals as a model of human cilia-related diseases. C. reinhardtii is also a model to study hydrogen production by green algae. In 2007, the complete genome of C. reinhardtii was sequenced, and microRNAs were also found in this species. In this study, high-throughput sequencing technique was used to scan the whole genome of C. reinhardtii, and 1,440 high confident long non-coding RNAs (936 lincRNAs, 310 intronic lncRNAs and 194 anti-sense lncRNAs) were identified. These lncRNAs have a median length of 509 nucleotides, and usually have 1~2 exons. In all, lncRNAs is shorter than mRNA, with fewer exons than mRNA. The transcription level of lncRNA is significantly lower than that of mRNAs, and the FPKM of most C. reinhardtii lncRNAs (85% of the 1,440) are less than 10. Compared with 98% lncRNAs spliced in human30, however, only 48.1% of C. reinhardtii lncRNAs were found spliced. This significant difference suggests the possible different origins and patterns of lncRNAs in C. reinhardtii and human.
Mercer et al. using tiling array successfully identified and characterized transcripts which were not detected and annotated by conventional sequencing approaches, because of their low or transient expression38. This suggested that our high throughput sequencing may also not include all lncRNAs in C. reinhardtii, and rare or transient lncRNAs and some lncRNAs responsive to special external stimuli were not identified under our experimental conditions. In this study, 111 lncRNAs can only be detected under sulfur-replete condition and 28 lncRNAs can only be detected under sulfur-deprived condition. Transcripts with long ORFs are considered more likely to encode proteins, and hence some filter principles exclude transcripts longer than 100 amino acids18 or 80 amino acids39 to remove transcripts with long ORFs, which are more likely to encode proteins. However, this restrictions may leave out some possible lncRNAs. That is why we did not discard the lncRNA candidates with long ORFs in the process of filtering.
The conservation of C. reinhardtii lncRNAs compared with other species was greatly affected by the integrality of genome assembly and the size of reference genomes. The C. reinhardtii have 64 lncRNAs conserved with C. subellipsoidea, and 169 lncRNAs conserved with V. carteri (Table 1). The longest C. reinhardtii conserved sequence length of lncRNA when compared with C. subellipsoidea and V. carteri is 433 and 485 nt, respectively. The number of conserved sequences possibly related to the genome size or/and sequencing and assembling completeness of the genome, whereas longer conserved sequences in C. subellipsoidea and V. carteri indicated that C. reinhardtii may have some lncRNAs that have higher conservation with C. subellipsoidea and V. carteri due to their close evolutionary relationship.
Even though some lncRNAs have verified functions, the molecular mechanism of how lncRNAs participate in bioprocesses is still largely unknown. For instance, lncRNAs can modulate protein-coding genes at transcription, post-transcription, and post-translation levels8,10,13,19,20. They can also affect the nearby genes positively or negatively by inducing chromatin remodeling or inhibiting RNA polymerase II recruitment13,19,20. What’s more, lncRNAs modulate alternative splicing by hybridization with targeted sense RNAs and block the recognition of the splice site of spliceosome36,37. In addition, some lncRNAs act as the precursor of miRNA, and can also interact with miRNA as a competing endogenous RNA10,13. Some lncRNAs are also able to bind with proteins to form RNA-protein complex to modulate protein activity or alter protein subcellular localization40,41. Therefore, by functions lncRNAs are classified as signaling, decoying, guiding and scaffolding lncRNAs36. At the same time, some lncRNAs have both Cis (acting on neighboring target genes) and Trans (identifying each other by the expression level) roles in regulating target genes. However, current prediction of Trans role of target gene needs more than 5 samples, so in this study only Cis targeted genes were considered. Further exploratory need more sequencing samples.
LncRNA is reported to modulate alternative splicing regulators in Arabidopsis37,42. Similar study on alternative splicing events was carried out to investigate C. reinhardtii. Surprisingly, the numbers and classification of mRNAs alternative splicing showed no difference. Future investigation of C. reinhardtii lncRNAs will continue with the integrative analysis of lncRNA, miRNA, mRNA and proteins, prediction and verification of lncRNA targets, and functions of differential expressed lncRNAs in the unicellular green alga C. reinhardtii.
The hydrogen production from green algae is a promising way to solve the global energy and environment problems. However, the main problem of hydrogen production from green algae is the inhibition of hydrogenase activity by oxygen, which results in the releasing of hydrogen from algal cells continuously for only a few seconds to a few minutes. Sulfur deprivation leads to sustained hydrogen production by C. reinhardtii, so the transcriptome and proteome of sulfur-deprived C. reinhardtii had been analyzed24,25,26,31. Transcriptome analysis revealed that sulfur deprivation resulted in repression of most transcripts encoding photosynthetic genes, except for LHCBM9 (encoding a major light-harvesting polypeptide), and this indicated a remodeling of the photosystem II light-harvesting complex under sulfur deprivation24. Photosynthetic machinery was also one of the most changed components under sulfur deprivation in proteomic analysis, and other major changes consist of protein biosynthetic apparatus, molecular chaperones, and 20 S proteasomal components26. However, more regulatory mechanisms should be retrieved from other regulatory system, such as miRNAs, and lncRNAs. Thus lncRNAs responsive to sulfur deprivation investigated in this study.
Electrons which are necessary in green algae hydrogen production source from photosynthetic electron transport chain, and hydrogenase links with photosynthetic electron transport chain by ferredoxin (Fd). In addition, the oxygen produced by photosynthesis can inhibit enzyme activity of hydrogenase. Therefore, hydrogen production in green algae is closely related to photosynthesis. Our study discovered 36 lncRNAs targeting to 23 photosynthesis-related mRNAs responsive to sulfur deprivation. Obviously, this indicates that the lncRNAs may modulate the photosynthesis-related mRNAs. Sulfur deprivation repressed the expression of most photosynthesis-related mRNAs (22 of all the 23 photosynthesis-related mRNAs), except for LHCSR3. As to photosynthesis-related lncRNAs, 22 lncRNAs were up-regulated and 14 lncRNAs were down-regulated. LncRNAs regulated mRNAs in diverse ways. For instance, lncRNA XLOC_069036 was up-regulated after sulfur deprivation, but its target gene PSBQ was repressed; lncRNA XLOC_064073 was down-regulated, and its target gene LHCBM1 was also down-regulated. Moreover, a considerable amount of mRNAs modulated by multiple lncRNAs, for example, the down-regulated mRNA PSBW was predicted to be regulated by tow up-regulated lncRNAs. On the other hand, lncRNAs always also had multiple targets, for example, the lncRNA XLOC_037244 was predicted to regulate LHCBM2 as well as LHCBM7. These diverse expression patterns of lncRNAs and mRNAs indicated complicated regulation mechanism and various functions of C. reinhardtii lncRNAs. Thus, the 36 lncRNAs possibly regulate photobiological hydrogen production in C. reinhardtii. Our further research will focus on these sulfur deprivation-responsive and photosynthesis-related lncRNAs.
In summary, in this study we reported the first genome wide lncRNA profiling from a photosynthetic microorganism Chlamydomonas reinhardtii. Moreover, we identified 367 lncRNAs responsive to a promising simple hydrogen induction treatment, i.e., sulfur deprivation, including 36 photosynthesis-related lncRNAs with sulfur deprivation-responsive target genes. The lncRNA investigation may provide new insights into complicate regulations of biofuel production and thus extensive metabolic engineering could be conducted for potential improvements in the field of microalgal biofuels. Based on the predication of lncRNA and their targets, genetic manipulations focusing on these target genes will be employed for further potential improvements of hydrogen production in this model green microalga.
Materials and Methods
Growth and treatments of the algae
C. reinhardtii CC849 were obtained from Chlamydomonas Genetic Centre (Duke University, Durham, USA). The algae was grown in a Tris-Acetate-Phosphate (TAP) medium at 25 °C and under continuous cool-white fluorescent lamps (≈100 μmol photons·m−2·s−1). The sulfur deprivation treatment was performed according to Shu and Hu26. The algae were grown in liquid TAP until mid-log phase and algal cells were collected by centrifugation and washed twice with liquid TAP medium without sulfur (TAP-S, for 1 L of Medium: 2X Filner’s Beijernicks Solution 25 ml; 1 M Potassium Phosphate 1 ml; Trace mineral solution 1 ml; Tris-Base 2.42 g; adjust pH to 7.0 by Glacial Acetic Acid. Sulfur-deprivation media TAP-S were prepared by replacement of the S-salts by their chloride counterparts). Algal cells of equal numbers were resuspended in TAP or TAP-S under continuous illumination for 24 h, and then cell aliquots were collected for RNA isolation.
Preparation of total RNA
Total RNA was extracted using RNAiso Plus reagent (Takara, Dalian, China) according to the manufacturer’s protocol. The algal cells cultured at 25 °C for 24 h in TAP and TAP-S were collected by centrifugation and treated with RNAiso Plus (Takara) immediately. Then rRNAs were removed by Ribo-zero™ rRNA Removal Kit (Epicentre, CA, USA) according to the manufacturer’s protocol. The quality of RNA was examined by using an Agilent 2100 Bioanalyzer.
LncRNA library construction and high-throughput sequencing
Equivalent total RNAs from TAP and TAP-S cultured algal cells were used to construct the sulfur-replete and sulfur-deprived libraries by NEB Next® Ultra™ Directional RNA Library Prep Kit for Illumina® (NEB, USA) following manufacturer’s recommendations. Briefly, RNA was broken into fragments by divalent cations under elevated temperature in NEBNext First Strand Synthesis Reaction Buffer, and converted to first strand cDNA using random hexamer primer and M-MuLV Reverse Transcriptase. Second strand cDNA was synthesized subsequently using DNA Polymerase I and RNase H. dNTPs with dTTP were replaced by dUTP in the reaction buffer. Remaining overhangs were converted into blunt ends by exonuclease/polymerase. Adaptors with hairpin loop structure were ligated to prepare for hybridization after adenylation of DNA 3′ ends. For selecting 150~200 nucleotides cDNA fragments, the library were purified with AMPure XP system (Beckman Coulter, Beverly, USA)12. Then size-selected, adaptor-ligated cDNA was incubated with 3 μl USER Enzyme (NEB, USA) at 37 °C for 15 min followed by 5 min at 95 °C. Then PCR was performed to obtain enriched cDNA library. At last, products were purified (AMPure XP system) and assessed (Agilent Bioanalyzer 2100 system). The clustering of the index-coded samples was performed on a cBot Cluster Generation System using TruSeq PE Cluster Kit v3-cBot-HS (Illumia) according to the manufacturer’s instructions. After cluster generation, sequencing of the two libraries was performed on the Illumina HiSeq 2500 platform. Reads with more than 10% N (Unable to determine base information), with adapter sequence, or of low quality were removed from the raw reads to obtain clean reads. Finally, clean reads were compared with C. reinhardtii genome from NCBI using Tophat243. The libraries preparation and deep sequencing were performed by Novogene Bioinformatics Technology Cooperation (Beijing, China).
Bioinformatics analysis for identifying lncRNAs
The transcripts including mRNA, lncRNA and rRNA were assembled using Cufflinks44 and scripture45. The assembled transcripts detected in two or more samples or by two or more assemblers were selected for further analysis. Then transcripts less than 200 nucleotides were sorted out. The transcripts that have three or more reads coverage are chosen for further analyses. The sequences of remained transcripts were compared with the known non-coding RNAs (rRNA, tRNA, snRNA, snoRNA, pre-miRNA and pseudogenes) using Cuffcompare. The transcript sequences were also compared with the known mRNAs, and the candidate lincRNA, intronic lncRNA, anti-sense lncRNA were determined by class code obtained from Cuffcompare.
The transcripts were then aligned to NCBI protein database (NRDB) by CPC (Coding Potential Calculator)46 Transcripts with known protein domains were excluded by Pfam Scan47 according to Pfam HMM48. The intersection of transcripts filtered by CPC and Pfam Scan were considered as the lncRNAs.
Quantitative Real-Time PCR (qRT-PCR) validation of lncRNAs
21 lncRNAs (FPKM range from 0 to 1423) were randomly chosen to validate the RNA-seq data. Total RNA were isolated respectively from algae cells cultivated in TAP and TAP-S for qRT-PCR using the RNAiso Plus reagent (Takara, Dalian, China) as previously described. First-strand cDNA was reverse transcribed by PrimeScriptTM RT reagent Kit with gDNA Eraser (Takara, Dalian, China). The qRT-PCR was performed using SYBR® Premix Ex TaqTM (Perfect Real Time) and ROX plus (Takara, Dalian, China). The U4 snRNA was used as the reference gene and all the primers used were as listed in Supplementary Table S2. The conditions for the PCR amplification were as follows: polymerase activation was conducted at 95 °C for 30 s; followed by 40 cycles at 95 °C for 5 s, 60 °C for 34 s. The specificity of the primer amplicons was tested by analysis of a melting curve and the PCR products were verified by gel purification and sequencing. This experiment was performed on QuantStudioTM 6 Flex Real-Time PCR System (Life technologies) containing three technical replicates and three biological replicates.
Distribution of lncRNAs along each chromosome
The C. reinhardtii lincRNAs, intronic lncRNAs and anti-sense lncRNAs were aligned to the genome of C. reinhardtii separately to obtain the lncRNA chromosome distribution. The lncRNAs were aligned by short blast, and the best hits were chosen to do subsequent analysis, with a summarized size of every 500 kb. The start sites of the lncRNA in the chromosome decided which zone this lncRNA was counted in. C. reinhardtii v5.5 genome from Phytozome10.2 was used for analysis of chromosome distribution.
LncRNA conservation in different species
The full length of all identified 1440 C. reinhardtii lncRNAs were used to blast against the genomes of Arabidopsis thaliana, Homo sapiens, Coccomyxa subellipsoidea, Oryza sativa, Sorghum bicolor and Volvox carteri, with the word-size = 5, and E value < 10E-5.
KEGG enrichment analysis
KEGG is a bioinformatics database resource integrates genomic, chemical and systemic functional information to understand high-level functions and utilities of the biological system from molecular-level information, especially large-scale molecular datasets generated by high-throughput experimental technologies (http://www.genome.jp/kegg/). We used KOBAS software for testing the statistical enrichment of differential expressed lncRNA target genes in KEGG pathways12.
Additional Information
How to cite this article: Li, H. et al. Genome-wide long non-coding RNA screening, identification and characterization in a model microorganism Chlamydomonas reinhardtii. Sci. Rep. 6, 34109; doi: 10.1038/srep34109 (2016).
References
Pauli, A., Rinn, J. L. & Schier, A. F. Non-coding RNAs as regulators of embryogenesis. Nat. Rev. Genet. 12, 136–149 (2011).
Shu, L. F. & Hu, Z. L. Small silencing RNAs in Chlamydomonas reinhardtii . Minerva Biotecnologica 22, 29–37 (2010).
Woo, C. J. & Kingston, R. E. HOTAIR lifts noncoding RNAs to new levels. Cell 129, 1257–1259 (2007).
Rinn, J. L. et al. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell 129, 1311–1323 (2007).
Guttman, M. et al. lincRNAs act in the circuitry controlling pluripotency and differentiation. Nature 477, 295–300 (2011).
Fatica, A. & Bozzoni, I. Long non-coding RNAs: new players in cell differentiation and development. Nat. Rev. Genet. 15, 7–21 (2014).
Dimond, A. & Fraser, P. Long Noncoding RNAs Xist in Three Dimensions. Science 341, 720–721 (2013).
Ponting, C. P., Oliver, P. L. & Reik, W. Evolution and functions of long noncoding RNAs. Cell 136, 629–641 (2009).
Wapinski, O. & Chang, H. Y. Long noncoding RNAs and human disease. Trends Cell Biol 21, 354–361 (2011).
Yang, G. D., Lu, X. Z. & Yuan, L. J. LncRNA: a link between RNA and cancer. Biochim Biophys Acta 1839, 1097–1109 (2014).
Carpenter, S. et al. A Long Noncoding RNA Mediates Both Activation and Repression of Immune Response Genes. Science 341, 789–792 (2013).
Wang, Y. Y. et al. Analyses of Long Non-Coding RNA and mRNA profiling using RNA sequencing during the pre-implantation phases in pig endometrium. Sci Rep 6, 20238 (2016).
Kim, E. D. & Sung, S. Long noncoding RNA: unveiling hidden layer of gene regulatory networks. Trends Plant Sci. 17, 16–21 (2012).
Liu, J. et al. Genome-wide analysis uncovers regulation of long intergenic noncoding RNAs in Arabidopsis . Plant Cell 24, 4333–4345 (2012).
Wang, H. et al. Genome-wide identification of long noncoding natural antisense transcripts and their responses to light in Arabidopsis . Genome Res. 24, 444–453 (2014).
Zhang, Y. C. et al. Genome-wide screening and functional analysis identify a large number of long noncoding RNAs involved in the sexual reproduction of rice. Genome Biology 15 (2014).
Ding, J. H. et al. A long noncoding RNA regulates photoperiod-sensitive male sterility, an essential component of hybrid rice. Proc. Natl. Acad. Sci. USA 109, 2654–2659 (2012).
Li, L. et al. Genome-wide discovery and characterization of maize long non-coding RNAs. Genome Biology 15, R40 (2014).
van Werven, F. J. et al. Transcription of two long noncoding RNAs mediates mating-type control of gametogenesis in budding yeast. Cell 150, 1170–1181 (2012).
Hiriart, E. & Verdel, A. Long noncoding RNA-based chromatin control of germ cell differentiation: a yeast perspective. Chromosome Res 21, 653–663 (2013).
Donaldson, M. E. & Saville, B. J. Natural antisense transcripts in fungi. Mol. Microbiol 85, 405–417 (2012).
Arthanari, Y., Heintzen, C., Griffiths-Jones, S. & Crosthwaite, S. K. Natural antisense transcripts and long non-coding RNA in Neurospora crassa . Plos One 9, e91353 (2014).
Merchant, S. S. et al. The Chlamydomonas Genome Reveals the Evolution of Key Animal and Plant Functions. Science 318, 245–250 (2007).
Melis, A., Zhang, L., Forestier, M., Ghirardi, M. L. & Seibert, M. Sustained photobiological hydrogen gas production upon reversible inactivation of oxygen evolution in the green alga Chlamydomonas reinhardtii . Plant Physiol. 122, 127–136 (2000).
Nguyen, A. V. et al. Transcriptome for photobiological hydrogen production induced by sulfur deprivation in the green alga Chlamydomonas reinhardtii . Eukaryot Cell 7, 1965–1979 (2008).
González-Ballester, D. et al. RNA-seq analysis of sulfur-deprived Chlamydomonas cells reveals aspects of acclimation critical for cell survival. Plant Cell 22, 2058–2084 (2010).
Chen, M. et al. Proteomic Analysis of Hydrogen Photoproduction in Sulfur-Deprived Chlamydomonas Cells. Journal of Proteome Research 9, 3854–3866 (2010).
Li, H. et al. Sustainable photosynthetic H2-production mediated by artificial miRNA silencing of OEE2 gene in green alga Chlamydomonas reinhardtii . International Journal of Hydrogen Energy 40, 5609–5616 (2015).
Molnár, A., Schwach, F., Studholme, D. J., Thuenemann, E. C. & Baulcombe, D. C. miRNAs control gene expression in the single-cell alga Chlamydomonas reinhardtii . Nature 447, 1126–1129 (2007).
Zhao, T. et al. A complex system of small RNAs in the unicellular green alga Chlamydomonas reinhardtii . Genes Dev. 21, 1190–1203 (2007).
Shu, L. F. & Hu, Z. L. Characterization and differential expression of microRNAs elicited by sulfur deprivation in Chlamydomonas reinhardtii . BMC Genomics 13, 108 (2012).
Jakab, G. et al. Chlamydomonas U2, U4 and U6 snRNAs. An evolutionary conserved putative third interaction between U4 and U6 snRNAs which has a counterpart in the U4atac-U6atac. biochimie 79, 387–395 (1997).
Ma, L., Bajic, V. B. & Zhang, Z. On the classification of long non-coding RNAs. RNA Biol. 10, 925–933 (2013).
St Laurent, G., Wahlestedt, C. & Kapranov, P. The Landscape of long noncoding RNA classification. Trends Genet. 31, 239–251 (2015).
Derrien, T. et al. The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res. 22, 1775–1789 (2012).
Wang, K. C. & Chang, H. Y. Molecular mechanisms of long noncoding RNAs. Mol. Cell 43, 904–914 (2011).
Bardou, F. et al. Long noncoding RNA modulates alternative splicing regulators in Arabidopsis . Dev. Cell 30, 166–176 (2014).
Mercer, T. R. et al. Targeted RNA sequencing reveals the deep complexity of the human transcriptome. Nat. Biotechnol 30, 99–104 (2012).
Zhang, W. et al. Identification of Maize Long Non-Coding RNAs Responsive to Drought Stress. Plos One 9, e98958 (2014).
Wilusz, J. E., Sunwoo, H. & Spector, D. L. Long noncoding RNAs: functional surprises from the RNA world. Genes Dev. 23, 1494–1504 (2009).
Mercer, T. R., Dinger, M. E. & Mattick, J. S. Long non-coding RNAs: insights into functions. Nature Reviews Genetics 10, 155–159 (2009).
Kornblihtt, A. R. A long noncoding way to alternative splicing in plant development. Dev. Cell 30, 117–119 (2014).
Kim, D. et al. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14, R36 (2013).
Trapnell, C. et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol 28, 511–515 (2010).
Guttman, M. et al. Ab initio reconstruction of cell type-specific transcriptomes in mouse reveals the conserved multi-exonic structure of lincRNAs. Nat. Biotechnol 28, 503–510 (2010).
Kong, L. et al. CPC: assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 35, W345–W349 (2007).
Mistry, J., Bateman, A. & Finn, R. D. Predicting active site residue annotations in the Pfam database. BMC Bioinformatics 8, 298 (2007).
Bateman, A. et al. The Pfam protein families database. Nucleic Acids Res. 32, D138–D141 (2004).
Acknowledgements
We thank Pr. Xuemei Chen from University of California, Riverside for her helpful comments and suggestions. This work was supported by the National Natural Science Foundation of China (31470431, 31100262, 31470389), Guangdong Natural Science Foundation (2014A030308017 and 2016A030313052), Project of DEGP (2015KTSCX125), Shenzhen special funds for Bio-industry development (NYSW20140327010012) and SRF for ROCS, SEM.
Author information
Authors and Affiliations
Contributions
H.L., Y.W. and M.C. participated in all experiments and drafted the manuscript. P.X. carried out the bioinformatics’ analysis and sequence comparisons. C.H. carried out the RNA extraction and reverse transcription. Z.Z. and C.W. participated in the qRT-PCR validation and English editing. J.W. participated in drafting the manuscript and performed the statistical analysis. Z.H. participated in the design of the study and guide the study. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Li, H., Wang, Y., Chen, M. et al. Genome-wide long non-coding RNA screening, identification and characterization in a model microorganism Chlamydomonas reinhardtii. Sci Rep 6, 34109 (2016). https://doi.org/10.1038/srep34109
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep34109
- Springer Nature Limited
This article is cited by
-
Different transcription of novel, functional long non-coding RNA genes by UV-B in green algae, Volvox carteri
International Microbiology (2023)
-
Identification and Characterization of Long Non-Coding RNAs Involved in Sex-Related Gene Regulation in Kelp Saccharina japonica
Journal of Ocean University of China (2023)
-
The response of LncRNAs associated with photosynthesis-and pigment synthesis-related genes to green light in Chlamydomonas reinhardtii
Photosynthesis Research (2023)
-
Identification and functional prediction of long non-coding RNAs of rice (Oryza sativa L.) at reproductive stage under salinity stress
Molecular Biology Reports (2021)
-
Roles of long non-coding RNAs in plant virus interactions
Journal of Plant Biochemistry and Biotechnology (2021)