
Abstract
Dimethylsulfoniopropionate (DMSP), which is the major source of organic sulfur in the world’s oceans, plays a significant role in the global sulfur cycle. This compound is rapidly degraded by marine bacteria either by cleavage to dimethylsulfide (DMS) or demethylation to 3-methylmercaptopropionate (MMPA). The diversity of genes encoding bacterial demethylation (dmdA) and DMS production (dddL and dddP) were measured in Arctic Kongsfjorden. Both dmdA and dddL genes were detected in all stations along a transect from the outer to the inner fjord, while dddP gene was only found in the outer and middle parts of the fjord. The dmdA gene was completely confined to the Roseobacter clade, while the dddL gene was confined to the genus Sulfitobacter. Although the dddP gene pool was also dominated by homologs from the Roseobacter clade, there were a few dddP genes showing close relationships to both Alphaproteobacter and Gammaproteobacter. The results of this study suggest that the Roseobacter clade may play an important role in DMSP catabolism via both demethylation and cleavage pathways in surface waters of Kongsfjorden during summer.
Similar content being viewed by others


Introduction
Dimethylsulfoniopropionate (DMSP) is an organic sulfur compound that occurs worldwide in large amounts (109 tons or more per year) in marine algae and plants1, where it may function as an osmolyte, cryoprotectant, predator deterrent, and antioxidant2,3,4,5. When released into seawater, DMSP is rapidly catabolised by bacteria via two enzymatic pathways: demethylation and cleavage6,7,8. The demethylation pathway converts a large fraction (50 to 90%) of dissolved DMSP to 3-methylmercaptopropionate (MMPA), which is subsequently incorporated into the biomass of microbial cells7,9. Over half of the bacterioplankton cells in ocean surface waters are capable of conducting demethylation of the phytoplankton metabolite DMSP10. The alternative cleavage pathway transforms the remaining part of the dissolved DMSP via various DMSP lyases to dimethylsulfide (DMS) and acrylate (or 3-hydroxypropionate)11,12. DMS represents the most important natural source of sulfur to the atmosphere, where its oxidation products cause cloud nucleation and may affect weather and climate13. DMS emissions can exert a greater influence on climate in regions in which low aerosol concentrations prevail, such as in the Arctic during summer14.
The only gene found to encode the first step in the DMSP demethylation pathway (dmdA) is known to be harboured by the Roseobacter clade, SAR11, SAR116 member “Candidatus Puniceispirillum marinum” IMCC1322, and Gammproteobacteria10,15,16. To date, seven DMSP lyase genes involved in the DMSP cleavage pathway, dddD, dddK, dddL, dddP, dddQ, dddY, and dddW, have been identified from bacterial isolates1,17. Among the DMSP lyase genes, dddP and dddQ are the most abundant ddd genes in the Global Ocean Sampling (GOS) data set18,19. The dddP gene occurs frequently in marine Alphaproteobacteria (e.g., the Roseobacter clade, SAR11 and SAR116), as well as in Gammaproteobacteria and some Ascomycete fungi1,17,18,20,21. The dddQ gene is found in the Roseobacter clade and SAR111,17. To date, most of our understanding of microbial DMSP metabolism in marine environments has come from studies conducted at low and mid-latitudes. Recent studies in Arctic waters indicate that bacteria primarily use DMSP as a carbon source, resulting in conversion efficiencies of DMSP into DMS of up to 31%22,23. However, studies of the abundance, diversity and biogeography of DMSP degradation genes in high-latitude marine environments remain insufficient.
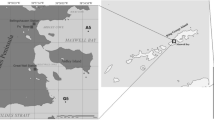
Kongsfjorden is a glacial fjord in the Arctic situated on the western side of Spitsbergen in the Svalbard archipelago that is influenced by the inflow of transformed Atlantic water as well as by meltwater of glacial origin (Fig. 1)24. In early summer in a post-spring bloom environment, DMSP, DMS, and chlorophyll a concentrations in water are 10 nmol l−1, 1.5 nmol l−1 and 0.21 μg l−1, respectively25. To describe the distribution of DMSP degradation genes relative to surface water and identify the bacterial communities potentially associated with these genes, we collected seawater samples following transect from the outer to the inner part of Kongsfjorden and studied the diversity of dmdA, dddL and dddP genes using a clone library method.

Maps of sampling sites in Kongsfjorden, Svalbard.
The left image was created using CorelDRAW 8 software, and the right image was created using Ocean Data View 4.1.3 (https://odv.awi.de/).
Results
Microbial cell abundance
Based on DAPI analysis, the microbial cell abundance in Kongsfjorden ranged from 1.40 × 109 to 3.26 × 109 cells l−1 and was highest at inner fjord station K5 (Table 1). To investigate the abundance of eubacteria and archaebacteria in surface seawater, sample K3 was analyzed using oligoFISH probes EUB338 and ARCH915. Remarkably, eubacterial abundance (on average 1.20 × 109 cells l−1) was one order of magnitude higher than archaebacterial abundance (on average 1.43 × 108 cells l−1). High detection of DAPI-stained cells by EUB338 using oligoFISH has been reported from surface samples collected from the California coast26. The percentage of bacteria detected by probe EUB338 was 85% of the cells detected with DAPI for sample K3. OligoFISH probe ARCH915 can unspecifically stain a population of marine bacteria27. The results of this study are consistent with those of a previous study of the prokaryotic community in surface waters that showed the population in Kongsfjorden was dominated by bacteria, while archaea only accounted for 1% of the DAPI-stained cells28.
Primer test for dmdA gene amplification
To check the validity of primers for dmdA gene amplification, primer pairs dmdAUF160 and dmdAUR697, and dmdA282F and dmdA591R were used on reference strain Ruegeria pomeroyi DSS-3, which contains the identified dmdA gene29. Using primer pair dmdAUF160 and dmdAUR697, two PCR product bands (580 bp and 373 bp) were obtained with significant similarity (99.7–100%) to the 23S rRNA gene and trimethylamine methyltransferase gene of Ruegeria pomeroyi DSS-3. Non-dmdA sequences captured by the universal dmdA primer set dmdAUF160 and dmdAUR697 have also been found in Gulf of Mexico seawater microcosoms10. In contrast, analysis with primer pair dmdA282F and dmdA591R produced a single band of 309 bp with 99.6% sequence similarity to the dmdA gene of Ruegeria pomeroyi DSS-3. Therefore, primers dmdA282F and dmdA591R were used in this study for dmdA gene clone library construction.
Bacterial dmdA gene clone libraries and distribution of dmdA genes
Three dmdA gene clone libraries were constructed from the environmental DNA of surface water samples along a transect from the outer (K1) to the inner (K5) part of the fjord (Table 2). A total of 263 clones showing a single band of approximately 306 bp resulted in107 operational taxonomic units (OTUs). Among them, three OTUs shared in the three libraries, accounted for 17%, 18% and 14% of clone library K1, K3 and K5, respectively. Shannon diversity index was higher at outer fjord station K1. However, pairwise comparison revealed no significant differences among the three libraries.
Based on 90% amino acid identity, a phylogenetic tree of these DmdA clone sequences (Fig. 2) yielded 19 major clusters (termed A1 to A8, B1 to B8, and C1 to C3), accounting for 95% of the total clone sequences. All sequences were distributed within the putative dmdA genes of strains belonging to the Roseobacter clade within the Rhodobacteraceae family. Clones affiliated with the genera Sulfitobacter (including Clusters A1, A2, A3, A4, B1, B2, B3 and C1) and Roseovarius (i.e., Cluster C2), and a member of the Roseobacter clade (i.e., Cluster C3) accounted for 28%, 25%, and 18% of the total clones, respectively. In addition, dmdA genotypes within Clusters C1, C2 and C3 found in all samples accounted for more than 44% of the total clones. Sequences affiliated with the genera Rhodobacter and Thalassobius were only observed in sample K1. However, sequences within Cluster B5 were more frequently observed at the inner station K5 than at the outer station K1.

Phylogenetic tree of deduced DmdA sequences from three clone libraries of seawater in Kongsfjorden as well as those from known bacterial species available in NCBI.
Cluster A indicates genotypes only observed in station K1, Cluster B indicates genotypes shared by stations K1 and K3 (or K5), and Cluster C genotypes shared among all stations. Bootstrap values of <50 have been removed for clarity. Numbers in parentheses following clone names or cluster names indicate the number of sequences found in stations K1, K3, and K5, respectively. The scale bar indicates evolutionary distance.
Bacterial dddL gene clone libraries and distribution of dddL genes
A total of 200 clones showing a single band of 143–339 bp were sequenced. Most of these were annotated as hypothetical protein, methionine-tRNA ligase and other enzymes, while only a total of ten clones showing a single band of 244 bp were distributed within the putative dddL genes of the genus Sulfitobacter (Fig. 3). The dddL gene sequences in genus Sulfitobacter were found in all investigated stations.

Phylogenetic tree of dddL gene sequences from three clone libraries of seawater in Kongsfjorden as well as those from known bacterial species available in NCBI.
Bootstrap values of <50 have been removed for clarity. Numbers in parentheses following clone name indicate the number of sequences found in stations K1, K3, and K5, respectively. The scale bar indicates evolutionary distance.
Bacterial dddP gene clone libraries and distribution of dddP genes
Two dddP gene clone libraries were constructed from samples from the outer (K1) and middle (K3) stations. No PCR product or a concentration of product too low was observed in sample K5, resulting in a failure of dddP gene clone library construction. A total of 87 clones showing a single band of approximately 835 bp resulted in 37 OTUs. Among them, three OTUs shared by the two libraries accounted for 52% and 69% of clone libraries K1 and K3, respectively. Pairwise comparison showed that there was no significant difference between the two libraries.
Based on 81% amino acid identity, a phylogenetic tree of these DddP clone sequences (Fig. 4) yielded three clusters (termed P1 to P3), which were shared by the two libraries. All sequences were distributed within the putative dddP genes. Sequences within Cluster P1 accounted for 96% and 84% of clone libraries K1 and K3, respectively, and showed high sequence similarities to DddP of Roseovarius nubinhibens (NCBI: WP_009813101). Genotypes within Clusters P2 and P3 were also affiliated with the Roseobacter clade. The DddP sequences formed a cluster separately from the cluster of DddD sequences from Pseudomonas species (Fig. 4).

Phylogenetic tree of deduced DddP sequences from two clone libraries of seawater in Kongsfjorden plus those from known bacterial species available in NCBI.
DddD sequences from Pseudomonas species were used as an outgroup. Bootstrap values of <50 have been removed for clarity. Numbers in parentheses following cluster names indicate the number of sequences found in stations K1 and K3, respectively. The scale bar indicates evolutionary distance.
Discussion
In Kongsfjorden, maximum bacterial abundance occurs in May and July30. Consistent with a previous study that showed the prokaryotic community in Kongsfjorden was dominated by bacteria28, the results of this study showed that bacteria was present in levels at least one order of magnitude more abundant than archaea, indicating that they may play a more important ecological role in the planktonic microbial community in Kongsfjorden. A 16S rRNA gene-based pyrosequencing survey revealed that sequences affiliated with the Roseobacter clade, including Loktanella, Octadecabacter, Paracoccus, Phaeobacter, Roseobacter and Sulfitobacter, comprise only a small percentage (2.3–6.9%) of the total clones at each station in the fjord31. However, Roseobacter clade members accounted for 97.7% of the Alphaproteobacteria31. In the present study, dmdA genotypes, and the identified dddL genotype were all clustered into the Roseobacter clade. In addition, sequences affiliated with the Roseobacter clade dominated dddP genotypes in the fjord. Previous study has shown that the Roseobacter groups play a dominant role in DMS production via DddP lyases in coastal waters of the northwestern Pacific Ocean20. These findings suggest that, despite its low abundance, the Roseobacter clade plays an important role in degradation of DMSP in surface waters of Kongsfjorden.
The DMSP demethylase gene (dmdA) is highly abundant in surface ocean waters15,29, while the DMSP lyase genes, including dddD, dddL and dddP, are in low abundance in bacteria from surface ocean waters15,18. When compared to dmdA sequences, which were detected at all investigated stations in Kongsfjorden, no dddP sequences were found at the inner station, and only a few identified dddL sequences were detected in this study. Previous studies have shown that the major route of bacterially-mediated DMSP catabolism in marine environments is via a demethylation process7,32,33, suggesting that most of the DMSP sulfur may also be assimilated by bacteria in Kongsfjorden. However, further study is required to perform quantitative analysis of the DMSP degradation genes to ascertain which catabolism pathway contributes more to the DMSP degradation in Kongsfjorden. A study using a 35S DMSP tracer to track the partitioning of DMSP sulfur to various products in oceanic and coastal seawater demonstrated that 60% of DMSP was assimilated by cells in coastal samples, while only a small portion of the DMSP was routed through the DMSP-cleavage pathway and DMS production34. Similar results have reported that DMS production from DMSP is a minor bacterial metabolic pathway in the Canadian High Arctic from northern Baffin Bay to the Beaufort Sea, as well as in Lancaster Sound22,23. In contrast, only 16% of DMSP was assimilated in ocean samples, while the remainder was transformed to dissolved non-volatile products34, which were probably formed by oxidation to DMSO and sulfate35. Cells in the coastal areas are likely to have higher growth rates and therefore increased sulfur demand, causing more sulfur to be incorporated into protein35. The primary sources of DMSP in marine surface waters are micro and macro-algae36. In Kongsfjorden, the maximum abundance of plankton occurs in July30. The capacity of the microbial community to take up dissolved DMSP and convert it into DMS increased with increasing phytoplankton biomass22. In Kongsfjorden, elevated DMSP production at high hydrogen concentration was largely a consequence of increased dinoflagellate biomass, and in particular, the increased abundance of the species Heterocapsa rotundata25. However, the increased DMSP production was not transformed into higher DMS concentrations. Because bacteria can both release DMS from DMSP and consume DMS as a major sink for DMS in the ocean37,38, this may have been a function of both increased demand for DMSP driven by elevated bacterial production and increased bacterial DMS consumption25. Bacteria consume DMSP in proportion to their abundance23. DMSP consumed by bacteria can then be routed through either one of two competing pathways, the demethylation or cleavage pathways. Experiments conducted with radiolabeled 35S-DMSP showed that bacteria switch from the demethylation pathway to the production of DMS when their sulfur demand is satisfied39.
The dmdA gene is largely confined to two groups of marine bacteria, the Roseobacters and the SAR11 clade1,35. Like the Roseobacters (e.g. Ruegeria pomeroyi DSS-3), Pelagibacterales strains of SAR11 have both the DMSP demethylation and cleavage pathways17. However, in Kongsfjorden, the dmdA genes in surface seawaters were entirely confined to the Roseobacters, showing a difference from the tropical and subtropical Pacific Ocean, where SAR11 bacterioplankton dominate the dmdA gene pool40,41. No sequence affiliated with SAR11 was detected in Kongsfjorden by pyrosequencing of 16S rRNA genes, whilst the Roseobacters dominated Alphaproteobacteria in bacterioplankton community31. Sequences affiliated with the genera Sulfitobacter and Roseovarius dominated the dmdA genotypes in Kongfjorden. At the same time, dddL sequences were confined to the genus Sulfitobacter, while sequences in the genus Roseovarius dominated dddP genotypes. Previous study of bacterioplankton communities in Kongsfjorden shows that sequences affiliated with the genus Sulfitobacter accounted for 55.8% of the Alphaproteobacteria31. However, sequence affiliated with the genus Roseovarius was not detected in bacterioplankton community31. Members of the Roseobacter clade, including the genera Roseobacter, Roseovarius and Ruegeria, have been found to can both demethylate and cleave DMSP11,18,42. Further study is required to examine whether both demethylation and cleavage pathways for DMSP occur in the same bacterium of the genus Sulfitobacter.
The dddP gene is one of the most frequently detected ddd genes in marine bacteria and is mainly found in the Roseobacter and SAR116 clades of Alphaproteobacteria18,20,43. However, evidence for horizontal gene transfer of dddP to some Gammaproteobacteria21 and fungal species18 has been reported. In the present study, the DddP sequence of Pseudomonas sp. DMSP-1, which is able to cleave DMSP into DMS and acrylate (data not shown), was found to be closely related to Cluster P2 and Roseicitreum antarcticum ZS2-28 (Fig. 4), suggesting a possible inter-class horizontal gene transfer of dddP between Alpha and Gammaproteobacteria. In addition, comparing with the absence of the genus Roseovarius, which dominated dddP genotypes in this study, the genera Sulfitobacter and Loktanella were the dominant members of the Alphaproteobacteria in bacterioplankton community in Kongsfjorden31, suggesting a possible inter-genus horizontal gene transfer of dddP in Alphaproteobacteria. Further study is required to verify the horizontal gene transfer of dddP to some bacteria in the seawater of Kongsfjorden.
Previous study shows that cloned sequences amplified with primer pair dddP_277F and dddP_1112R are distributed within Alphaproteobacteria and fungi43. Our study on Antarctic samples (unpublished) also showed that cloned dddP sequences were distributed within Alphaproteobacteria and Gammproteobacteria. In addition, amplified with primer set dddLf and dddLr, dddL genes were detected in Pseudomonas within the Gammaproteobacteria44 and Sulfitobacter within the Alphaproteobacteria in this study. Those results indicate that the primer sets used for amplification of dddL and dddP genes are not too biased towards the Roseobacter clade gene forms. Amplified with a universal dmdA primer set dmdAUF160 and dmdAUR697, a total of 239 sequences affiliated with Ruegeria pomeroyi DSS-3 were found in Gulf of Mexico10. However, in this study dmdA gene was not detected in R. pomeroyi DSS-3 using primer set dmdAUF160 and dmdAUR697. Though dmdA gene was successfully detected in R. pomeroyi DSS-3 using primer set dmdA282F and dmdA591R in this study, further study is required to check the specificity and coverage of the newly designed primers in the future, because that primer set might only capture dmdA genes of the marine Roseobacter clade.
The diversity of dmdA, dddL and dddP genes in surface seawaters of Kongsfjorden showed that planktonic bacteria in this Arctic fjord possess DMSP degradation genes for both demethylation and cleavage pathways. The Roseobacter clade (represented by the genera Sulfitobacter and Roseovarius) may play an important role in bacterial DMSP consumption. As PCR amplification is not quantitative in the same way as metagenomic data sets, a catalyzed reporter deposition-fluorescence in situ hybridization (CARD-FISH) approach should be considered for quantification of planktonic marine bacteria harbouring different DMSP degradation genes in further studies. In the marine environment, the prevalence of different bacterial DMSP degradation pathways is regulated by a complex set of factors including UV-A dose, temperature, carbon supply, and DMSP availability in relation to bacterial sulfur demand6,45,46. However, regulation mechanisms of the competing DMSP degradation pathways have not yet been thoroughly understood. A previous study shows that the best correlate with shifts of the dominant dmdA clades in response to an induced phytoplankton bloom was chlorophyll a concentration11. Thus, further studies of the abundance and distribution of DMSP degradation genes and the corresponding bacterial community structure in Kongsfjorde before, during, and after the phytoplankton bloom would help our understanding of DMSP catabolism pathways and environmental factors regulating these pathways in Arctic coastal waters.
Methods
Sample collection
Surface water samples were collected along a transect from the outer (K1) to the inner part (K5) of Kongsfjorden during the Chinese Arctic Yellow River Station Expedition in July of 2011 (Fig. 1). Sampling locations, physical factors and biogeochemical properties of the samples are summarized in Table 1. After collection, 50 ml of each water sample were fixed with buffered glutaraldehyde to a final concentration of 2% for 18 h at 4 °C, then filtered onto 0.2 μm pore-sized black polycarbonate membrane (Millipore, Germany) under low vacuum (<150 mm Hg). In addition, a 50 ml water subsample of K3 fixed with buffered glutaraldehyde was filtered onto 0.2 μm pore-sized white polycarbonate membrane (Millipore, Germany). Filters were air dried and stored at −20 °C until further processing. Microorganisms present in the sample were collected by filtration of 500 ml water onto 0.2 μm pore-sized Nucleopore filters (Whatman, UK). The filters were placed in sterile 2 ml centrifuge tubes and covered with 1.5 ml lysis buffer47. After processing, the tubes were immediately frozen and stored at −80 °C until DNA extraction.
Microscopic microbial enumeration
Prokaryote abundance was determined by direct microscopic counts using the 4′,6-diamidino-2-phenylindole (DAPI) staining technique48. Black polycarbonate membrane filters were stained with the fluorochrome DAPI for 5 min at a concentration of 10 μg ml−1 in the dark. Two carbonindocyanine 3 (CY3)-labelled oligonucleotide probes, EUB338 and ARCH915, which are targeted to bacteria and to archaea, respectively49,50, were used for fluorescence in situ hybridization (FISH) counts of environmental sample K3. Sections of white polycarbonate membrane filters were hybridized as previously described51 using the CY3-labeled probes EUB338 and ARCH915. Microscopic counting was conducted using an epifluorescence microscope (Nikon ECLIPSE 80i, Japan) fitted with a digital camera (Nikon DXM1200F, Japan) under green light (detection of CY3-labelled cells) and UV light (DAPI-stained cells).
DNA extraction
Total community DNA extraction was conducted as described by Bosshard et al.47. The purity and concentration of the extracted DNA were evaluated by use of a Nanodrop 2000 spectrophotometer (Thermo Scientific, Denmark). The molecular weight of DNA in extracts was determined by electrophoresing portions of extracts on 1.5% agarose gels using a 1 kb ladder (Promega, USA) as a size marker.
Amplification of dmdA and ddd genes
Two universal primer pairs, dmdAUF160 and dmdAUR69716, and dmdA282F (5′-TGCTSTSAACGAYCCSGT-3′) and dmdA591R (5′-ACRTAGAYYTCRAAVCCBCCYT-3′), were used for dmdA gene amplification. Additionally, the universal primer pair dddLf and dddLr44 was used for dddL gene amplification and the universal primer pair dddP_277F and dddP_1112R43 was used for dddP gene amplification. Genomic DNA from the reference strain Ruegeria pomeroyi DSS-3, which possesses a dmdA gene of 1095 bp and a dddP gene of 1182 bp, was used as a template for standards. Both distilled water and genomic DNA from Escherichia coli DH5α were used as a template for negative control.
Amplification was conducted in 25 μl reactions with a Mastercycle Gradient Instrument (Eppendorf, Germany). All PCRs were performed in duplicate using 10–100 ng template DNA, and products were pooled before purification. dmdA gene amplification was conducted using primer pair dmdAUF160 and dmdAUR697 as described by Varaljay et al.16. PCR using the dmdA282F and dmdA591R primer set was conducted using a reaction mixture composed of 4.0 μl of template DNA, 2.5μl of 10 × PCR buffer (Mg2+ plus; TaKaRa, China), 100 μM deoxynucleoside triphosphates (dNTPs), 5 μg bovine serum albumin (BSA), 1 U of Taq DNA polymerase (TaKaRa, China), and 0.4 μM of each described primer. PCR conditions were as follows: initial denaturation at 94 °C for 3 min, followed by 35 cycles of denaturing at 94 °C for 1 min, annealing at 54 °C for 30 s, extension at 72 °C for 45 s, and final extension at 72 °C for 10 min.
dddL gene amplification was performed as described by Raina et al.44. The PCR reagent concentrations were identical to those described for dmdA gene amplification using primer pair dmdA282F and dmdA591R except that BSA was absent. The PCR reaction mixture for the dddP gene consisted of 4.0 μl of template DNA, 12.5 μl of DreamTaq Green PCR Master mix (2×; Thermo Scientific, Germany), 5 μg BSA, and 0.4 μM of each described primer. PCR conditions were identical to those described above except that annealing was conducted at 55 °C for 30 s and extension at 72 °C for 1 min. PCR products were visualized by electrophoresis on a 1% agarose gel stained with ethidium bromide.
Clone library construction and sequencing
PCR products from seawater samples were purified using a gel purification kit (TaKaRa, China). Purified DNA was ligated into a pMD18-T cloning vector (TaKaRa, China) according to the manufacturer’s instructions, then used to transform Escherichia coli DH5α competent host cells. Only plasmid clones containing a target insert were sent to Majorbio Bio-pharm Technology Co., Ltd (Shanghai, China) for sequencing. Clones with inserts of the wrong size or chimeric sequences were removed from subsequent analyses.
Data analysis
As the known DddPs with a similarity >81% from different bacteria belong to the same genus43, sequences showing more than 81% amino acid identity with each other were grouped in the same cluster. For DmdAs, sequences showing more than 90% amino acid identity with each other were grouped in the same cluster. Coverage, Simpson’s diversity index and Shannon’s diversity index were generated using the SpadeR program (https://chao.shinyapps.io/SpadeR). Amplicon sequences were annotated using a BLASTX search against NCBI databases (http://www.ncbi.nlm.nih.gov). This analysis was used to distinguish correct target sequences from closely related paralogous sequences and to classify amplicons by cluster. Sequence alignment and phylogenetic tree building were completed using the MEGA 5.05 program with the Poisson model. Neighbour-joining bootstrap tests of phylogeny were run with 1000 replicates.
Nucleotide sequence accession numbers
The nucleotide sequences obtained in the present study have been deposited in the GenBank database under accession numbers KC864796 (dddL gene), KU162982-163012 (dmdA genes), KU177047-177209 (dmdA genes), KU215540-215580 (dmdA genes), KU215581-215594 (dddP genes), and KU233485-233520 (dddP genes).
Additional Information
How to cite this article: Zeng, Y.-X. et al. Diversity of bacterial dimethylsulfoniopropionate degradation genes in surface seawater of Arctic Kongsfjorden. Sci. Rep. 6, 33031; doi: 10.1038/srep33031 (2016).
References
Curson, A. R., Todd, J. D., Sullivan, M. J. & Johnston, A. W. Catabolism of dimethylsulphoniopropionate: microorganisms, enzymes and genes. Nat. Rev. Microbiol. 9, 849–859 (2011).
Sunda, W., Kieber, D., Kiene, R. & Huntsman, S. An antioxidant function for DMSP and DMS in marine algae. Nature 418, 317–320 (2002).
Strom, S., Wolfe, G., Slajer, A., Lambert, S. & Clough, J. Chemical defense in the microplankton II: Inhibition of protist feeding by β-dimethylsulfoniopropionate (DMSP). Limnol. Oceanogr. 48, 230–237 (2003).
Karsten, U., Kirst, G. & Wiencke, C. Dimethylsulphoniopropionate (DMSP) accumulation in green macioalgae from polar to temperate regions: interactive effects of light versus salinity and light versus temperature. Polar Biol. 12, 603–607 (1992).
Dickson, D. & Kirst, G. The role of β-dimethylsulphoniopropionate, glycine betaine and homarine in the osmoacclimation of Platymonas subcordiformis. Planta 167, 536–543 (1986).
Kiene, R. P., Linn, L. J. & Bruton, J. A. New and important roles for DMSP in marine microbial communities. J. Sea Res. 43, 209–224 (2000).
Kiene, R. P., Linn, L. J., González, J., Moran, M. A. & Bruton, J. A. Dimethylsulfoniopropionate and methanethiol are important precursors of methionine and protein-sulfur in marine bacterioplankton. Appl. Environ. Microbiol. 65, 4549–4558 (1999).
Ledyard, K. M., Dacey, J. W. & Dacey, J. Microbial cycling of DMSP and DMS in coastal and oligotrophic seawater. Limnol. Oceanogr. 41, 33–40 (1996).
Reisch, C. R. et al. Novel pathway for assimilation of dimethylsulphoniopropionate widespread in marine bacteria. Nature 473, 208–211 (2011).
Howard, E. C. et al. Changes in dimethylsulfoniopropionate demethylase gene assemblages in response to an induced phytoplankton bloom. Appl. Environ. Microbiol. 77, 524–531 (2011).
Gonzalez, J. M., Kiene, R. P. & Moran, M. A. Transformation of sulfur compounds by an abundant lineage of marine bacteria in the alpha-subclass of the class Proteobacteria. Appl. Environ. Microbiol. 65, 3810–3819 (1999).
Johnston, A. W. et al. Molecular diversity of bacterial production of the climate-changing gas, dimethyl sulphide, a molecule that impinges on local and global symbioses. J. Exp. Bot. 59, 1059–1067 (2008).
Todd, J. D. et al. Structural and regulatory genes required to make the gas dimethyl sulfide in bacteria. Science 315, 666–669 (2007).
Woodhouse, M., Mann, G., Carslaw, K. & Boucher, O. Sensitivity of cloud condensation nuclei to regional changes in dimethyl-sulphide emissions. Atmos. Chem. Phys. 13, 2723–2733 (2013).
Howard, E. C., Sun, S., Biers, E. J. & Moran, M. A. Abundant and diverse bacteria involved in DMSP degradation in marine surface waters. Environ. Microbiol. 10, 2397–2410 (2008).
Varaljay, V. A., Howard, E. C., Sun, S. & Moran, M. A. Deep sequencing of a dimethylsulfoniopropionate-degrading gene (dmdA) by using PCR primer pairs designed on the basis of marine metagenomic data. Appl. Environ. Microbiol. 76, 609–617 (2010).
Sun, J. et al. The abundant marine bacterium Pelagibacter simultaneously catabolizes dimethylsulfoniopropionate to the gases dimethyl sulfide and methanethiol. Nat. Microbiol. 16065, doi: 10.1038/nmicrobiol.2016.65 (2016).
Todd, J., Curson, A., Dupont, C., Nicholson, P. & Johnston, A. The dddP gene, encoding a novel enzyme that converts dimethylsulfoniopropionate into dimethyl sulfide, is widespread in ocean metagenomes and marine bacteria and also occurs in some Ascomycete fungi. Environ. Microbiol. 11, 1376–1385 (2009).
Todd, J. D. et al. DddQ, a novel, cupin-containing, dimethylsulfoniopropionate lyase in marine roseobacters and in uncultured marine bacteria. Environ. Microbiol. 13, 427–438 (2011).
Choi, D. H. et al. Pyrosequencing revealed SAR116 clade as dominant dddP-containing bacteria in oligotrophic NW Pacific Ocean. Plos One 10, e0116271 (2015).
Curson, A. R., Fowler, E. K., Dickens, S., Johnston, A. W. & Todd, J. D. Multiple DMSP lyases in the γ-proteobacterium Oceanimonas doudoroffii. Biogeochemistry 110, 109–119 (2012).
Luce, M. et al. Distribution and microbial metabolism of dimethylsulfoniopropionate and dimethylsulfide during the 2007 Arctic ice minimum. J. Geophys. Res: Oceans 116 (2011).
Motard-Côté, J. et al. Distribution and metabolism of dimethylsulfoniopropionate (DMSP) and phylogenetic affiliation of DMSP‐assimilating bacteria in northern Baffin Bay/Lancaster Sound. J. Geophys. Res: Oceans 117 (2012).
Svendsen, H. et al. The physical environment of Kongsfjorden–Krossfjorden, an Arctic fjord system in Svalbard. Polar Res. 21, 133–166 (2002).
Archer, S. et al. Contrasting responses of DMS and DMSP to ocean acidification in Arctic waters. Biogeosciences 10, 1893–1908 (2013).
Cottrell, M. T. & Kirchman, D. L. Community composition of marine bacterioplankton determined by 16S rRNA gene clone libraries and fluorescence in situ hybridization. Appl. Environ. Microbiol. 66, 5116–5122 (2000).
Pernthaler, A., Preston, C. M., Pernthaler, J., DeLong, E. F. & Amann, R. Comparison of fluorescently labeled oligonucleotide and polynucleotide probes for the detection of pelagic marine bacteria and archaea. Appl. Environ. Microbiol. 68, 661–667 (2002).
De Corte, D., Sintes, E., Yokokawa, T. & Herndl, G. J. Comparison between MICRO–CARD–FISH and 16S rRNA gene clone libraries to assess the active versus total bacterial community in the coastal Arctic. Environ. Microbiol. Rep. 5, 272–281 (2013).
Howard, E. C. et al. Bacterial taxa that limit sulfur flux from the ocean. Science 314, 649–652 (2006).
Iversen, K. R. & Seuthe, L. Seasonal microbial processes in a high-latitude fjord (Kongsfjorden, Svalbard): I. Heterotrophic bacteria, picoplankton and nanoflagellates. Polar Biol. 34, 731–749 (2011).
Zeng, Y.-X. et al. Bacterioplankton community structure in the Arctic waters as revealed by pyrosequencing of 16S rRNA genes. Antonie Van Leeuwenhoek 103, 1309–1319 (2013).
Zubkov, M. V. et al. Linking the composition of bacterioplankton to rapid turnover of dissolved dimethylsulphoniopropionate in an algal bloom in the North Sea. Environ. Microbiol. 3, 304–311 (2001).
Niki, T., Kunugi, M. & Otsuki, A. DMSP-lyase activity in five marine phytoplankton species: its potential importance in DMS production. Mar. Biol. 136, 759–764 (2000).
Kiene, R. P. & Linn, L. J. The fate of dissolved dimethylsulfoniopropionate (DMSP) in seawater: tracer studies using 35S-DMSP. Geochim. Cosmochim. Ac. 64, 2797–2810 (2000).
Reisch, C. R., Moran, M. A. & Whitman, W. B. Bacterial catabolism of dimethylsulfoniopropionate (DMSP). Front Microbiol. 2, 172 (2011).
Yoch, D. C. Dimethylsulfoniopropionate: its sources, role in the marine food web, and biological degradation to dimethylsulfide. Appl. Environ. Microbiol. 68, 5804–5815 (2002).
Levasseur, M. Impact of Arctic meltdown on the microbial cycling of sulphur. Nature Geosci. 6, 691–700 (2013).
Kiene, R. P. & Bates, T. S. Biological removal of dimethyl sulphide from sea water. Nature 345, 702–705 (1990).
Pinhassi, J. et al. Dimethylsulfoniopropionate turnover is linked to the composition and dynamics of the bacterioplankton assemblage during a microcosm phytoplankton bloom. Appl. Environ. Microbiol. 71, 7650–7660 (2005).
Varaljay, V. A. et al. Bacterial dimethylsulfoniopropionate degradation genes in the oligotrophic north pacific subtropical gyre. Appl. Environ. Microbiol. 78, 2775–2782 (2012).
Cui, Y. et al. Abundance and distribution of dimethylsulfoniopropionate degradation genes and the corresponding bacterial community structure at dimethyl sulfide hot spots in the tropical and subtropical pacific ocean. Appl. Environ. Microbiol. 81, 4184–4194 (2015).
Moran, M. et al. Ecological genomics of marine Roseobacters. Appl. Environ. Microbiol. 73, 4559–4569 (2007).
Peng, M. et al. Phylogenetic diversity of the dddP gene for dimethylsulfoniopropionate-dependent dimethyl sulfide synthesis in mangrove soils. Can. J. Microbiol. 58, 523–530 (2012).
Raina, J.-B., Tapiolas, D., Willis, B. L. & Bourne, D. G. Coral-associated bacteria and their role in the biogeochemical cycling of sulfur. Appl. Environ. Microbiol. 75, 3492–3501 (2009).
Levine, N. M. et al. Environmental, biochemical and genetic drivers of DMSP degradation and DMS production in the Sargasso Sea. Environ. Microbiol. 14, 1210–1223 (2012).
Simó, R., Pedrós-Alió, C., Malin, G. & Grimalt, J. O. Biological turnover of DMS, DMSP and DMSO in contrasting open-sea waters. Mar. Ecol. Prog. Ser. 203, 1–11 (2000).
Bosshard, P. P., Santini, Y., Grüter, D., Stettler, R. & Bachofen, R. Bacterial diversity and community composition in the chemocline of the meromictic alpine Lake Cadagno as revealed by 16S rDNA analysis. FEMS Microbiol. Ecol. 31, 173–182 (2000).
Robarts, R. D. & Sephton, L. M. The enumeration of aquatic bacteria using DAPI. J. Limnol. Soc. S. Afr. 7, 72–74 (1981).
Amann, R. I., Krumholz, L. & Stahl, D. A. Fluorescent-oligonucleotide probing of whole cells for determinative, phylogenetic, and environmental studies in microbiology. J. Bacteriol. 172, 762–770 (1990).
Sanders, R. W., Porter, K. G., Bennett, S. J. & DeBiase, A. E. Seasonal patterns of bacterivory by flagellates, ciliates, rotifers, and cladocerans in a freshwater planktonic community. Limnol. Oceanogr. 34, 673–687 (1989).
Comte, J. et al. Microbial community structure and dynamics in the largest natural French lake (Lake Bourget). Microb. Ecol. 52, 72–89 (2006).
Acknowledgements
We appreciate the assistance of the Chinese Arctic and Antarctic Administration (CAA) who organized the Chinese Arctic Yellow River Station Expedition in 2011. We are also grateful to Prof. Hai-Yan Jin of Second Institute of Oceanograghy of State Oceanic Administration for kindly providing chlorophyll a and nutrient data. This work was supported by the National Natural Science Foundation of China (Grant No. 41476171) and the Chinese Polar Environment Comprehensive Investigation and Assessment Program (Grant No. CHINARE2016-02-01).
Author information
Authors and Affiliations
Contributions
Y.-X.Z. contributed to the design of the study and preparation of the manuscript. Z.-Y.Q. constructed the amplicon libraries and performed data analysis. Y.Y., H.-R.L. and W.L. contributed equally to the discussion of results.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Zeng, YX., Qiao, ZY., Yu, Y. et al. Diversity of bacterial dimethylsulfoniopropionate degradation genes in surface seawater of Arctic Kongsfjorden. Sci Rep 6, 33031 (2016). https://doi.org/10.1038/srep33031
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep33031
- Springer Nature Limited
This article is cited by
-
Top abundant deep ocean heterotrophic bacteria can be retrieved by cultivation
ISME Communications (2023)
-
The role of a changing Arctic Ocean and climate for the biogeochemical cycling of dimethyl sulphide and carbon monoxide
Ambio (2022)
-
Distribution of dissolved organic carbon linked to bacterial community composition during the summer melting season in Arctic fjords
Polar Biology (2022)
-
Biogeographic traits of dimethyl sulfide and dimethylsulfoniopropionate cycling in polar oceans
Microbiome (2021)
-
High diversity of planktonic prokaryotes in Arctic Kongsfjorden seawaters in summer 2015
Polar Biology (2021)