
Abstract
Magnesium chloride (MgCl2) with the rhombohedral layered CdCl2-type structure (α-MgCl2) has been studied experimentally using synchrotron angle-dispersive powder x-ray diffraction and Raman spectroscopy using a diamond-anvil cell up to 100 GPa at room temperature and theoretically using first-principles density functional calculations. The results reveal a pressure-induced second-order structural phase transition to a hexagonal layered CdI2-type structure (β-MgCl2) at 0.7 GPa: the stacking sequence of the Cl anions are altered resulting in a reduction of the c-axis length. Theoretical calculations confirm this phase transition sequence and the calculated transition pressure is in excellent agreement with the experiment. Lattice dynamics calculations also reproduce the experimental Raman spectra measured for the ambient and high-pressure phase. According to our experimental results MgCl2 remains in a 2D layered phase up to 100 GPa and further, the 6-fold coordination of Mg cations is retained. Theoretical calculations of relative enthalpy suggest that this extensive pressure stability is due to a low enthalpy of the layered structure ruling out kinetic barrier effects. This observation is unusual, as it contradicts with the general structural behavior of highly compressed AB2 compounds.
Similar content being viewed by others
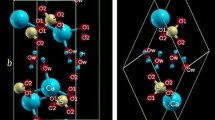
Introduction
According to Pauling’s first rule, the ambient pressure crystal structure of divalent metal halides and oxides AX2 is mainly determined by the cation-anion radius ratio R = rc/ra1,2. For R values > 1, 9-fold coordinated cations ionic structures like cotunnite (α-PbCl2-type) are formed while for R > 0.73, 8-fold coordinated cations ionic structures like fluorite (CaF2 and SrCl2) are formed3. In the case of 0.73 > R > 0.41 the well-known rutile-type structure (e.g. TiO2 or MnF2) with 6-fold coordinated cations is mainly adopted4. For 0.35 < R < 0.41 cations remain in 6-fold coordination; however, 2D layered, in contrast to 3D fluorite and rutile, Lawrencite-type structures are favored like the CdCl2-type structure5. Thus, these layered structures hold a very interesting position in the AX2 compound phase diagram as intermediate between 3D structures with 4-fold coordinated cations for 0.35 < R (quartz-SiO2) and rutile-type (stishovite-SiO2). Lawrencite-type structures are characterized by edge sharing AX6 octahedra forming a “ sandwich” of X-A-X layers (Fig. 1) where the stacking of halide atoms may be cubic-close packed ABCABC along the c-axis (e.g. rhombohedral CdCl2-type6) or hexagonal close-packed ABABAB along the c-axis (e.g. hexagonal CdI2-type7,8) or consist of mixed stacking faults with a random succession of hexagonal and cubic close-packed layers9. In contrast to the fluorite or rutile type metal halides and oxides, studies of the structural behavior of compressed Lawrencite-type compounds appear to be very limited and to the best of our knowledge only FeCl210,11 has been systematically studied by X-ray diffraction at pressures up to 65 GPa. At ambient conditions, MgCl2 with a R = 0.4012 crystalizes into alternative crystalline polymorphs, which is a common aspect of this class of inorganic X-A-X layered compounds: (a) the most common is α-MgCl2, CdCl2-type SG R-3m (166) Z = 35,13,14 followed by the less common (b) β-MgCl2, CdI2-type SG P-3m1 (164) Z = 19. The computed density-functional theory energy difference between these phases is on the order of vibrational contributions to the structure (e.g., only 0.02 eV per formula unit volume)2. Mechanically milling MgCl2 can easily shift the polymorphic concentration ratio. The latter effect is industrially important given the role of MgCl2 to support titanium halide propagation centers (nucleation sites), prepared by milling, in Ziegler-Natta catalysts used to accelerate the polymerization of olefins15. To the best of our knowledge, no high pressure experimental structural study has been reported on MgCl2, plausibly because of its extremely high hygroscopicity. A previous theoretical study2 suggests that pressurized MgCl2 (α or β phases) transforms to the rutile structure at ≈17 GPa and the fluorite structure at ≈77 GPa i.e. trending towards higher connectivity or 3D-like structures as normally would be expected for AX2 inorganic compounds.

Schematic representations of: (a) CdCl2-type and (b) CdI2-type crystal structures of MgCl2. Blue and green spheres indicate Mg and Cl anions respectively.
There are technological interests that motivate the characterization of extreme condition properties of MgCl2. It is well-known that a number of metals, metal oxides and halide materials have extraordinarily effective antimicrobial properties. Halogen, interhalogen and halogen oxide gases oxidize and disrupt the action of bacterial cells: membrane function related genes are repressed, primary metabolism related genes and protein synthesis genes are repressed and amino acid synthesis genes become activated. Detonation chemistry may generate halide species and physical conditions that are especially effective at destroying/neutralizing bio-agents; however, large-scale tests can be relatively time consuming and expensive. Laboratory-scale measurements generate data required to semi-empirically calculate detonation performance including products16. Confident semi-empirical thermochemical calculations of chemistry under extreme pressure-temperature conditions are made using equations of state data and phase diagrams of formulated reactants and likely high concentration product materials. Magnesium (Mg) is an excellent candidate for use as an explosive additive due to its high heat of combustion and relatively low boiling point temperature17,18,19,20. In addition, MgO and halogen adducts show significant biocidal activity21. The reaction of Mg with chlorine (Cl) leading to MgCl2 production may occur for example in Mg-loaded energetic formulations with significant Cl content, e.g. due to the use of an oxidizer such as ammonium perchlorate (AP) e.g. ref. 22. The use of MgCl2 as an additive in energetic formulations is also a plausible route to achieving high biocidal activity. In both scenarios, knowledge of the MgCl2 equation of state (EOS) is crucial toward understanding and computing the reactive shock behavior of energetic systems that involve a significant amount of MgCl2.
In order to examine the high pressure structural behavior of MgCl2 and also to expand knowledge of the high pressure structural behavior of layered-structured metal halide compounds we have carried out a detailed synchrotron angle-dispersive powder x-ray diffraction and Raman spectroscopy studies up to 100 GPa and compared these results with first-principles density functional calculations. We report a second-order phase transition to the CdI2-type structure at ≈0.7 GPa that remains stable up to 1 Mbar. Remarkably, the high pressure MgCl2 phase retains a 2D layered-structure with a 6-fold coordination of Mg cations up to the highest pressure of this study. The results are discussed within the context of the well established structural behavior of highly compressed AX2 compounds.
Results
Structural properties under pressure
In Fig. 2 we present integrated diffraction patterns of MgCl2 at selected pressures up to 100 GPa. The evolution of the XRD data shows discontinuous changes beginning at approximately 0.7 GPa thus revealing the occurrence of a phase transition. Above 50 GPa, Bragg peak intensity decreases and peak widths increase, which may signal the onset of amorphization; however, the most intense high pressure phase (HP) Bragg peaks can be followed up to 100 GPa without any sign of a subsequent phase transition. The HP phase Bragg peaks can be systematically indexed with the hexagonal CdI2-type structure. To determine the structural parameters the diffraction patterns were analyzed by performing Rietveld refinements using the GSAS23 software. In both structures, the only free positional parameter is the z-coordinate of Cl− anions. In detail, Mg atoms occupy the 3a(0, 0, 0) and 1a(0, 0, 0,) while Cl atoms the 6c(0, 0, z) and 2d(1/3, 2/3, z) Wyckoff Positions (WP) in ambient and HP phases respectively. Examples of refined profiles are plotted in the Supplementary information Fig. 1.

XRD patterns of MgCl2 at selected pressures.
The patterns at 2.4 and 3.5 GPa correspond to a phase mixture of CdCl2- and CdI2-type phases.
We determined pressure dependent lattice parameters, cell volumes per formula unit (Vp.f.u.) and interatomic distances for both MgCl2 structures. Experimental and theoretical results are plotted in Fig. 3. The experimental and theoretical lattice parameters and the (Vp.f.u.) values match quite well for both phases. The maximum difference between calculated and measured c- and a- values is less than 2.9% and 1.0% respectively (Fig. 3a). These results increase confidence in the accuracy of the theoretical methods used in this work. The corresponding structural details are summarized in Table 1. The observed continuity of lattice parameter values (with cHP = camb/3) and Vp.f.u. before and after the phase transition signals a second-order phase transition much like the case of FeCl210. However, the two-phase coexistence pressure range of ≈3 GPa appears to be much more extensive than in the case of FeCl2. The compressibility of the HP phase c-axis (Fig. 3(b) inset) exceeds the a-axis during initial compression up to 10 GPa thus reflecting, as normally expected due to the weak Van der Waals interlayer forces, higher compressibility in the direction perpendicular to the layers that reduces the free interlayer spacing (Fig. 1). This phenomenon is also reflected by the decreasing c/a axial ratio, which abruptly becomes pressure invariant at 10 GPa (Fig. 4(a)). We note that the initial c/a axial ratio is very close to the ideal hexagonal ratio 1.633 and it strongly decreases with increasing pressure until reaching an effectively pressure independent value of ≈1.51 above 10 GPa and up to 50 GPa. A similar trend is reported in the case of FeCl211. However, in the present study the c/a ratio remains constant in this pressure range; there is no indication of an abrupt change of lattice parameters or discontinuous change of the pressure dependence of the axial ratio11. This can be attributed to the absence of an electronic phase transition in MgCl2 as opposed to FeCl2, i.e. in this study we observe the pure physical effect of compression on the crystal structure, that is, pressure homogenizes the structure. The constant value (~1.51) of c/a ratio above 10 GPa reflects the reduced compressibility of the c-axis, which becomes equal to the a-axis at least below 60 GPa in the experimental data (Fig. 3(b) inset). The origin of this effect can be understood in terms of the three different pressure dependent Cl-Cl interatomic distances plotted in Fig. 4(b). Initial compression mainly affects the interlayer Cl-Cl distance while Cl-Cl distances within individual Cl-Mg-Cl “sandwich” are only slightly affected. At 10 GPa, the interlayer Cl-Cl distance becomes shorter than the internal Cl-Mg-Cl “sandwich” Cl-Cl separation; here it is plausible to assume that the increased charge repulsion between interlayer Cl-Cl anions serves to markedly reduce the c-axis compressibility. We also note that the data plotted in Fig. 4 stops at 16 GPa. Although initial samples consisted of nearly perfect fine powdered grains yielding uniform intensity ring-like 2D XRD images, the phase transition increased the sample grain size distribution resulting in much less uniform (spotty) diffraction ring intensity profiles. Higher intensity large diffraction spots spatially broaden Bragg peaks. In addition to grain size induced broadened diffraction rings, pressure induced peak broadening also occurred thus preventing us from confidently determining Cl-Cl distances above 16 GPa. It is interesting that the c/a ratio increases above 20 GPa in the calculated and above 50 GPa in the experimental results. It is plausible that pressure dependent repulsion between the Cl− anions reaches a threshold; a significant reduction in compressibility along the c-axis could be signaled by the inflected (increasing) c/a ratio.

(a) Pressure dependence of the lattice parameters and (b) volume-pressure data for CdCl2- and CdI2-type phases of MgCl2. The solid red line is a third-order BM equation of state fit of the HP phase experimental data. The inset in (b) shows the normalized lattice parameters of the CdI2-type phase. The asterisks denote the values of the lattice parameters at the onset pressure of the HP phase. Experimental results are represented by solid and open symbols during compression and decompression respectively. Calculated values are represented by solid green and blue lines for CdCl2- and CdI2-type phases respectively.

(a) The pressure dependent c/a axial ratio of MgCl2. The horizontal dashed black line represents the ideal hexagonal c/a ratio value. Experimental results are represented by solid symbols and the calculated values are represented by a solid blue line. (b) Three different pressure dependent Cl-Cl distances: (i) interlayer between Cl anions of different Cl-Mg-Cl “sandwich” (black), (ii) intralayer Cl-Cl (blue) and (iii) internal Cl anions within the same Cl-Mg-Cl “sandwich” (red).
Consistent with most high-pressure EOS studies, we conducted unweighted fits of the pressure-volume data to a third-order Birch-Murnaghan (B-M) equation of state24 and determined the bulk modulus Ko and its first derivative K′ at zero pressure for the CdCl2-type phase and at the experimental onset pressure for the CdI2-type phase. The elastic parameters obtained this way are given in Table 1 and are comparable with those reported for FeCl211. The Ko values of both MgCl2 phases are about three times lower than the respective ones for the rutile-type fluoride counterparts, like MgF2 and MnF225,26, although in both structures cations are 6-fold coordinated. This highlights the lower dimensionality-connectivity of the Lawrencite-type compounds in comparison to the 3D rutile-type compounds. Upon pressure release, the phase transition appears to be reversible with negligible hysteresis and MgCl2 returns back to the CdCl2-type structure.
To gain deeper insight into how MgCl2 responds under quasi-static compression, we conducted weighted fits and used the reduced  goodness-of-fit formalism to compare the effectiveness of three EOS models to represent the P-V data. The model that generates minimal parameter error and has a reduced
goodness-of-fit formalism to compare the effectiveness of three EOS models to represent the P-V data. The model that generates minimal parameter error and has a reduced  value closest to 1 represents the “ best-fit model”. We tested the Birch-Murnaghan24, (B-M), 2nd to 5th orders, the Vinet27 and the F-f 28 finite strain 1st to 3rd order EOS models for the ambient phase MgCl2. The first-order F-f model was sufficient to represent the data; however, the 2nd order B-M model is statistically better at representing the ambient phase MgCl2 data (see Table 2 and supplementary Figure 3). The same analytical approach was applied to the high-pressure phase except here we replace the F-f model with a corresponding linearized G-g stress-strain model where an arbitrary reference V0 value is chosen and the ambient pressure properties are then determined at g = go (strain at ambient pressure) using the G-g relation29 (see Supplementary Figure 4). Again the 2nd order B-M model was found to reasonably represent the data, see Table 2 and Fig. 5(a), although the G-g EOS model estimated standard deviation (esd) values are lower in magnitude. The computed G and g errors are relatively low and thus lead to more significantly weighted differences between the model and the measured pressure. This is why the G-g model
value closest to 1 represents the “ best-fit model”. We tested the Birch-Murnaghan24, (B-M), 2nd to 5th orders, the Vinet27 and the F-f 28 finite strain 1st to 3rd order EOS models for the ambient phase MgCl2. The first-order F-f model was sufficient to represent the data; however, the 2nd order B-M model is statistically better at representing the ambient phase MgCl2 data (see Table 2 and supplementary Figure 3). The same analytical approach was applied to the high-pressure phase except here we replace the F-f model with a corresponding linearized G-g stress-strain model where an arbitrary reference V0 value is chosen and the ambient pressure properties are then determined at g = go (strain at ambient pressure) using the G-g relation29 (see Supplementary Figure 4). Again the 2nd order B-M model was found to reasonably represent the data, see Table 2 and Fig. 5(a), although the G-g EOS model estimated standard deviation (esd) values are lower in magnitude. The computed G and g errors are relatively low and thus lead to more significantly weighted differences between the model and the measured pressure. This is why the G-g model  value is comparatively large despite the small maximum pressure difference from the data. The high pressure V0 value is just slightly smaller than the ambient pressure volume. What is perhaps unusual is that the low pressure phase is less compressible than the high pressure phase. If one takes into account the respective K0 esd’s, particulary in the case of the α-MgCl2, then the strength of this observation is not highly convincing. The relatively high K0 esd value for α-MgCl2 is a direct consequence of both the low Pc of the phase transition and the low number of the P-V data points for α-MgCl2. Moreover, the use of second-order B-M EOS models, K′ is fixed, further affects the K0 values. Refer to Supplementary Information for a description of the complete statistical analysis including fitting procedures. For the best-fit model, we plot corresponding two-dimensional confidence ellipses to reveal two-variable correlation information (Fig. 5(b)).
value is comparatively large despite the small maximum pressure difference from the data. The high pressure V0 value is just slightly smaller than the ambient pressure volume. What is perhaps unusual is that the low pressure phase is less compressible than the high pressure phase. If one takes into account the respective K0 esd’s, particulary in the case of the α-MgCl2, then the strength of this observation is not highly convincing. The relatively high K0 esd value for α-MgCl2 is a direct consequence of both the low Pc of the phase transition and the low number of the P-V data points for α-MgCl2. Moreover, the use of second-order B-M EOS models, K′ is fixed, further affects the K0 values. Refer to Supplementary Information for a description of the complete statistical analysis including fitting procedures. For the best-fit model, we plot corresponding two-dimensional confidence ellipses to reveal two-variable correlation information (Fig. 5(b)).

(a) MgCl2 high-pressure phase data fits, unweighted with green line and weighted with red line, using a 2nd order B-M EOS model. (b) The experimentally weighted V0 versus K0 confidence ellipses. The magenta colored ellipse is 0.607-σ (50.3% confidence), blue is 1-σ (68.3% confidence), green is 2-σ (95.4% confidence) and the black ellipse is 3-σ (99.7% confidence).
The calculated pressure dependent enthalpies for the α-MgCl2 and β-MgCl2 are shown in Fig. 6(a) over the pressure range 0–25 GPa. The calculation of the ambient pressure structure correctly predicts the α-MgCl2 polymorph as the thermodynamic ground state of MgCl2. The β-MgCl2 structure becomes more thermodynamically stable than the α-MgCl2 structure at ≈0.9 GPa, which agrees well with the experimentally measured transition pressure of 0.7 GPa.

(a) Calculated pressure dependent enthalpy differences for α-MgCl2 and β-MgCl2 phases of MgCl2 up to 20 GPa. (b) Calculated enthalpy differences for α-MgCl2 and β-MgCl2 phases of MgCl2 together with the hypothetical rutile, fluorite and cotunnite phases as a function of pressure up to 100 GPa. The enthalpy of the α-MgCl2 phase is taken as the reference.
Raman scattering under pressure
Two Raman-active zone-center modes are predicted from group theory for both the α-MgCl2 and β-MgCl2 phases with the symmetries: A1g + Eg30. A1g corresponds to the displacement of Cl along the c-axis and the Eg mode corresponds to lateral Cl displacements within the Cl-Mg-Cl layers31, see inset of Fig. 7(b). At ambient pressure, with MgCl2 encapsulated within the unpressurized DAC sample chamber, the observed Raman mode frequencies are in excellent agreement with previous studies30 (Fig. 7). The calculated frequencies for the A1g and Eg modes are 245 cm−1 and 157 cm−1, respectively, which closely match with the experimental values of 242.6 cm−1 and 154 cm−1. Above 1 GPa, the number of observed Raman modes remains the same (Fig. 7(a)) and no apparent discontinuity of the Raman frequencies is observed (Fig. 7(b)). This observation is congruent with a second-order α-MgCl2 → β-MgCl2 phase transition and is moreover consistent with XRD measurements and relative enthalpy calculations. We calculated room-temperature phonon dispersion curves for α-MgCl2 at 0 and 0.67 GPa (see Supplementary information Fig. 3). Soft modes are clearly revealed at the T symmetry point, which is consistent with a second-order phase transition to the beta phase. Above 70 GPa, it is not possible to measure the low-frequency broad Eg mode. On the other hand, the A1g mode is easily observed up to 100 GPa and its frequency varies smoothly with increased pressure (Fig. 7(b)). The continuity of the A1g together with the absence of any new intense Raman peaks further justifies the argument that MgCl2 remains in the β-MgCl2 phase up to 100 GPa. The mode Grüneisen parameters (γT) determined using the experimental results of this work are shown in Supplementary Table 2. The γT parameters of modes of the β-MgCl2 phase are common for materials with mixed ionic-covalent bonding (such as within the Cl-Mg-Cl layers)32, due to the presence of weaker interlayer bonds, which initially are more compressible.

(a) Raman spectra of MgCl2 at various pressures. The low intensity Eg modes are marked with vertical red arrows. (b) Pressure dependent phonon frequencies. Experimental results are represented by open symbols and calculated values with solid lines, see supplementary information Fig. 2 for the low pressure data. The inset shows the eigenvectors of the Raman-active zone-center modes A1g and Eg.
Discussion
As discussed in the introduction, we would normally expect MgCl2 to undergo a pressure-induced phase transitions towards higher connectivity (3D) and coordination number structures. According to the general systematics of pressurized AX2 compounds33 a typical α-quartz (rhombohedral S.G. P3221 (154) CN = 4) → rutile (tetragonal S.G. P42/mnm (136) CN = 6) → CaF2-type (cubic fluorite SG Fm-3m (225) CN = 8) → α-PbCl2-type (cotunnite orthorhombic SG Pnma (62) CN = 9) → Ni2In (hexagonal S.G. P63/mmc (194) CN = 11) sequence of phase transitions is expected34,35,36,37 with an overall increase in cation coordination number from 4 (α-quartz) to 11 (Ni2In). In this sequence, only the prototypical structures are noted because of the plethora of closely related alternative structural types. For instance one can group the CaCl2-type and the α-PbO2-type in the rutile family and the PdF2-type and FeS2-type (pyrite) in the fluorite family, see discussion in ref. 26 and Fig. 1 of ref. 38. So, the application of pressure results in higher coordination number (4 to 11) and phase transitions resulting in structures with characteristically higher R = rc/ra values due to the well known higher compressibility of anions in comparison to cations39. Here we summarize the pressure induced phase transitions of few AX2 systems relevant to MgCl2, i.e. with low R = rc/ra values at ambient conditions. SiO2, which is considered as the AB2 compound with the highest pressure phase transition, crystallizes in the α-quartz structure at ambient conditions and transforms to rutile (Stishovite) above 10 GPa40. MgF2 crystallizes to the rutile structure at ambient conditions and transforms to a modified fluorite structure (PdF2-type) above 14 GPa and to the cotunnite structure above 35 GPa. Although there are no experimental data on BeX2 compounds a very recent theoretical study of BeF241 suggests that the ambient pressure α-quartz structure transforms to rutile above 27 GPa. Finally, CaCl2 crystallizes in the CaCl2-type (rutile family) at ambient pressure and transforms to cotunnite above 10 GPa.
As evident from our XRD and Raman spectroscopy results, MgCl2 remains in the β-MgCl2 2D layered structure up to 100 GPa A kinetic barrier could be inhibiting the expected phase transformation. This phenomenon has been observed in the case of the SiO2 for the α-PbO2-type phase transitioning to the PdF2-type phase above 268 GPa42 and also in the cases of CaF2 and SrF243 transitioning from the cotunnite-type phase to the Ni2In-type structure. In the aforementioned examples higher temperatures, using laser heating, were needed to overcome the kinetic barriers. In order to elucidate if the same scenario applies to MgCl2 we performed pressure dependent first-principles enthalpy calculations for the β-MgCl2 phase and the hypothetical rutile, fluorite and cotunnite phases of MgCl2. The results are plotted in Fig. 6(b). As it can be clearly seen from the enthalpy vs pressure plot, β-MgCl2 remains the more stable phase up to 100 GPa. Moreover, the enthalpy difference between the β-MgCl2 and the candidate rutile, fluorite and cotunnite phases increases with pressure. Thus, it is not expected that these structures will become energetically favorable even above 100 GPa. Although slight modifications of the prototypical candidate structures might have lower enthalpies than the prototypical ones it is expected that the difference between them should be much lower than the difference with β-MgCl2. Moreover, it is normally expected that the enthalpy difference between the β-MgCl2 and these slight modifications will also increase with pressure26,38,44.
From the above discussion it is clear that MgCl2 is a rare exception; it does not follow the general structural trend of highly compressed AX2 compounds up to the maximum pressure of this study. One possible scenario is that in order for Mg to have an increased coordination number a precondition must first be met, what we may call, for the sake of brevity, layer mixing, which is inhibited by charge repulsion between interlayer Cl-Cl anions. However, this scenario implies that a kinetic barrier is at play, the existence of which is not supported by the results of our first-principles enthalpy calculations. It does appear that once a layered structure is established (α-MgCl2 and β-MgCl2 have similar structural characteristics and enthalpies) with anions positioned in an ideal close-packed arrangement, as opposed to buckled hcp layers in rutile, then the enthalpy of the system reaches a profoundly deep minimum well. A closer look at the hypothetical rutile-type structure of MgCl2 reveals that the MgCl6 octahedron in this structure is axially distorted. Specifically, two Mg-Cl bonds in the MgCl6 octahedron are compressed along the axial direction, forming a D4h point group that is similar to the coordination of high-spin manganese (III). For example, at 100 GPa, the two shorter Mg-Cl bonds are 1.84 Å and the four longer ones are 1.907 Å, respectively, in the rutile structure. Such a distortion is energetically unfavorable. On the other hand, in the R-3m and P-3m1 structures the MgCl6 octahedra maintain the Oh symmetry even at the Mbar region, thus these two structures are energetically more favorable than the rutile structure. The axial compression of the MgCl6 octahedron in the rutile structure is opposite to what one would expect from the Jahn-Teller stabilization, which is another interesting topic but clearly beyond the current scope of this paper. Electronic transitions do seem to affect structural stability or instability. In the case of the sister compound FeCl2, a second-order phase transition from the ambient CdCl2-type phase to the CdI2-type has been observed at very low pressure (ca 0.6 GPa)10. This phase remains stable without any sign of a structural phase transition, although two electronic phase transitions were reported, up to 65 GPa11, i.e., FeCl2 remained in a layered motif up to this pressure. The high energy of the 3d orbital level in Mg, prohibits the s to d electron transition to occur in MgCl2 in the Mbar regions. Such electron transition would otherwise induce transition-metal behaviors for the metal and lead to new structures, for example, those observed in CaCl2 above 10 GPa. Pressure induced structural phase transitions, to yet unidentified structures, have been reported in the case of ZrS2 (at 8 GPa) and PbI2 (at 0.9 and 5 GPa)45, both have the CdCl2-type structure at ambient pressure. To the best of our knowledge, these are the only cases of reported pressure induced structural phase transitions of CdCl2-type compounds.
The equation of state of MgCl2, determined here from experimental data, is used to constrain thermochemical equilibrium calculations46 for Mg-containing energetic formulations that likely produce Mg, MgO, Mg(OH)2 and MgCO3 products. The addition of relevant EOS product species data improves the confidence of semi-empirical calculations of extreme condition thermochemistry. Chemical formulations can be in silico tuned to optimize conditions and products required to more efficiently neutralize biological threats. The EOS data determined in the present study will, in part, enable the development of thermochemical prediction tools to guide the development of efficient bio-agent defeat energetic formulations by optimizing the production of chosen specific biocidal products at detonation conditions.
In conclusion, the quasi-hydrostatic high-pressure structural dynamics of α-MgCl2 have been characterized by a combined experimental and first-principles study up to 100 GPa. A second-order phase transition to the β-MgCl2 phase has been observed to occur at approximately 0.7 GPa. We report a complete pressure dependent structural analysis including a systematic determination of pressure dependent Cl-Cl interatomic distances, which provides a clear understanding of the observed anisotropic compliance along the c-axis during initial compression and after its abrupt pressure invariance occurs above 10 GPa. Our results reveal that β-MgCl2 is unexpectedly stable up to the highest pressure of this study. The ideally positioned closed-packed Cl anion arrangement and lack of interfering electronic transitions can be partially attributed to the robustness of the β-MgCl2 hexagonal primitive cell structure. These results provide valuable insight into the high-pressure response and surprising stability of 2D layered CdI2-type compounds.
Methods
Experimental study
High purity (>99.99%) commercially available (Sigma-Aldrich) MgCl2 was ground to fine powder for x-ray diffraction (XRD) measurements. The sample including pressure sensors were loaded into diamond-anvil cell (DAC) sample chambers. For each of two x-ray studies, rhenium gaskets (preindented to 40–45 μm thick using 400 μm or 100 μm diameter beveled culets) were used to radially confine the pressurized samples. Initial sample chamber diameters were nominally 150 μm or 30 μm for the smaller culet. Silicone oil was utilized as a pressure-transmitting medium (PTM) for XRD at low pressures and Ne for XRD at higher pressures including Raman measurements. Pressure was determined using a known ambient temperature EOS of gold47 and also using a calibrated ruby luminescence scale48. Integration of powder diffraction images to yield scattering intensity versus 2θ patterns and initial analysis were performed using the DIOPTAS49 program.
Image plate CCD detectors were used to collect pressure dependent X-ray diffraction data at the Advanced Light Source Beamline 12.2.2. An X-ray wavelength of λ = 0.4959Å was selected using a Si(111) double-crystal monochromator. Exposures time varied between 10 and 30 secs. The sample to detector distance of 300 mm was determined using a CeO2 (or LaB6) diffraction pattern. The X-ray beam was focused to 10 × 10 μm using Kirkpatrick-Baez mirrors. More details on the experimental set up are given in Kunz et al.50. XRD data at pressures above 30 GPa were collected at the Extreme Conditions Beamline P02.2 at DESY (Germany)using a PerkinElmer detector51. The monochromatic x-ray beam (wavelength λ = 0.2898 Å) was focused to a nominal diameter of 4μm using Kirkpatrick-Baez mirrors.
First-Principles Theoretical Calculations
To determine the equation of state of MgCl2, we performed density functional calculations of pressure dependent enthalpies for a number of different crystal structures including the experimentally determined α-MgCl2 and β-MgCl2 phases. The additional calculated structures include fluorite (Fm-3m), rutile (P42/mnm) and cotunnite (Pnma). The Vienna ab initio Simulation Package (VASP) program52 was used for total energy and lattice dynamics calculations combined with the projected augmented wave (PAW) potential53,54, the Perdew-Burke-Ernzerhof (PBE) exchange-correlation functional55 and a kinetic energy cutoff of 500 eV. The 2p63s2 for Mg and 3s23p5 for Cl were treated as valence states. Dense k-point grids56 were employed to sample the first Brillouin zone (BZ) for candidate structures, which yielded energies that converged to within 1 meV/atom. Specifically, the k-point grids used in the total-energy calculations are, 12 × 12 × 12 for the α-MgCl2, 12 × 12 × 8 for the β-MgCl2, 12 × 12 × 12 for the fluorite structure, 8 × 8 × 12 for the rutile structure and 8 × 12 × 6 for the cotunnite structure, respectively. Lattice dynamics calculations were performed employing the density functional perturbation method, where the Hessian matrix and the vibrational frequencies were determined at the BZ center. A 12 × 12 × 12 and 12 × 12 × 8 k-point mesh was used for α-MgCl2 and β-MgCl2, respectively in these lattice dynamics calculations. Room temperature phonon dispersion relations were calculated employing the self-consistent ab initio lattice dynamical (SCAILD) method57 through a combination of VASP52 and PHON58 programs. SCAILD calculations were carried out employing supercells of 81 atoms for both α and β phases to yield the phonon frequencies that are converged within 0.05 THz (1.67 cm−1).
Thermochemical calculations
Chemical equilibrium thermochemical calculations, based on a statistical mechanics theory of multi-component multi-phase reactive mixtures parameterized by experimental and simulation data46,59 typically yields thermodynamic predictions at extreme conditions that differ by less than 1–2% from the experimental results for a wide range of energetic materials.
Additional Information
How to cite this article: Stavrou, E. et al. High-pressure X-ray diffraction, Raman and computational studies of MgCl2 up to 1 Mbar: Extensive pressure stability of the β-MgCl2 layered structure. Sci. Rep. 6, 30631; doi: 10.1038/srep30631 (2016).
References
Burdett, J., Price, G. & Price, S. Factors influencing solid-state structure-an analysis using pseudopotential radii structural maps. Phys. Rev. B 24, 2903 (1981).
Barrera, G. D., Allan, N. L. & Soriano, M. R. The stability of polymorphs of MgCl2 - an ab initio study. Chem. Phys. Lett. 278, 267–271 (1997).
Bragg, W. L. The analysis of crystals by the x-ray spectrometer. Proceedings of the Royal Society of London A: Mathematical, Physical and Engineering Sciences 89, 468–489 (1914).
Vegard, L. Lv . results of crystal analysis. Philosophical Magazine Series 6 32, 505 (1916).
Ferrari, A., Braibanti, A. & Bigliardi, G. Refinement of the crystal structure of NiCl2 and of unit-cell parameters of some anhydrous chlorides of divalent metals. Acta Crystallogr. 16, 845–847 (1963).
Pauling, L. & Hoard, J. The crystal structure of cadmium chloride. Z. Kristallogr. 74, 546 (1930).
Bozorth, R. M. The crystal structure of cadmium iodide. J. Am. Chem. Soc. 44, 2232 (1922).
Madelung, O., Rssler, U. & Schulz, M. CdCl2, CdBr2, CdI2, HgCl2, HgBr2, HgI2 crystal structure, chemical bond. In Madelung, O., Rssler, U. & Schulz, M. (eds.) Non-Tetrahedrally Bonded Elements and Binary Compounds I, vol. 41C of Landolt-Brnstein-Group III Condensed Matter (Springer, Berlin Heidelberg, 1998).
Bassi, I. W., Polato, F., Calcaterra, M. & Bart, J. C. J. A new layer structure of MgCl2 with hexagonal close packing of the chlorine atoms. Z. Kristallogr. 159, 297 (1982).
Vettier, C. & Yelon, W. The structure of FeCl2 at high pressures. J. Phys. Chem. Solids 36, 401 (1975).
Rozenberg, G. et al. Pressure-induced structural, electronic and magnetic phase transitions in FeCl2 studied by x-ray diffraction and resistivity measurements. Phys. Rev. B 79, 214105 (2009).
Shannon, R. D. Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Crystallogr. Section A 32, 751 (1976).
Partin, D. & O’Keeffe, M. The structures and crystal chemistry of magnesium chloride and cadmium chloride. J. Solid State Chem. 95, 176 (1991).
Pauling, L. On the crystal structure of the chlorides of certain bivalent elements. Proc. Natl. Acad. Sci. USA 15, 709 (1929).
Galli, P. et al. The activation of MgCl2-supported Ziegler-natta catalysts - a structural investigation. Eur. Polym. J. 19, 19 (1983).
Pita, R. Toxin weapons: from World War I to Jihadi terrorism. Toxin Rev. 28, 219–237 (2009).
Frost, D., Zhang, F., Murray, S. & McCahan, S. Critical conditions for ignition of metal particles in a condensed explosive. Proc. Int. Det. Symp. 12th. ONR 693 (2002).
Frost, D., Goroshin, S., Ripley, R. & Zhang, F. Effect of scale on the blast wave from a metalized explosive. Proc. Int. Det. Symp. 13th. ONR 97 (2006).
Whittaker, M., Cutler, R. & Anderson, P. Boride-based materials for energetic applications. MRS Proceedings 1405 (2012).
Trzcinski, W. A., Paszula, J. & Grys, S. Detonation and blast wave characteristics of nitromethane mixed with particles of an aluminium - magnesium alloy. Propellants Explos. Pyrotech. 35, 85–92 (2010).
Stoimenov, P. K., Zaikovski, V. & Klabunde, K. J. Novel halogen and interhalogen adducts of nanoscale magnesium oxide. J. Am. Chem. Soc. 125, 12907–12913 (2003).
Dolgoborodov, A. Y. Mechanically activated oxidizer-fuel energetic composites. Combust. Explos. Shock Waves 51, 86–99 (2015).
Larson, A. C. & Dreele, R. B. V. Gsas: General structure analysis system report LAUR 86-748. Tech. Rep., Los Alamos National Laboratory (2000).
Birch, F. Finite strain isotherm and velocities for single-crystal and polycrystalline nacl at high-pressures and 300k. Journal Of Geophysical Research 83, 1257–1268 (1978).
Gerlich, D., Hart, S. & Whittal, D. Pressure derivatives of the elastic moduli of the rutile-structure difluorides. Phys. Rev. B 29, 2142 (1984).
Stavrou, E., Yao, Y., Goncharov, A. F., Konôpková, Z. & Raptis, C. High-pressure structural study of MnF2 . Phys. Rev. B 93, 054101 (2016).
Vinet, P., Ferrante, J., Smith, J. R. & Rose, J. H. A universal equation of state for solids. J. Phys. C 19, L467–L473 (1986).
Birch, F. Finite elastic strain of cubic crystals. Phys. Rev. 71, 809–824 (1947).
Jeanloz, R. Finite-strain equation of state for high-pressure phases. Geophys. Res. Lett. 8, 1219–1222 (1981).
Capwell, R. Raman spectra of crystalline and molten MgCl2 . Chem. Phys. Lett. 12, 443 (1972).
Sano, H. et al. Raman spectra of magnesium chloride thin films. Surface Science 502503, 70 (2002).
Sherman, W. Bond anharmonicities, gruneisen parameters and pressure-induced frequency-shifts. J. Phys. c:solid state physics 13, 4601 (1980).
Leger, J. & Haines, J. Crystal chemistry of the AX(2) compounds under pressure. Eur. J. Solid State Inorg. Chem. 34, 785 (1997).
Haines, J., Leger, J. M. & Schulte, O. The high-pressure phase transition sequence from the rutile-type through to the cotunnite-type structure in pbo2. J. Ph. Condens. Matter. 8, 1631–1646 (1996).
Haines, J. et al. X-ray diffraction and theoretical studies of the high-pressure structures and phase transitions in magnesium fluoride. Phys. Rev. B 64, 134110 (2001).
Ming, L., Manghnani, M., Matsui, T. & Jamieson, J. Phase transformations and elasticity in rutile-structured difluorides and dioxides. Physics of the Earth and Planetary Interiors 23, 276 (1980). Special Issue Physico-Chemical Properties of Mantle Materials at High Pressure.
Leger, J., Haines, J., Atouf, A., Schulte, O. & Hull, S. High-pressure x-ray-diffraction and neutron-diffraction studies of BaF2 - an example of a coordination-number of 11 in AX2 compounds. Phys. Rev. B 52, 13247 (1995).
Barreda-Argüeso, J. A. et al. Pressure-induced phase-transition sequence in CoF2: An experimental and first-principles study on the crystal, vibrational and electronic properties. Phys. Rev. B 88, 214108 (2013).
Fukunaga, O. & Yamaoka, S. Phase transformations in ABO4 type compounds under high pressure. Phys. Chem. Miner. 5, 167–177 (1979).
Stishov, S. & Belov, N. Crystal structure of a new dense modification of silica sio2 . Dokl. Akad. Nauk SSSR 143, 951-& (1962).
Rakitin, M. S. et al. A novel phase of beryllium fluoride at high pressure. Phys. Chem. Chem. Phys. 17, 26283 (2015).
Kuwayama, Y., Hirose, K., Sata, N. & Ohishi, Y. The pyrite-type high-pressure form of silica. Science 309, 923 (2005).
Dorfman, S. M. et al. Phase transitions and equations of state of alkaline earth fluorides CaF2, SrF2 and BaF2 to mbar pressures. Phys. Rev. B 81, 174121 (2010).
López-Moreno, S., Romero, A. H., Mejía-López, J., Muñoz, A. & Roshchin, I. V. First-principles study of electronic, vibrational, elastic and magnetic properties of FeF2 as a function of pressure. Phys. Rev. B 85, 134110 (2012).
Hiue, M., Akahama, Y., Kobayashi, M. & Kawamura, H. Pressure-induced structural phase transition of CdI2-type compounds. The Review Of High Pressure Science and Technology 7, 344 (1998).
Bastea, S. & Fried, L. Chemical equilibrium detonation. In Zhang, F. (ed.) Shock Waves Science and Technology Library, Vol. 6, Shock Wave Science and Technology Reference Library (Springer, Berlin Heidelberg, 2012).
Matsui, M. High temperature and high pressure equation of state of gold. International Conference On High Pressure Science and Technology, Joint Airapt-22 and Hpcj-50215, 012197 (2010).
Syassen, K. Ruby under pressure. High Pres. Res. 28, 75 (2008).
Prescher, C. & Prakapenka, V. B. Dioptas: a program for reduction of two-dimensional x-ray diffraction data and data exploration. High Pres. Res. 35, 223–230 (2015).
Kunz, M. et al. A beamline for high-pressure studies at the advanced light source with a superconducting bending magnet as the source. J. Synchrotron Radiat. 12, 650 (2005).
Liermann, H.-P. et al. The extreme conditions beamline P02.2 and the extreme conditions science infrastructure at PETRAIII. Journal of Synchrotron Radiation 22, 908–924 (2015).
Kresse, G. & Furthmuller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169 (1996).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758 (1999).
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953 (1994).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865 (1996).
Monkhorst, H. & Pack, J. Special points for brillouin-zone integrations. Phys. Rev. B 13, 5188 (1976).
Souvatzis, P., Eriksson, O., Katsnelson, M. & Rudin, S. The self-consistent ab initio lattice dynamical method. Computational Materials Science 44, 888–894 (2009).
Alfé, D. Phon: A program to calculate phonons using the small displacement method. Comput. Phys. Commun. 180, 2622–2633 (2009).
Fried, L. & Souers, P. Cheetah: A next generation thermochemical code. Tech. Rep. UCRL-ID-117240, Lawrence Livermore National Laboratory (1994).
Anderson, O. Equations of State of Solids in Geophysics and Ceramic Science (Oxford University Press Inc, 1995).
Acknowledgements
This work was performed under the auspices of the U. S. Department of Energy by Lawrence Livermore National Security, LLC under Contract DE-AC52-07NA27344. We gratefully acknowledge support for this research by the Defense Threat Reduction Agency under contract number HDTRA1-11-1-4538I (program managers are Dr. D. Allen Dalton and Dr. Suhithi Peiris). The work at the University of Saskatchewan was supported by Natural Sciences and Engineering Research Council of Canada (NSERC). Y. Yao gratefully acknowledges the Information and Communications Technology group at the University of Saskatchewan Westgrid and Comput Canada for providing computing resource. B.K. acknowledges financial support from LLNL on subcontract number B608474 and additional support provided by The Scientific and Technological Research Council of Turkey fellowship under Contract No: 114C120. The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231. Portions of this research were carried out at the light source PETRA III at DESY, a member of the Helmholtz Association. The research leading to these results has received funding from the European Community’s Seventh Framework Programme (FP7/2007-2013) under grant agreement no. 12284.
Author information
Authors and Affiliations
Contributions
E.S. and J.M.Z. designed the study, J.M.Z., E.S. and B.K. performed the experiments and analyzed the experimental data, Y.Y. performed the first-principles calculations, S.B. performed thermochemical calculations, M.K. and Z.K. performed experiments and contributed to the experimental methods. E.S., J.M.Z. and Y.Y. contributed in writing the manuscript. All authors contributed to interpretation, discussion of the data and revised the paper critically for important intellectual content.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Stavrou, E., Yao, Y., Zaug, J. et al. High-pressure X-ray diffraction, Raman and computational studies of MgCl2 up to 1 Mbar: Extensive pressure stability of the β-MgCl2 layered structure. Sci Rep 6, 30631 (2016). https://doi.org/10.1038/srep30631
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep30631
- Springer Nature Limited