
Abstract
Spectroscopic properties of a series of (Sr0.98-xBaxEu0.02)Si2O2N2 (0 ≤ x ≤ 0.98) compounds has been studied under high hydrostatic pressure applied in a diamond anvil cell up to 200 kbar. At ambient pressure the crystal structures of (Sr0.98-xBaxEu0.02)Si2O2N2 (0 ≤ x ≤ 0.98) are related to the ratio of strontium to barium and three different phases exists: orthorhombic Pbcn(0.78 ≤ x ≤ 0.98), triclinic P1 (0 < x ≤ 0.65) and triclinic P1 (0.65 < x < 0.78). It was found that Eu2+ luminescence reveals abrupt changes under pressure (decay time, energy and shape) which indicate the variation of the local symmetry and crystal field strength in Eu2+ sites. These changes are attributed to the reversible pressure-induced structural phase transitions of triclinic (Sr0.98-xBaxEu0.02)Si2O2N2 into orthorhombic structure. Pressure in which phase transition occurs decreases linearly with increasing of Ba composition in (Sr0.98-xBaxEu0.02)Si2O2N2 series. Additionally, very different pressure shifts of the Eu2+ luminescence in different phases of (Sr0.98-xBaxEu0.02)Si2O2N2:Eu from −40 cm−1/kbar to 0 cm−1/kbar have been observed. This effect is explained by different interaction of the Eu2+ 5d electron with the second coordination sphere around the impurity cations.
Similar content being viewed by others

Introduction
Rare earth activated oxonitridosilicate compounds have been widely explored as advanced phosphors in white emitting diodes (WLEDs) due to very efficient and thermally stable luminescence that covers a broad spectral range, under excitation of blue and/or UV light1,2,3,4,5. Moreover, since these materials have been synthesized under ambient pressure they have a lower production cost than high-pressure synthesized nitridosilicates materials such as M2Si5N8:Eu (M = Ca, Sr, Ba), MAlSiN3:Eu (M = Ca, Sr)5,6,7,8,9,10. Among oxonitridosilicate phosphors, there is an increasing interest in layered MSi2O2N2:Eu (M = Ca, Sr, Ba) which show efficient yellow, green and cyan emission, respectively11,12,13,14,15,16,17. The present research in rare-earth ions doped MSi2O2N2 (M = Ca, Sr, Ba) mainly focuses on the development of an appropriate host composition by mixing alkaline earth metal cations to adjust their emission properties. For example, structure variations in MSi2O2N2:Eu (M = Ba, Sr) with gradually substitution of Ba with Sr leads to continous or abrupt changes in luminescence properties of these compounds14,15,18,19.
Based on high resolution synchrotron XRD measurements, it has been shown that crystal structures of the (Sr0.98-xBaxEu0.02)Si2O2N2 (0 ≤ x ≤ 0.98) are related to the ratio of strontium to barium. Depending on barium content three different phases exist: Phase A (0.78 ≤ x ≤ 0.98), Phase B (0 < x ≤ 0.65) and Phase C (0.65 < x < 0.78)18. Phase A and Phase B are isostructural to well-known crystal structures of BaSi2O2N2 and SrSi2O2N2, respectively, whereas Phase C adopts a distorted variation of the BaSi2O2N2 type structure14. The crystal structure of Phase A with orthorhombic unit cell is described by Pbcn space group, while Phase B with a triclinic unit cell is described by P1 space group. Phase C is a new crystal structure that is different from Phase A and Phase B and can be regarded as a “transition state” between them, since it combines the unit cell-metrics of the Phase A type with corrugated metal-ion layers found in the Phase B. Nominally Phase C with triclinic unit cell is attributed to P1 space group. Detailed studies on the high resolution transmission electron microscopy and scanning electron diffraction analysis of selected areas, have shown that Phase C usually contains a small amount of nanocrystalline domains of Phase B with a triclinic unit cell18,19. This could be due to inhomogenous distribution of Ba and Sr components in the mixed (Sr0.98-xBax)Si2O2N2 crystals. In (Sr0.98-xBaxEu0.02)Si2O2N2, the barium, strontium and europium ions are in the channels, which are formed by [SiON3] tetrahedra and forms linear chains in the structure. In the case of Phase A and Phase C structures, cation chains are located in a small distance from each other, which is almost equal to the intra-chain distance forming a plane in the structure, while in the case of Phase B cation chains are separated from each other much more than the intra-chain distance. Thus the second coordination sphere around Eu2+ ions in Phase A and Phase C consist of Sr2+/Ba2+ cations forming rectangle while in the case of Phase B the Sr2+/Ba2+ cations form a line. Cation distribution in the second coordination sphere can results in a very different spectroscopic properties of Eu2+ luminescence. Poort et al.20 have suggested that electron–lattice interaction energy, manifested by large Stokes shift between emission and absorption spectra is large when Eu2+ ions occupy the lattice sites that belongs to the linear cationic chain.
The high pressure study of the (Sr0.98-xBaxEu0.02)Si2O2N2 doped with Eu2+ presented in this contribution is continuation and extension of the research work presented in ref. 18. where the relations between the luminescence properties and the content of Ba and Sr have been discussed.
It is well known that pressure is an important physical parameter that can induce the structural phase transitions21,22. X-ray diffraction study of (Sr0.98Eu0.02)Si2O2N2 under hydrostatic pressure has shown that in pressure range up to 96.5 kbar triclinic structure was conserved23. The main motivation on performing high hydrostatic pressure was to investigate whether high pressure influences the structural stability of (Sr0.98-xBaxEu0.02)Si2O2N2 (0 < x ≤ 0.98) to induce phase transitions. Phase transitions were identified trough a comparison of the luminescence and Raman properties under pressure. High pressure, which permits us to continuously vary structural parameters, allow to answer question whether phase transitions are related to structural parameters like bond length and lattice constants. To achieve that, complementary studies of chemical pressure (through replacement of Sr2+ by larger Ba2+) and external high hydrostatic pressure up to 200 kbar (20 GPa) applied in diamond anvil cell (DAC) have been carried out. Finally, we have developed the phase diagram of (Sr0.98-xBaxEu0.02)Si2O2N2 which shows the relation between chemical and external pressure compression effects.
Properties of complex consisting of Eu2+ ions and its negatively charged coordination sphere can be described using crystal field approximation in such case high hydrostatic pressure causes the increase of the energies of the 4f 7, 4f 65d1 electronic configuration with respect to the host bands energies and the increase of the 4f 65d1 levels splitting24,25. The superposition of these effects are seen as a pressure-induced diminishing of the energy of luminescence related to the 4f 65d1 → 4f 7 transition. The influence of the second coordination sphere may bring about drastic changes in this effect, since it can magnify or cancel the influence of the first coordination sphere. As a result, different pressure behavior of the luminescence band related to the 4f 65d → 4f 7 transition of Eu2+ ions in different phases is expected.
The study of luminescence under high hydrostatic pressure results in better understand the relationship between pressure and crystal phases of oxyntridosilicate phosphors and further clarify the influences of these phase transitions on photoluminescence properties, which might be general to oxynitride materials and will be useful in tuning optical and other properties that are sensitive to local coordination environments.
Results
For spectroscopic studies the representatives of three different phases were selected: (Ba0.98Eu0.02)Si2O2N2 (Phase A; orthorhombic; Pbcn), (Sr0.49Ba0.49Eu0.02)Si2N2O2 (Phase B; triclinic; P1) and (Sr0.23Ba0.75Eu0.02)Si2O2N2 (Phase C; triclinic; P1).
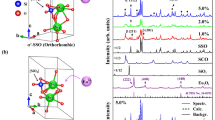
Photoluminescence excitation (PLE) and photoluminescence (PL) spectra of the (Sr0.98-xBaxEu0.02)Si2O2N2 system for x = 0.98 (Phase A) x = 0.49 (Phase B) and x = 0.75 (Phase C) are presented in Fig. 1a–c, respectively. To interpret the spectra the first and the second coordination sphere in the vicinity of Eu2+ ions in different crystal phases were considered. The first and the second coordination of Eu2+ in Sr2+/Ba2+ sites in Phase A, B and C are presented in Fig. 2a–c, respectively.

PLE and PL spectra of (Sr0.98-xBaxEu0.02)Si2O2N2 samples (a) x = 0.98 (b) x = 0.49 (c) x = 0.75. Emission spectra are excited at 442 nm (and additionally at 488 nm for x = 0.75). Excitation spectra were monitored at maximum of luminescence intensity.

The first and second coordination of Eu2+ central ions in three phases of (Sr0.98-xBaxEu0.02) Si2O2N2 samples (a) x = 0.98 (Phase A; orthrorhombic; Pbcn), (b) x = 0.49 (Phase B; triclinic; P1) (c) x = 0.75 (Phase C; triclinic; P1). Eu2+/Sr2+/Ba2+-ions are drawn in light grey spheres, O-ions in red spheres and N-ions in blue spheres.
PLE spectra consist of several bands related to the parity allowed transitions from the ground state 4f 7(8S7/2) to excited states of the 4f 65d1 electronic configuration of Eu2+ ions. The PL spectra consist of broad band(s) which is (are) related to the 4f 65d1 → 4f 7 (d-f) transition of Eu2+ ions. The maximum of luminescence band varies from blue to green spectral region, depending on the ratio of Ba2+ to Sr2+. The PL spectrum of the (Ba0.98Eu0.02)Si2O2N2 consists of a single band peaking at 495 nm with a full width at half maximum (FWHM) equal to 32 nm (see Fig. 1a). In the orthorhombic phase (space group Pbcn, Phase A)18 of (Ba0.98Eu0.02)Si2O2N2, Eu2+ ions can occupy Ba2+ site which is coordinated by eight oxygen ions creating cuboid and two long ranged nitrogen ions (see Fig. 2a)13,14. The Ba-O bond lengths range from 2.74 Å to 3.10 Å and the average distance is equal to 2.89 Å whilst Ba-N distances are equal for booth nitrogen and the distance is equal to 3.31 Å.
We consider this to be the first coordination sphere. The Ba2+ ions form linear chains in the lattice. The distance between two separate Ba2+ chain is almost equal to distance between Ba2+ ions in a given chain, thus the second coordination sphere consist of an Ba2+ cations forming rectangle around Eu2+ central ion (see Fig. 2a).
The PL spectrum of the (Sr0.49Ba0.49Eu0.02)Si2N2O2 is presented in Fig. 1b. The spectrum consists of a broad band with a maximum at 560 nm and FWHM equal to 80 nm. In the triclinic phase (space group P1, Phase B)12,14,16 of the (Sr0.49Ba0.49Eu0.02)Si2N2O2there are four different Sr2+/Ba2+ crystallographic sites, which can be occupy by Eu2+ ions. These four sites are very similar and have nearly the same surrounding. Each of them can be depicted as a distorted trigonal prism of six oxygen ions around Sr2+/Ba2+ which is capped by one N ion (see Fig. 2b)12,16. The Sr/Ba-O bond lengths range from 2.50 Å to 3.05 Å (the average distance is equal to 2.67 Å) and Sr/Ba-N distance is equal to 2.58 Å.
The Sr2+/Ba2+ ions in this case are in channels, which forms almost linear chains and the distance between two separate Ba2+/Sr2+ chains are longer than between Ba2+/Sr2+ ions in a given chain. Thus the second coordination sphere around central ions consist of two Sr2+/Ba2+ cations forming the line (see Fig. 2b). When Eu2+ ion is built into this chain the attractive potential of the close positive charges orients and elongates the 5d orbital of Eu2+ into the chain direction partially out of the first coordination sphere20. As the result, the 5d electron is more delocalized than in the case of Phase A, where no preferential orientation and elongation of 5d orbital occurs. Such delocalization of the 5d1 electron in Eu2+ is responsible for the fact that the energy of electron-lattice interaction in the excited state of Eu2+ is larger than in Phase A and results in a lower energy of the luminescence band and larger FWHM.
The PL spectra of the (Sr0.23Ba0.75Eu0.02)Si2O2N2 obtained with different excitation wavelength are presented in Fig. 1c. Under UV to blue excitation, the emission spectrum consists of two broad bands (solid curve). The first band peaking at 470 nm with FWHM equal to 41 nm is attributed to the d-f emission of Eu2+ occupying Sr2+/Ba2+ positions in Phase C (triclinic P1). In this phase there is one Ba2+/Sr2+ site, which can be depicted as a distorted cuboid of eight O2− ions (mean distance between oxygen ions and central ion is equal to 2.92 Å) and two long-ranged N3− ions 3.14 Å and 3.57 Å.
It is worth noting that among these three environments of Eu2+ ions (Phase A, B, C) the average distance between the Eu2+ ion and nearest O/N ions (first coordination sphere) is the largest in Phase C and smallest in Phase B. Similar but smaller is in the phase A. The differences in the average distances result in the differences in the crystal field splitting of the 4f 65d electronic manifold of Eu2+ ion. The smallest crystal field splitting hence the maximum of Eu2+ luminescence has the highest energy in Phase C. The strong shift of Eu2+ luminescence into red in Phase B is the result of the largest crystal field splitting.
The second coordination sphere around central ions consists of the Ba2+ cations forming rectangle around central ion (see Fig. 2c). This situation is similar to the case of Phase A, and there is no preferential orientation in the space and elongation of d orbitals, thus the Eu2+ luminescence is observed at high energy with a small FWHM. The second band in the yellow range of the spectrum is attributed to the occurrence of a small amount of nanocrystalline domains that contains less Ba than x = 0.75 (domains of Phase B). This could be due to inhomogenous distribution of a Ba and Sr components in the mixed (Sr0.98-xBax)Si2O2N2 crystals.
Emission spectra related to Eu2+ in domains of Phase B were obtained when the (Sr0.23Ba0.75Eu0.02)Si2O2N2 was excited with wavelength 488 nm (dotted curve in Fig. 1c). This emission consists of a broad band peaked at 575 nm with FWHM equal to 83 nm. Position of the maximum of the luminescence band allows to relate this emission to the Phase B domains of (Sr0.98-xBaxEu0.02)Si2O2N2 with concentration x = 0.6318.
Photoluminescence spectra of the (Sr0.98-xBaxEu0.02)Si2O2N2 (x = 0.98, 0.49, 0.75) measured at different high hydrostatic pressures are presented in Fig. 3(a–d). Energies of the (Sr0.98-xBaxEu0.02)Si2O2N2(x = 0.98, 0.49 and 0.75) emission maxima versus pressure are presented in Fig. 4. In Fig. 4 the green solid circles refers to the (Ba0.98Eu0.02)Si2O2N2, the orange squares refers to the (Sr0.49Ba0.49Eu0.02)Si2O2N2 and solid and open blue triangles refer to the (Sr0.23Ba0.75Eu0.02)Si2O2N2 samples.

PL spectra of (Sr0.98-xBaxEu0.02)Si2O2N2 under different pressures (a) x = 0.98 excited at 442 nm (d), x = 0.49 excited at 442 nm, (c,d) x = 0.75 excited at 442 nm and 488 nm, respectively.

Pressure dependence of the maximum of PL spectrum (Sr0.98-xBaxEu0.02)Si2O2N2 for x = 0.98 –green solid circles, x = 0.49 –orange solid squares and x = 0.75 –blue solid and open triangles.
In the case of the (Ba0.98Eu0.02)Si2O2N2 (Phase A) a red shift of the 4f 65d1 −4f 7 emission band is observed (see Fig. 3a). The energy of the emission maximum shifts linearly with increasing pressure with a rate of about-20 cm−1/kbar (see Fig. 4, green circles).
Figures 3(b) and 4 (orange solid squares) present the pressure evolution of the (Sr0.49Ba0.49Eu0.02)Si2O2N2 luminescence. Up to 40 kbar the PL spectrum with maximum at 560 nm does not change. At pressures above 40 kbar additional luminescence band with maximum at about 500 nm appears. The band with maximum at 560 nm is quenched with increasing pressure and vanishes at 50 kbar. Above 50 kbar only the band with maximum at about 500 nm is seen. Further increase of pressure up to 200 kbar causes the shift of emission maximum toward to the lower energies with rate approximately equal to −20 cm−1/kbar (see Fig. 4 orange solid squares). Such behavior of the PL of the (Sr0.49Ba0.49Eu0.02)Si2O2N2 versus pressure can be attributed to the pressure induced structural transformation of the Eu2+-ligand system. At ambient condition the (Sr0.49Ba0.49Eu0.02)Si2O2N2 has a triclinic SrSi2O2N2-type structure (Phase B). The new emission signal that appears at pressure above 40 kbar can be related to beginning of a structural transformation of the (Sr0.49Ba0.49Eu0.02)Si2O2N2. Since from 40 to 50 kbar the PL spectrum of the (Sr0.49Ba0.49Eu0.02)Si2O2N2 consist of two bands we propose that two phases coexist in this pressure range. One notices that at 50 kbar the luminescence band of the (Sr0.49Ba0.49Eu0.02)Si2O2N2 has maximum at the same energy as the (Ba0.98Eu0.02)Si2O2N2. Moreover, when pressure increases above 50 kbar the luminescence maximum shifts with the same rate (−22.8 cm−1/kbar–orange solid squares) as it has been observed for (Ba0.98Eu0.02)Si2O2N2 (−21.3 cm−1/kbar-green solid circles). This indicates that pressure-induced phase transition from triclinic (Sr0.49Ba0.49Eu0.02)Si2O2N2 (Phase B) to orthorhombic phase (Phase A) takes place.
The PL spectra of the (Sr0.23Ba0.75Eu0.02)Si2O2N2 under different pressure are presented in Fig. 3(c,d). In Fig. 3(c) the 4f 65d1 − 4f 7Eu2+ emission, obtained under excitation with wavelength 442 nm is presented. When pressure increases up to 20 kbar, energy of the maximum of emission band decreases (the red shift is approximately equal to −40 cm−1/kbar). Above 20 kbar, the red shift rate diminishes and is approximately equal to −20 cm−1/kbar (see solid blue triangles in Fig. 4) which is similar to that observed for orthorhombic (Ba0.98Eu0.02)Si2O2N2 and high pressure orthorhombic (Sr0.49Ba0.49Eu0.02)Si2O2N2 structures. We propose that this change in the rate of the PL pressure shift, is the result of phase transition from triclinic (Phase C) to orthorhombic (Phase A) phase of the (Sr0.23Ba0.75Eu0.02)Si2O2N2 system.
Figure 3(d) presents pressure evolution of the emission of the (Sr0.23Ba0.75Eu0.02)Si2O2N2 in Phase B domains, excited with wavelength 488 nm. It is expected that these domains would transform into Phase A under pressure and this actually happened (see Figs 3d and 4). Within the pressure range from ambient to 20 kbar the PL maximum does not change and the luminescence intensity gradually decrease. Above 20 kbar this luminescence is completely quenched. The phase transition in this case occurs for a lower pressure (approximately 20 kbar) than it is observed for (Sr0.49Ba0.49Eu0.02)Si2O2N2 (approximately 50 kbar) as the ratio of Ba to Sr in the domains is higher (x = 0.63).
Room temperature luminescence decays of (Sr0.98-xBaxEu0.02)Si2O2N2 for x = 0.98 (Phase A), x = 0.49 (Phase B) and x = 0.75 (Phase C and domains of Phase B) recorded at different pressures were monitored at the maximum of the 5d14f 6 → 4f 7transition. The example of the PL decays for the (Sr0.49Ba0.49Eu0.02)Si2O2N2 are shown in Fig. 5. All decays related to the Eu2+ emission in compounds under study were close to single exponential. The decay times versus pressure for all considered samples are collected in Fig. 6. The PL decay time obtained for the (Ba0.98Eu0.02)Si2O2N2 (green circles), at ambient pressure is equal to 0.38 μs and increase gradually to 0.54 μs at 200 kbar. In the absence of nonradiative processes this behavior can be expected because the luminescence lifetime elongates as the emission is red shifted with increasing pressure26.

PL decays of the (Sr0.49Ba0.49Eu0.02)Si2O2N2 for different pressures collected at maximum of luminescence under excitation of 442 nm.

Pressure dependence of the PL decay times of (Sr0.98-xBaxEu0.02)Si2O2N2 for x = 0.98 –green circles, x = 0.49 –orange squares and x = 0.75 –open and solid blue triangles.
The PL decay time obtained for the (Sr0.49Ba0.49Eu0.02)Si2O2N2 (orange squares) at ambient pressure is equal to 1.13 μs. The decay time slightly decrease with increasing pressure to 0.89 μs at 50 kbar which is accompanied by the decrease of emission intensity and at 50 kbar luminescence with maximum at 560 nm vanishes completely.
Across the phase transition at about 40 kbar, new luminescence band appeared (see Fig. 3b) and this emission decays with the time equal to 0.42 μs. Increase of pressure slightly elongates the luminescence decay like it was observed for the (Ba0.98Eu0.02)Si2O2N2 sample. The pressure-dependence of the PL decay time for the (Sr0.49Ba0.49Eu0.02)Si2O2N2 confirms our previous consideration of the structural transformation of Phase B into Phase A induced at about 40~50 kbar. The PL decay of the (Sr0.23Ba0.75Eu0.02)Si2O2N2 (Phase C) monitored at maximum of luminescence is represented by closed blue solid triangles. The decay time at ambient pressure is equal to 0.36 μs. Above 20 kbar the decay time elongates with pressure and reach the value of 0.67 μs at 200 kbar. The same behavior is observed for the (Sr0.49Ba0.49Eu0.02)Si2O2N2 above 50 kbar (after phase transition) and for the (Ba0.89Eu0.02)Si2O2N2 in all considered pressure. Pressure dependence of the PL decay time of the Phase B domains in the (Sr0.23Ba0.75Eu0.02)Si2O2N2 are represented by open blue triangles. At ambient pressure Phase B domains have a decay time equals 1.19 μs, which is close to the value obtained for the (Sr0.49Ba0.49Eu0.02)Si2O2N2 in Phase B. Pressure slightly shortens the decay time to a value of 0.99 μs for 20 kbar which is accompanied by decrease of luminescence intensity. Above 20 kbar the luminescence from Phase B domains is not present in the emission spectra of the (Sr0.23Ba0.75Eu0.02)Si2O2N2 system.
One should discuss the phenomenon of the Eu2+ luminescence in Phase B. In contrast to Phases A and C the Eu2+ emission in Phase B consists of broad band characterized by FWHM equal to 83 nm, which decays with the decay time 1.2 μs, whereas the Eu2+ emission in A and C phases is represented by much sharper band, characterized by FWHM equal to 41 nm decaying with the lifetime ~0.36 μs. Additionally very different pressure shifts of the 4f 65d → 4f 7 luminescence of Eu2+ in the Phases A and C (where it is equal to −20 cm−1/kbar and −40 cm−1/kbar, respectively) and in Phase B (where it is equal zero) have been observed. This different pressure dependences of the emission can be attributed to different interaction of the Eu2+ system in the 4f 65d1 state with the second coordination sphere of positive ions. In the case of Eu2+ in (Sr0.98-xBaxEu0.02)Si2O2N2 in Phase B, the 5d electron of Eu2+ is attracted by the positively charged chain and therefore is delocalized and can be partly located out of the first coordination sphere of negative ions. This results in large Stokes shift and FWHM of the luminescence, actually much larger than in the case of the 4f 65d → 4f 7 luminescence of Eu2+ in (Sr0.98-xBaxEu0.02)Si2O2N2 in Phase A and Phase C.
Since pressure decrease distance between Eu2+ion and ligand ions, it is expected that the pressure induces the red shift of the d-f luminescence due to the increase of crystal field splitting of 5d electronic manifold. This effect has been previously observed in many different compounds27. The first coordination sphere of the Eu2+ in (Sr0.98-xBaxEu0.02) Si2O2N2 is build with negative ions having approximately cubic symmetry. These causes the splitting of the 5d orbital into doubly degenerated the lower state (e) and triply degenerated the higher state (t)28. In the (Sr0.98-xBaxEu0.02)Si2O2N2 having Phase A the second coordination sphere is built with four positive ions (approximately C4v symmetry) which can be considered as an octahedron without vertical ions. This configuration magnifies the splitting caused by first configurational sphere and additionally splits the double degenerated the lower state (e) into the non-degenerated a1 and b1 states. As the result large pressure shift of the luminescence related to the 4f 65d → 4f 7 luminescence Eu2+ emission in Phases A and C is observed. Due to the delocalization of the 5d electron in (Sr1-xBaxEu0.02)Si2O2N2 in Phase B the energy of the lowest state of the 5d14f 6 electronic manifold can be not sensitive to actual positions of negative ions forming the first coordination sphere and finally does not dependent on pressure.
Raman spectroscopy has been applied to confirm structure changes of (Sr0.98-xBaxEu0.02)Si2O2N2 for x = 0.1, 0.3, 0.49, 0.75 and 0.98 and pressure-induced phase transition of the (Sr0.98-xBaxEu0.02)Si2O2N2. The ambient pressure Raman spectra of (Sr0.98-xBaxEu0.02)Si2O2N2 for different Ba concentrations are presented in Fig. 7. The crystal structure of oxynitridosilicates (Sr0.98-xBaxEu0.02)Si2O2N2 are built of SiON3 tetrahedral layers with the same general topology for all samples. It is expected that internal stretching and bending vibrations of the tetrahedral SiON3 groups contribute to Raman peaks in the range 400–1200 cm−1 for all samples29,30. In this spectral region, the Raman spectrum of (Sr0.98-xBaxEu0.02)Si2O2N2 for x = 0.98 is dominated by an intense lines with energies equal to 415 cm−1 (labeled νa) and 1020 cm−1 and for x ≤ 0.75, 401~405 cm−1 (labeled νc and νb) and 1020 cm−1. Difference between position of νa with respect to νb and νc peaks are due to different degree of distortion of SiON3 groups. The external vibrations involving the entire SiON3 as well as their neighboring Sr2+/Ba2+ ions are observed below 400 cm−1 29,30. Here the highest intensity peaks in the spectrum of (Sr0.98-xBaxEu0.02)Si2O2N2 (x = 0.1) are detected at 244 cm−1, 261 cm−1, 278 cm−1 and 298 cm−1. The substitution of Sr2+ by Ba2+ up to x = 0.49 causes broadening of observed peaks but the specific Raman peak pattern is preserved. For Ba2+ content x = 0.75 and x = 0.98 apparently different Raman pattern is observed below 400 cm−1. The highest intensity peaks in the spectrum (below 400 cm−1) for (Sr0.98-xBaxEu0.02)Si2O2N2 (x = 0.75) are observed at 275 cm−1 and 260 cm−1, whereas for (Ba0.98-xEu0.02)Si2O2N2 (x = 0.98) the highest intensity peaks are observed at 201 cm−1, 237 cm−1, 259 cm−1 and 289 cm−1. Difference in position and relative intensity of Raman peaks of studied samples clearly illustrate that three different phases exist across (Sr0.98-xBaxEu0.02)Si2O2N2 series and confirmed the previous XRD results18.

Raman spectra of (Sr0.98-xBaxEu0.02)Si2O2N2 for different Ba content x.
To verify our hypothesis about pressure-induced phase transformations from triclinics (Phase B and C) to orthorhombic phase (Phase A) Raman spectra of the (Sr0.98-xBaxEu0.02)Si2O2N2 for x = 0.49, 0.75 and 0.98 have been measured at different pressures up to ~80 kbar and the results are presented in Fig. 8a–c. It should be noted here that in our experiments the Eu2+ luminescence with laser beam excitation of 632 nm contributes greatly to the background level causing the Raman spectra were very difficult or even impossible to measure under pressure greater than 80 kbar. In the case of (Ba0.98Eu0.02)Si2O2N2 (see Fig. 8a). It is seen that due to the decrease of bonding length, energy of all Raman peaks increases with pressure. This increase is observed with a slightly different pressure rate for all lines causing that some of unresolved peaks at ambient pressure (see for example νa) start to be resolved at higher pressure, but the main Raman pattern and number of peaks does not change with pressure. These result confirms findings from luminescence studies, that the orthorhombic phase of (Ba0.89Eu0.02)Si2O2N2 is preserved in measured pressure range. Figure 8b shows high-pressure Raman spectra of (Sr0.49Ba0.49Eu0.02)Si2O2N2. The most interesting effect occurs above 32 kbar where a significant change in the Raman lines structure is observed. Let us focus on the most intensive vibration peak νb related to the internal stretching and bending vibrations of the SiON3 groups. It is seen that due to the increase of Si–O/N bonding strength energy of νb increases when pressure increases. In the range of pressure between 40 kbar and 50 kbar additional peak labeled νa appears and simultaneously intensity of νb decreases. Above 50 kbar peak νa remains exclusively. The νa Raman line can be attributed to an orthorhombic structure of the (Ba0.98Eu0.02)Si2O2N2. The appearance of νa peak at ~42 kbar confirms the phase transition of the (Sr0.49Ba0.49Eu0.02)Si2O2N2 from triclinic to orthorhombic phase. Figure 8c shows high-pressure Raman spectra of (Sr0.23Ba0.75Eu0.02)Si2O2N2 which is representative of Phase C. Also in this case we have observed significant changes in Raman spectra but at lower pressure of about 26 kbar suggesting a structural transition occurs at this point. At this pressure additional peak labeled νa appears and simultaneously intensity of νc decreases. Also obvious changes in Raman spectra below 400 cm−1 are observed. Upon decompression, the luminescence and structural properties of the (Sr0.98-xBaxEu0.02)Si2O2N2 (x = 0.98, 0.49 and 0.75) of these crystals returns to ambient state. It means that the high-pressure induced phase transition of the (Sr0.98-xBaxEu0.02)Si2O2N2 (0.49 and 0.75) is reversible.

Raman spectra of the (Sr0.98-xBaxEu0.02)Si2O2N2 for (a) x = 0.98 (b) x = 0.49 and (c) x = 0.75 at different pressures.
In summary we developed the phase diagram of (Sr0.98-xBaxEu0.02)Si2O2N2 which has been shown on Fig. 9. The dotted lines represent the phase boundaries between different phases in the pressure and Ba content space. Two points corresponding to pressures of phase transitions for (Sr0.98-xBaxEu0.02)Si2O2N2 x = 0.3 and x = 0.1 (black squares in Fig. 9) were obtained at high pressure luminescence experiments, however the respective luminescence and Raman spectra are not presented in this contribution. The phase diagram shows that pressure in which phase transition between Phase B and A occurs decreases linearly with increasing Ba concentration in (Sr0.98-xBaxEu0.02)Si2O2N2. The Boundary between Phase B and A is indicated by dotted red line. For concentration rage 0.65 < x < 0.78 and pressure lower than 20 kbar the (Sr0.98-xBaxEu0.02)Si2O2N2 system exists in Phase C. The boundary between Phase B and C is indicated by dotted black line.

Phase diagram of (Sr0.98-xBaxEu0.02)Si2O2N2.
Methods
Chemicals and Materials
SrCO3 (≥99.9%), SiO2 (≥99.995%), Si3N4 (≥99.9%) and Eu2O3 (≥99.99%) were purchased from Aldrich Corporation. BaCO3 (≥99.9%) was purchased from J.T. Baker Corporation. All of the initial chemicals were used without further purification. Aluminum oxide crucibles and cylindrical molybdenum crucibles (20 mm × 50 mm) were used in the sintering process of the samples.
Synthesis
A series of oxonitridosilicate compounds, (Sr0.98-xBaxEu0.02)Si2O2N2 (0.1 ≤ x ≤ 0.98), was prepared using two-step solid-state reaction processes. Nonstoichiometric amounts of SrCO3, BaCO3, SiO2 and Eu2O3 powders, in which the molar ratio of (Sr, Ba)/Si = 0.8 were ground in an agate mortar for 30 min to form a homogeneous mixture. The mixture was then placed in aluminum oxide crucible and fired at 1250 to 1350 °C for 6 h under flowing 95%N2−5% H2 atmosphere in a horizontal tube furnace. The sintered products were ground, yielding crystalline powder (Sr0.98-xBaxEu0.02)Si2O2N2 (0.1 ≤ x ≤ 0.98). The crystalline powder was mixed with stoichiometric amounts of Si3N4 and reground for 30 min in an agate mortar. After forming a homogeneous mixture, the mixture was loaded into a cylindrical molybdenum crucible with a screw-cap and fired again at 1500 to 1550 °C for 6 h under flowing 95% N2−5%H2 atmosphere in the horizontal tube furnace. The second sintered products were ground again, yielding the resulting phosphor powder. The details of samples preparation and ambient pressure structural characterization were presented and discussed in paper18.
Spectroscopic characterization
Raman spectra were recorded at room temperature and ambient pressure on a Horiba JobinYvon Lab Ram Aramis spectrometer with a He-Ne laser providing excitation light at 633 nm with the 1200 l/mm grating.
Photoluminescence excitation spectra were acquired using a FluoroMax-4P TCSPC spectrofluorometer produced by Horiba, containing Czerny-Turner monochromators for excitation and emission. An excitation source in this system was a 150-W ozone-free Xenon lamp. Fluorescence intensity was measured using a R928 Side-on photomultiplier. Steady state luminescence spectra were excited with He-Cd laser with the wavelength of 442 nm. The photoluminescence spectra were recorded on a SR-750-D1 luminescence spectrometer with an Andor CCD camera DU420A-OE type, which can detect signal on large wavelength range 250–1000 nm with an accuracy of 0.5 nm. The spectra were corrected for instrumental spectral response using a standard lamp.
The experimental setup for luminescence kinetics consists of a PL 2143 A/SS laser as the excitation source and a PG 401/SH parametric optical generator. This system can generate 30 ps laser pulses, with the frequency of 10 Hz with wavelengths ranging from 220 to 2200 nm. The emission signal was analyzed by a Bruker Optics 2501S spectrometer and the Hamamatsu Streak Camera model C4334-01 with a final spectral resolution 0.47 nm. Luminescence decays were obtained by the integration of streak camera images over the wavelength intervals. Details of the experimental setup are described in the paper31. High hydrostatic pressure was applied in a Merrill Bassett type DAC32. Polydimethylsiloxane oil was used as the pressure-transmitting medium and pressure was measured by the shift of the R1 luminescence line of ruby.
Additional Information
How to cite this article: Lazarowska, A. et al. Structural phase transitions and photoluminescence properties of oxonitridosilicate phosphors under high hydrostatic pressure. Sci. Rep. 6, 34010; doi: 10.1038/srep34010 (2016).
References
Xie, R.-J. & Hirosaki, N. Silicon-based oxynitride and nitride phosphors for white LEDs—A review. Sci. Technol. Adv. Mater. 8, 588–600 (2007).
Xie, R.-J., Hirosaki, N., Li, Y. & Takeda, T. Rare-earth activated nitride phosphors: synthesis, luminescence and applications. Materials 3, 3777–3793 (2010).
Yamamoto, H. & Yamamoto, T. Phosphors for white LEDsInNitride Semiconductor Light-Emitting Diodes (LEDs) Materials, Technologies and Applications (Woodhead, Cambridge, 2014).
Zeuner, M., Pagano, S. & Schnick, W. Nitridosilicates and oxonitridosilicates: from ceramic materials to structural and functional diversity. Angew. Chem., Int. Ed. 50, 7754–7775 (2011).
Lin, C. C. & Liu, R. S. Advances in phosphors for light-emitting diodes. J. Phys. Chem. Lett. 2, 1268–1277 (2011).
Zeuner, M., Schmidt, P. J. & Schnick, W. One-pot synthesis of single-source precursors for nanocrystalline LED phosphors M2Si5N8:Eu2+ (M = Sr, Ba). Chem. Mater. 21, 2467–2473 (2009).
Lee, B., Lee, S., Jeong, H. G. & Sohn, K. S. Solid-state combinatorial screening of (Sr,Ca,Ba,Mg)2Si5N8:Eu2+ phosphors. ACS. Comb. Sci. 13, 154–158 (2011).
Zhang, Z. J. et al. Photoluminescence properties of Yb2+ in CaAlSiN3 as a novel red-emitting phosphor for white LEDs. J. Mater. Chem. 22, 23871–23876 (2012).
Li, Y. Q., Hirosaki, N., Xie, R. J., Takeda, T. & Mitomo, M. Yellow-orange-emitting CaAlSiN3:Ce3+ phosphor: structure, photoluminescence and application in white LEDs. Chem. Mater. 20, 6704–6714 (2008).
Uheda, K. et al. Luminescence properties of a red phosphor, CaAlSiN3:Eu2+, for white light-emitting diodes. Electrochem. Solid State Lett. 9, H22–H25 (2006).
Höppe, H. A., Stadler, F., Oeckler, O. & Schnick, W. Ca[Si2O2N2]-a novel layer silicate. Angew. Chem. Int. Ed. 43, 5540–5542 (2004).
Oeckler, O., Stadler, F., Rosenthal, T. & Schnick, W. Real structure of SrSi2O2N2 . Solid State Sci. 9, 205–212 (2007).
Kechele, J. A., Oeckler, O., Stadler, F. & Schnick, W. Structure elucidation of BaSi2O2N2–A host lattice for rare-earth doped luminescent materials in phosphor-converted (pc)-LEDs. Solid State Sci. 11, 537–543 (2009).
Seibald, M., Rosenthal, T., Oeckler, O. & Schnick, W. Highly efficient pc-LED phosphors Sr1-xBaxSi2O2N2:Eu2+ (0 ≤ x ≤ 1)-crystal structures and luminescence properties revisited. Crit. Rev. Solid State Mater. Sci. 39, 215–229 (2014).
Seibald, M. et al. Unexpected luminescence properties of Sr0.25Ba0.75Si2O2N2:Eu2+- a narrow blue emitting oxonitridosilicate with cation ordering. Chem. Eur. J. 18, 13446–13452 (2012).
Stadler, F. et al. Crystal structure, physical properties and HRTEM investigation of the new oxonitridosilicate EuSi2O2N2 . Chem.-Eur. J. 12, 6984–6990 (2006).
Li, Y. Q., Delsing, A. C. A., de With, G. & Hintzen, H. T. Luminescence properties of Eu2+-activated alkaline-earth silicon-oxynitride MSi2O2-δN2+2/3δ (M = Ca, Sr, Ba): a promising class of novel LED conversion phosphors. Chem. Mater. 17, 3242–3248 (2005).
Li, G. et al. Photoluminescence tuning via cation substitution in oxonitridosilicate phosphors: DFT calculations, different site occupations and luminescence mechanisms. Chem. Mater. 26, 2991–3001 (2014).
Joos, J. J., Botterman, J. & Smet, P. Evaluating the use of blue phosphors in white LEDs: the case of Sr0.25Ba0.75Si2O2N2:Eu2+. J. Solid State Lighting 1, 1–16 (2014).
Poort, S. H. M., Blokpoel, W. P. & Blasse, G. Luminescence of Eu2+ in barium and strontium aluminate and gallate. Chem. Mater. 7, 1547–1551 (1995).
Gong, C. et al. Structural phase transition and photoluminescence properties of YF3:Eu3+ nanocrystals under high pressure. J. Phys. Chem. C 118, 22739–22745 (2014).
Lazarowska, A., Mahlik, S., Krosnicki, M., Grinberg, M. & Malinowski, M. Pressure-induced phase transition in LiLuF4:Pr3+ investigated by an optical technique. J. Phys. Cond. Matter. 24, 115502 (2012).
Ermakova, O. et al. Equation of state for Eu-doped SrSi2O2N2 . J. Chem. Phys. 141, 014705 (2014).
Grinberg, M. Excited states dynamics under high pressure in lanthanide-doped solids. J. Lumin. 131, 433–437 (2011).
Mahlik, S., Lazarowska, A., Grinberg, M., Liu, T.-C. & Liu, R.-S. Luminescence spectra of β-SiAlON/Pr3+ under high hydrostatic pressure. J. Phys Chem. C 117, 13181–13186 (2013).
Poort, S. H. M., Meyerink, A. & Blasse, G. J. Phys. Chem. Solids 58, 1451–1456 (1997).
Grinberg, M. Excited states dynamics under high pressure in lanthanide-doped solids. J. Lumin. 131, 433–437 (2011).
Bersuker, I. B. Electronic Structure and Properties of Transition Metal Compounds, Introduction to Theory (John Wiley & Sons Inc, New York, 1996).
Kahlenberg, V. et al. Rietveld analysis and raman spectroscopic investigations on α-Y2Si2O7 . Anorg. Chem. 634, 1166–1172 (2008).
Nasdala, L., Smith, D., Kaindl, R. & Ziemann, M. Raman Spectroscopy: Analytical Perspectives in Mineralogical Research (Eotvos University Press, Budapest, 2004).
Kubicki, A. A., Bojarski, P., Grinberg, M., Sadownik, M. & Kukliński, B. Time-resolved streak camera system with solid state laser and optical parametric generator in different spectroscopic applications. Opt. Comm. 269, 275–280 (2006).
Merrill, L. & Bassett, W. A. Miniature diamond anvil pressure cell for single crystal x‐ray diffraction studies. Rev. Sci. Instr. 45, 290–294 (1974).
Acknowledgements
This study has been supported by the National Centre for Research and Development (projects: NOPLID, PL-TWII/8/2015 and TAZOLED, PBS3/A5/48/2015). R.S.L. would like to thank the Ministry of Science and Technology of Taiwan (contract numbers MOST 104-2113-M-002-012-MY3 and 104-2923-M-002-007-MY3). G.L. thanks the National Natural Science Foundation of China (Grant No. NSFC 21301162) for financially supporting this research. The contribution of S. M. was supported by the grant “Iuventus Plus” 0271/IP3/2015/73 from the Ministry of Science and Higher Education.
Author information
Authors and Affiliations
Contributions
A.L. and S.M. conceived and perform experiments. A.L. was primarily responsible for the experiments. R.S.L. and G.L. synthesized the Sr0.98-xBaxEu0.02)Si2O2N2 (0 ≤ x ≤ 0.98) samples. M.G., A.L. and S.M. interpreted the spectroscopic data. A.L. wrote the main manuscript text, M.G. and S.M. edited the manuscript. All the authors discussed the results and reviewed the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Lazarowska, A., Mahlik, S., Grinberg, M. et al. Structural phase transitions and photoluminescence properties of oxonitridosilicate phosphors under high hydrostatic pressure. Sci Rep 6, 34010 (2016). https://doi.org/10.1038/srep34010
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep34010
- Springer Nature Limited