
Abstract
The genetics of both syndromic (SHL) and non-syndromic hearing loss (NSHL) is characterized by a high degree of genetic heterogeneity. We analyzed whole exome sequencing data of 102 unrelated probands with apparently NSHL without a causative variant in known NSHL genes. We detected five causative variants in different SHL genes (SOX10, MITF, PTPN11, CHD7, and KMT2D) in five (4.9%) probands. Clinical re-evaluation of these probands shows that some of them have subtle syndromic findings, while none of them meets clinical criteria for the diagnosis of the associated syndrome (Waardenburg (SOX10 and MITF), Kallmann (CHD7 and SOX10), Noonan/LEOPARD (PTPN11), CHARGE (CHD7), or Kabuki (KMT2D). This study demonstrates that individuals who are evaluated for NSHL can have pathogenic variants in SHL genes that are not usually considered for etiologic studies.
Similar content being viewed by others


Introduction
Hearing loss (HL) affects approximately 1 in 650 children, more than half of which is attributed to genetic factors1. Based on distinctive clinical features, approximately 30% of genetic HL is syndromic (SHL) and 70% is non-syndromic (NSHL)2. To date, more than 145 NSHL loci have been identified3 and HL has been reported as a feature in more than 400 syndromes4.
Identifying the specific genetic cause of HL in an individual is difficult due to extensive genetic and clinical heterogeneity. The same pathogenic variant may present differently in either the severity of HL or in the overall clinical presentation5,6. In this study, we searched for causative DNA variants in a large cohort of individuals who apparently were diagnosed with NSHL after excluding variants in known NSHL genes (Supplementary Table S1).
Results
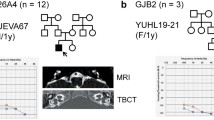
On average, each exome had 99%, 95% and 89% of mappable bases of the Gencode defined exome represented by coverage of 1X, 5X, and 10X reads, respectively. We detected heterozygous pathogenic variants in five different SHL genes that can explain HL in five (4.9%) Turkish families (Table 1). The pathogenic variants MITF (MIM 156845) c.328C > T7,8 (Waardenburg syndrome type II; MIM 193510), SOX10 (Kallmann syndrome) c.481C > T9, CHD7 (MIM 608892) c.5050G > A9,10,11,12 (CHARGE syndrome; MIM 214800 and Kallmann syndrome; MIM 612370), PTPN11 (MIM 176876) c.1492C > T13,14,15,16 (LEOPARD syndrome; MIM 151100), and KMT2D (MIM 602113) c.13259G > A17 (Kabuki syndrome; MIM 147920) had all been previously reported in patients with syndromic findings. All the identified variants were confirmed with Sanger sequencing in probands and tested for in available family members (Fig. 1).

Black squares and circles denote affected males and females with hearing loss, respectively. Proband 694 has a square face with prominent nasal bridge and columella. Other facial features of CHARGE syndrome such as facial palsy or outer ear findings are missing. Proband 713 has hypertelorism and slightly low-set, posteriorly rotated ears. Other facial features of Noonan/LEOPARD syndrome such as epicanthal folds or ptosis is missing. Proband 859 shows telechantus, epicanthal folds, slightly downslanting palpebral fissures with short columella. Facial features of Kabuki syndrome including long palpebral fissures with eversion of the lateral lower eyelid, arched and broad eyebrows with the lateral third displaying sparseness, and large or prominent ears are missing.
In family 78, along with the proband, the deaf brother and normal hearing mother both carried the MITF c.328C > T variant. Phenotypic features of the 11-year-old female proband and her 7-year-old brother included only congenital severe to profound HL without pigmentary changes in hair, eyes, or skin. The mother was not available for clinical evaluation but denied having pigmentary changes or HL.
In family 94, while both affected children carried the putative pathogenic variant (SOX10 c.481C > T), the mother did not and the father was unavailable. The mother denied HL in the deceased father. The proband was a 9-year-old female with profound HL. She did not have pigmentary skin, hair, or eye changes. She had a normal neurological examination. The 11-year-old brother had congenital profound deafness with no pigmentary alterations in eyes, skin, or hair.
The proband in family 694 (CHD7 c.5050G > A variant) had congenital profound deafness associated with inner ear anomalies. At age 18 years her height and weight measured 165 cm and 60 kg, respectively; both were between the 50th and 75th centiles. She was at Tanner 5 stage for puberty and had her menarche at age 14 years. She did not have eye anomalies or a heart defect. She reported normal smelling. She did not have a history of nasal obstruction, nasal discharge, or otitis media. A nasogastric catheter was successfully passed through both nasal openings. While she had mild facial features of CHARGE syndrome (Fig. 1), she did not have other features of CHARGE or Kallmann syndromes (Table 2).
In family 713, the proband and his father carried the PTPN11 c.1492C > T variant. The proband was a 4-year-old boy with congenital profound deafness. A thorough clinical re-evaluation of the proband detected minor craniofacial features without reaching diagnostic criteria of Noonan or LEOPARD syndromes (Fig. 1, Table 3)18,19,20,21,22. Father denied having HL and lentigines but was not available for clinical evaluation.
The proband in family 859 had the the KMT2D c.13259G > A variant and was a 1-year-old male with profound deafness associated with bilateral cochlear aplasia. The patient was on target for neuromotor development and growth except for speech delay. Further clinical evaluation detected only telecanthus with bilateral epicanthal folds without findings of Kabuki syndrome (Fig. 1). The mother carried the same variant but did not have HL, cognitive problems, or distinctive findings of Kabuki syndrome.
Discussion
In this study, we detected pathogenic variants in MITF, SOX10, CHD7, PTPN11, and KMT2D in patients who were diagnosed as having NSHL. While these genes have been implicated in various forms of SHL, they were not reported to cause NSHL. Re-evaluation of probands shows that some of them exhibit subtle syndromic findings not sufficient to make the clinical diagnosis of the associated syndrome (Supplementary Tables S2 to S5). Importantly, in several families, other individuals carried the same variant but had normal hearing.
Waardenburg syndrome (WS) is the most common type of autosomal dominant SHL with a frequency of 1/40,000 in the general population23. WS is primarily characterized by HL and pigmentation anomalies in eyes, skin, and hair23,24. While there are additional findings affecting face, limbs, or gastrointestinal system in WS types I, III, and IV, WS type II presents only with HL and pigmentary anomalies (Supplementary Table S2). One Chinese and one Caucasian patient with typical pigmentary changes and deafness consistent with WS type II have been reported to be heterozygous for the MITF c.328C > T variant7,8. SOX10 variants are known to cause WS types II and IV (Hirschsprung disease as an additional finding) (MIM 613266), PCWH (peripheral demyelinating neuropathy, central demyelinating leukodystrophy, Waardenburg syndrome, and Hirschsprung disease) (MIM 609136), and Kallmann syndrome with deafness25. It is interesting to note that two patients with SOX10 mutations resembling NSHL and inner ear anomalies were reported23. SOX10 c.481C > T has been previously reported in a patient with Kallmann syndrome9. Our patients with MITF and SOX10 variants did not have any of the pigmentary or neurological findings reported in WS and SOX10-related disorders. Formal evaluation of anosmia and hypogonadism reported in these syndromes are difficult to assess in young children and was not attempted.
CHARGE syndrome is autosomal dominant condition with a prevalence of 1 in 10,000 and is characterized by Coloboma of the eye, Heart defects, Atresia of the choanae, Retardation of growth and/or development, Genital and/or urinary tract abnormalities, and Ear anomalies and/or deafness26,27. Congenital anomalies are usually detected after birth. Overall psychomotor milestones from birth to age of 4 years can be severely delayed, although half of the children with CHARGE syndrome at primary-school age may have satisfactory intellectual ability28. Pathogenic variants in CHD7 cause CHARGE syndrome and hypogonadotropic hypogonadism 5 with anosmia (a.k.a. Kallmann syndrome) or without anosmia (MIM 612370). CHD7 c.5050G > A has been previously reported to cause CHARGE syndrome (Table 2)9,10,11,20. Studies have shown that semicircular canal atresia is a major finding of CHARGE and the patients with semicircular canal atresia should be screened for causative CHD7 variants29.
Noonan syndrome (NS) is an autosomal dominant condition seen 1 in 1,000–2,500 live births. It is characterized by distinctive facial features (low-set, posteriorly rotated ears with fleshy helices, hypertelorism, eyelid ptosis, downslanting palpebral fissures and epicanthal folds), postnatal growth reduction, congenital heart defects, hypertrophic cardiomyopathy, webbing of the neck, and skeletal anomalies30. The facial characteristics are usually prominent in early and middle childhood and more subtle in adulthood. Birth weight and length are usually normal. Growth failure is often apparent from the first year of life31. Cardiac abnormalities are present in 50–80% of individuals with NS. Median age at diagnosis for hypertrophic cardiomyopathy is five months32,33. LEOPARD syndrome (LS; multiple Lentigines and café-au-lait spots, Electrocardiographic-conduction abnormalities, Ocular hypertelorism/obstructive cardiomyopathy, Pulmonary stenosis, Abnormalities of the genitalia in males, Retardation of growth, and Deafness) is characterized by multiple lentigines and NS features. Pathogenic variants in PTPN11 are the most common cause of Noonan and LEOPARD syndromes. The c.1492C > T variant is located in exon 13 which is a hot spot region for this gene. This variant has previously been reported in individuals with LEOPARD syndrome with variable phenotypic features including multiple lentigines, hypertrophic cardiomyopathy, dysmorphic facial features and variable degree of cognitive deficits (Table 3)13,14,15,16,34.
Kabuki syndrome (KS) is a rare autosomal dominant disorder and is characterized by five cardinal manifestations: a distinctive face (elongated palpebral fissures with eversion of the lateral third of the lower eyelid, arched and broad eyebrows with the lateral third displaying sparseness or notching, short columella with depressed nasal tip, and large, prominent, or cupped ears), skeletal anomalies, digit abnormalities, mild-to-moderate intellectual disability and postnatal growth retardation. While growth is normal at birth, growth failure occurs during the first year of life and becomes more marked with increasing age20. Hearing loss is seen in up to 40% of patients, mostly due to recurrent otitis media; sensorineural hearing loss is a rare finding in patients with KS35. Pathogenic variants in KMT2D and KDM6A (MIM 300128) are only the known causes of the disorder17. The KMT2D c.13259G > A variant has been reported previously in a Korean 3-year-old female with typical facial features including long palpebral fissures with everted lower eyelids, large dysplastic ears, arched eyebrows, flat nasal tip, as well as prominent digital pads, developmental delay and hypotonia without HL17 (the variant’s identity was confirmed by the corresponding author via personal communication). Neither parent carried the same variant.
Known examples of genes that can cause both SHL and NSHL include MYO7A (MIM 276903), USH1C (MIM 605242), PCDH15 (MIM 605514), CDH23 (MIM 605516), CIB2 (MIM 605564), DFNB31 (MIM 607928) for Usher syndrome (MIM 276900; 276904; 602083; 601067; 614869; 611383), SLC26A4 (MIM 605646) for Pendred syndrome (MIM 274600), ACTG1 (MIM 102560) for Baraitser-Winter syndrome 2 (MIM 614583), TBC1D24 (MIM 613577) for DOOR syndrome (MIM 220500), KARS (MIM 601421) for Charcot-Marie-Tooth disease, recessive intermediate B (MIM 613641) and COL11A2 (MIM 120290) for Stickler syndrome (MIM 184840)36,37,38,39,40,41,42,43,44,45,46. These genes are usually included in gene panels developed for deafness. SLC26A4 is one of the most common genes causing autosomal recessive hearing loss with enlarged vestibular aqueduct or Pendred syndrome5,6,47. Even the same pathogenic SLC26A4 variant can cause nonsyndromic deafness or Pendred syndrome, exemplifying variable phenotypic expressivity of the same gene48. Numerous functional studies of SLC26A4 pathogenic variants suggest that different mechanisms, including endoplasmic reticulum retention and defective plasma membrane targeting of pendrin, residual function of the pendrin, and additional genetic and/or environmental factors might play role in pathogenesis49,50,51.
Our study shows that variants in at least five SHL genes can mimic NSHL. The origin of the variable phenotypes caused by the same variants may be genetic or environmental modifiers. It should be noted that some of the syndromic findings may be age dependent, such as retinitis pigmentosa in Usher syndrome or delayed puberty in Kallmann syndrome, and cannot be detected in a young child with HL. Some children may only have subtle phenotypic features of a syndrome, such as mild facial features of CHARGE or Noonan syndrome without meeting diagnostic criteria. Clinical investigation for every potential finding of over 400 syndromes is not practical during the etiological evaluation of an individual with HL. Moreover, laboratory evaluations would yield equivocal results as recently reported for Jervell and Lange-Nielsen syndrome (MIM 220400) in which interpretation of EKG findings was complicated by sinus arrhythmia52. We recommend that consideration be given to inclusion of all genes known to cause any form of HL in gene panels developed for hereditary deafness. Alternatively all-inclusive genome analysis such as whole exome or whole genome sequencing should be considered when deafness gene panel studies prove inconclusive.
Methods
Statement of Ethics
This study was approved by the University of Miami Institutional Review Board (USA), the Growth and Development Research Ethics Committee (Iran), and Ankara University Medical School Ethics Committee (Turkey). Methods were carried out in accordance with the approved guidelines. A signed informed consent form was obtained from each participant or, in the case of a minor, from parents.
Subjects
This study includes 102 probands selected from a larger cohort that has been found to be negative for all known NSHL genes initially by Sanger sequencing (for variants in GJB2 and mtDNA 1555A > G) and subsequently via whole exome sequencing (for a list of screened genes see Supplementary Table S1). Families were from Turkey (77), Iran (18) and the USA (7). Probands were the only affected individuals in 54 families (simplex); there were at least 2 affected siblings born to unaffected parents in the remaining 48 families (multiplex) suggesting autosomal recessive inheritance. In all cases, HL was congenital or prelingual in onset with a severity ranging from severe to profound. Clinical evaluations of all probands were performed by both an otorhinolaryngologist and a clinical geneticist. These evaluations included a thorough physical examination, otoscopy, and ophthalmoscopy. An EKG, urinalysis, and, when available, high resolution CT of the temporal bone or MRI were performed in probands. Blood was collected from probands and family members (parents and siblings) when available and DNA was extracted from peripheral leukocytes by standard protocols.
Whole-Exome Sequencing and Bioinformatic Analysis
Agilent SureSelect Human All Exon 50 Mb and a HiSeq 2000 instrument (Illumina) were used for capture and sequencing based on our previously published protocol47. Variants were called using FreeBayes53. After overall quality checks, Genesis 2.0 (https://www.genesis-app.com/) was used for variant filtering. We filtered variants in all morbid OMIM genes (except for the previously screened known NSHL genes -Supplemental Table S1) with minor allele frequency thresholds of 0.005 for recessive and 0.0005 for dominant variants as recommended54. For allele frequencies we used ExAC (http://exac.broadinstitute.org/) and EVS (http://evs.gs.washington.edu/EVS/) databases as well as our internal whole exome sequencing database that includes more than 300 individuals for each studied population. SIFT (http://sift.jcvi.org/), Polyphen2 (http://genetics.bwh.harvard.edu/pph2/) and MutationTaster (http://www.mutationtaster.org/) were used for in silico analysis. In addition, XHMM and Conifer were used for the Copy Number Variant (CNVs) detection55,56,57. Only genes that have been reported to have HL as a phenotypic finding in OMIM were further selected. Variants meeting these criteria were further annotated based on their presence and pathogenicity information in Human Gene Mutation Database (HGMD) and ClinVar58,59. In addition American College of Medical Genetics and Genomics (ACMG) Standards and Guidelines 2015 were used for the variant interpretation60. Probands who were found to carry variants in SHL genes were subsequently re-evaluated for syndromic findings.
Additional Information
How to cite this article: Bademci, G. et al. Variations in Multiple Syndromic Deafness Genes Mimic Non-syndromic Hearing Loss. Sci. Rep. 6, 31622; doi: 10.1038/srep31622 (2016).
References
Morton, C. C. & Nance, W. E. Newborn hearing screening–a silent revolution. N Engl J Med 354, 2151–2164 (2006).
Van Camp, G., Willems, P. J. & Smith, R. J. Nonsyndromic hearing impairment: unparalleled heterogeneity. Am J Hum Genet 60, 758–764 (1997).
Toriello et al. Hereditary Hearing Loss and Its Syndromes. Oxford University Press (2013).
Khan, M. R., Bashir, R. & Naz, S. SLC26A4 mutations in patients with moderate to severe hearing loss. Biochem Genet 51, 514–523 (2013).
Miyagawa, M., Nishio, S. Y., Usami, S. & Deafness Gene Study, C. Mutation spectrum and genotype-phenotype correlation of hearing loss patients caused by SLC26A4 mutations in the Japanese: a large cohort study. J Hum Genet 59, 262–268 (2014).
Chen, H. et al. Novel mutations of PAX3, MITF, and SOX10 genes in Chinese patients with type I or type II Waardenburg syndrome. Biochem Biophys Res Commun 397, 70–74 (2010).
Wildhardt, G. et al. Spectrum of novel mutations found in Waardenburg syndrome types 1 and 2: implications for molecular genetic diagnostics. BMJ Open 3, (2013).
Marcos, S. et al. The prevalence of CHD7 missense versus truncating mutations is higher in patients with Kallmann syndrome than in typical CHARGE patients. J Clin Endocrinol Metab 99, E2138–E2143 (2014).
Asakura, Y. et al. Endocrine and radiological studies in patients with molecularly confirmed CHARGE syndrome. J Clin Endocrinol Metab 93, 920–924 (2008).
Husu, E. et al. Phenotype in 18 Danish subjects with genetically verified CHARGE syndrome. Clin Genet 83, 125–134 (2013).
Fujita, K. et al. Abnormal basiocciput development in CHARGE syndrome. AJNR Am J Neuroradiol 30, 629–634 (2009).
Digilio, M. C. et al. RASopathies: Clinical Diagnosis in the First Year of Life. Mol Syndromol 1, 282–289 (2011).
Edwards, J. J. et al. A PTPN11 allele encoding a catalytically impaired SHP2 protein in a patient with a Noonan syndrome phenotype. Am J Med Genet A 164A, 2351–2355 (2014).
Kratz, C. P. et al. Lethal proliferation of erythroid precursors in a neonate with a germline PTPN11 mutation. Eur J Pediatr 165, 182–185 (2006).
Sarkozy, A. et al. Clinical and molecular analysis of 30 patients with multiple lentigines LEOPARD syndrome. J Med Genet 41, e68 (2004).
Cheon, C. K. et al. Identification of KMT2D and KDM6A mutations by exome sequencing in Korean patients with Kabuki syndrome. J Hum Genet 59, 321–325 (2014).
Blake, K. D. et al. CHARGE association: an update and review for the primary pediatrician. Clin Pediatr (Phila) 37, 159–173 (1998).
Liu, X. Z., Newton, V. E. & Read, A. P. Waardenburg syndrome type II: phenotypic findings and diagnostic criteria. Am J Med Genet 55, 95–100 (1995).
Niikawa, N. et al. Kabuki make-up (Niikawa-Kuroki) syndrome: a study of 62 patients. Am J Med Genet 31, 565–589 (1988).
van der Burgt, I. Noonan syndrome. Orphanet J Rare Dis 2, 4 (2007).
Verloes, A. Updated diagnostic criteria for CHARGE syndrome: a proposal. Am J Med Genet A 133A, 306–308 (2005).
Pingault, V. et al. SOX10 mutations mimic isolated hearing loss. Clin Genet 88, 352–359 (2015).
Kochhar, A., Hildebrand, M. S. & Smith, R. J. Clinical aspects of hereditary hearing loss. Genet Med 9, 393–408 (2007).
Pingault, V. et al. Loss-of-function mutations in SOX10 cause Kallmann syndrome with deafness. Am J Hum Genet 92, 707–724 (2013).
Sanlaville, D. & Verloes, A. CHARGE syndrome: an update. Eur J Hum Genet 15, 389–399 (2007).
Pagon, R. A., Graham, J. M. Jr., Zonana, J. & Yong, S. L. Coloboma, congenital heart disease, and choanal atresia with multiple anomalies: CHARGE association. J Pediatr 99, 223–227 (1981).
Raqbi, F. et al. Early prognostic factors for intellectual outcome in CHARGE syndrome. Dev Med Child Neurol 45, 483–488 (2003).
Green, G. E., Huq, F. S., Emery, S. B., Mukherji, S. K. & Martin, D. M. CHD7 mutations and CHARGE syndrome in semicircular canal dysplasia. Otol Neurotol 35, 1466–1470 (2014).
Tartaglia, M., Gelb, B. D. & Zenker, M. Noonan syndrome and clinically related disorders. Best Pract Res Clin Endocrinol Metab 25, 161–179 (2011).
Otten, B. J. & Noordam, C. Growth in Noonan syndrome. Horm Res 72 Suppl 2, 31–35 (2009).
Hickey, E. J. et al. Survival implications: hypertrophic cardiomyopathy in Noonan syndrome. Congenit Heart Dis 6, 41–47 (2011).
Wilkinson, J. D. et al. Outcomes in children with Noonan syndrome and hypertrophic cardiomyopathy: a study from the Pediatric Cardiomyopathy Registry. Am Heart J 164, 442–448 (2012).
Digilio, M. C. et al. LEOPARD syndrome: clinical diagnosis in the first year of life. Am J Med Genet A 140, 740–746 (2006).
Tekin, M., Fitoz, S., Arici, S., Cetinkaya, E. & Incesulu, A. Niikawa-Kuroki (Kabuki) syndrome with congenital sensorineural deafness: evidence for a wide spectrum of inner ear abnormalities. Int J Pediatr Otorhinolaryngol 70, 885–889 (2006).
Albert, S. et al. SLC26A4 gene is frequently involved in nonsyndromic hearing impairment with enlarged vestibular aqueduct in Caucasian populations. Eur J Hum Genet 14, 773–779 (2006).
Ahmed, Z. M. et al. PCDH15 is expressed in the neurosensory epithelium of the eye and ear and mutant alleles are responsible for both USH1F and DFNB23. Hum Mol Genet 12, 3215–3223 (2003).
Ouyang, X. M. et al. Mutations in the alternatively spliced exons of USH1C cause non-syndromic recessive deafness. Hum Genet 111, 26–30 (2002).
Chakchouk, I. et al. Novel mutations confirm that COL11A2 is responsible for autosomal recessive non-syndromic hearing loss DFNB53. Mol Genet Genomics (2015).
Riazuddin, S. et al. Mutation spectrum of MYO7A and evaluation of a novel nonsyndromic deafness DFNB2 allele with residual function. Hum Mutat 29, 502–511 (2008).
Kemerley, A., Sloan, C., Pfeifer, W., Smith, R. & Drack, A. A novel mutation in ACTG1 causing Baraitser-Winter syndrome with extremely variable expressivity in three generations. Ophthalmic Genet 1–5 (2016).
von Brederlow, B. et al. Identification and in vitro expression of novel CDH23 mutations of patients with Usher syndrome type 1D. Hum Mutat 19, 268–273 (2002).
Riazuddin, S. et al. Alterations of the CIB2 calcium- and integrin-binding protein cause Usher syndrome type 1J and nonsyndromic deafness DFNB48. Nat Genet 44, 1265–1271 (2012).
Ebermann, I. et al. A novel gene for Usher syndrome type 2: mutations in the long isoform of whirlin are associated with retinitis pigmentosa and sensorineural hearing loss. Hum Genet 121, 203–211 (2007).
Campeau, P. M. et al. The genetic basis of DOORS syndrome: an exome-sequencing study. Lancet Neurol 13, 44–58 (2014).
McLaughlin, H. M. et al. Compound heterozygosity for loss-of-function lysyl-tRNA synthetase mutations in a patient with peripheral neuropathy. Am J Hum Genet 87, 560–566 (2010).
Bademci, G. et al. Comprehensive analysis via exome sequencing uncovers genetic etiology in autosomal recessive nonsyndromic deafness in a large multiethnic cohort. Genet Med 18, 364–371 (2016).
Masmoudi, S. et al. Pendred syndrome: phenotypic variability in two families carrying the same PDS missense mutation. Am J Med Genet 90, 38–44 (2000).
Rotman-Pikielny, P. et al. Retention of pendrin in the endoplasmic reticulum is a major mechanism for Pendred syndrome. Hum Mol Genet 11, 2625–2633 (2002).
Taylor, J. P., Metcalfe, R. A., Watson, P. F., Weetman, A. P. & Trembath, R. C. Mutations of the PDS gene, encoding pendrin, are associated with protein mislocalization and loss of iodide efflux: implications for thyroid dysfunction in Pendred syndrome. J Clin Endocrinol Metab 87, 1778–1784 (2002).
Scott, D. A. et al. Functional differences of the PDS gene product are associated with phenotypic variation in patients with Pendred syndrome and non-syndromic hearing loss (DFNB4). Hum Mol Genet 9, 1709–1715 (2000).
Tekin, D. et al. Comprehensive genetic testing can save lives in hereditary hearing loss. Clin Genet 87, 190–191 (2015).
Erik Garrison, G. M. Haplotype-based variant detection from short-read sequencing. arXiv:1207.3907 [q-bio.GN] (2012).
Shearer, A. E. et al. Utilizing ethnic-specific differences in minor allele frequency to recategorize reported pathogenic deafness variants. Am J Hum Genet 95, 445–453 (2014).
Fromer, M. et al. Discovery and statistical genotyping of copy-number variation from whole-exome sequencing depth. Am J Hum Genet 91, 597–607 (2012).
Krumm, N. et al. Copy number variation detection and genotyping from exome sequence data. Genome Res 22, 1525–1532 (2012).
Bademci, G. et al. Identification of copy number variants through whole-exome sequencing in autosomal recessive nonsyndromic hearing loss. Genet Test Mol Biomarkers 18, 658–661 (2014).
Stenson, P. D. et al. The Human Gene Mutation Database: 2008 update. Genome Med 1, 13 (2009).
Landrum, M. J. et al. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res 42, D980–D985 (2014).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17, 405–424 (2015).
Acknowledgements
This work was supported by National Institutes of Health grants R01DC009645 and R01DC012836 to M.T.
Author information
Authors and Affiliations
Contributions
G.B. and M.T. designed the study. F.B.C., J.F., D.D., O.D.-H., G.S. and G.B. performed the experiments and analyzed the data. G.B., F.B.C., S.H.B. and I.L. drafted the manuscript. G.B., S.H.B. and M.T. interpreted the data and wrote the manuscript. L.S., T.A., T.K., L.O., H.A., I.M., G.S., S.T.-Y., Y.O., N.M., M.B., N.B., A.A., F.O., M.Y.-B. and M.T. participated in sample and data collection. All authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Bademci, G., Cengiz, F., Foster II, J. et al. Variations in Multiple Syndromic Deafness Genes Mimic Non-syndromic Hearing Loss. Sci Rep 6, 31622 (2016). https://doi.org/10.1038/srep31622
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep31622
- Springer Nature Limited
This article is cited by
-
Rethinking non-syndromic hearing loss and its mimics in the genomic era
European Journal of Human Genetics (2024)
-
CHD7 variants associated with hearing loss and enlargement of the vestibular aqueduct
Human Genetics (2023)
-
A comprehensive genotype–phenotype evaluation of eight Chinese probands with Waardenburg syndrome
BMC Medical Genomics (2022)
-
Comprehensive genetic testing improves the clinical diagnosis and medical management of pediatric patients with isolated hearing loss
BMC Medical Genomics (2022)
-
Genomic analysis of childhood hearing loss in the Yoruba population of Nigeria
European Journal of Human Genetics (2022)